")
Back to Journals » ImmunoTargets and Therapy » Volume 14
Adaptation of Natural Killer Cells to Hypoxia: A Review of the Transcriptional, Translational, and Metabolic Processes
Authors Chang TD, Chen YJ , Luo JL, Zhang C, Chen SY, Lin ZQ , Zhang PD, Shen YX, Tang TX, Li H, Dong LM, Tang ZH, Chen D, Wang YM
Received 21 August 2024
Accepted for publication 8 February 2025
Published 18 February 2025 Volume 2025:14 Pages 99—121
DOI https://doi.org/10.2147/ITT.S492334
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Flavio Salazar-Onfray
Te-Ding Chang,1,2 Yu-Jie Chen,1,2 Jia-Liu Luo,1,2 Cong Zhang,1,2 Shun-Yao Chen,1,2 Zhi-Qiang Lin,1,2 Pei-Dong Zhang,1,2 You-Xie Shen,1,2 Ting-Xuan Tang,3 Hui Li,1,2 Li-Ming Dong,1,2 Zhao-Hui Tang,1,2 Deng Chen,1,2 Yu-Man Wang4,5
1Division of Trauma Surgery, Emergency Surgery & Surgical Critical, Tongji Trauma Center, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 2Department of Emergency and Critical Care Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 3Department of Orthopedics, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China; 4Department of Geriatrics, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China; 5Key Laboratory of Vascular Aging, Ministry of Education, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China
Correspondence: Deng Chen, Email [email protected]; Yu-Man Wang, Email [email protected]
Abstract: As important innate immune cells, natural killer (NK) cells play an essential role in resisting pathogen invasion and eliminating transformed cells. However, the hypoxic microenvironment caused by disease conditions is an important physicochemical factor that impairs NK cell function. With the increasing prominence of NK cells in immunotherapy, there has been a surge of interest in developing biological means through which NK cells may overcome the inhibition caused by hypoxia in disease conditions. Although the effects of hypoxic conditions in shaping the functions of NK cells have been increasingly recognized and investigated, reviews have been scantly. A comprehensive understanding of how NK cells adapt to hypoxia can provide valuable insights into how the functional capacity of NK cells may be restored. This review focuses on the functional alterations of NK cells in response to hypoxia. It delineates the mechanisms by which NK cells adapt to hypoxia at the transcriptional, metabolic, translational levels. Furthermore, given the complexity of the hypoxic microenvironment, we also elucidated the effects of key hypoxic metabolites on NK cells. Finally, this review discusses the current clinical therapies derived from targeting hypoxic NK cells. The study of NK cell adaptation to hypoxia has yielded new insights into immunotherapy. These insights may lead to development of novel strategies to improve the treatment of infectious diseases and cancer.
Keywords: NK cells, hypoxia, immunotherapy, innate immunity, metabolism
Introduction
Natural killer (NK) cells are recognized as indispensable and important innate immune cells.1,2 NK cells develop from bone marrow progenitor cells and are distributed throughout the body in liver, bone marrow and blood.3–8 The number of NK cells has been estimated to be 2×106, comprising 1% of the total immune cell population, 2% of the lymphocyte population, and 10% of the peripheral lymphocyte population.9 As a family of innate lymphocyte cells (ILCs), NK cells are equipped with a repertoire of inhibitory and activating receptors.3 The combined action of these two types of receptors can selectively defense against infected and transformed cells while sparing normal cells.3–8 The sustained maintenance and enhancement of NK cell recognition and functionality have emerged as pivotal to NK cell-based immunotherapies over the past five years.3
However, hypoxia challenges the maintenance of normal NK cell function in various diseases. NK cells exert their functions through the combined action of surface germline-encoded activating and inhibitory receptors.10 The expression of NK cell surface receptors makes them susceptible to hypoxia.11 The equilibrium between NK cell receptor activation and inhibition is disrupted in the hypoxic environment, resulting in a series of hypoxia-induced adaptation in NK cells. The predominant trend is the downregulation of activating receptors, with minimal effects observed on inhibitory receptors (Box 1).11 This imbalance between activating and inhibitory receptors ultimately results in impaired cytotoxicity and cytokine secretion of NK cells in hypoxic conditions.12 The expression of perforin and granzyme in NK cells has been reported to decrease in hypoxia conditions.13 In experiments involving co-cultures of NK cell and tumors, the cytotoxic effect of NK cells on tumor cells was found to be significantly impaired under hypoxic conditions.14 In addition to cytotoxicity, the capacity of NK cells to secrete proinflammatory cytokines (eg, IFNγ and TNFα) and chemokines (eg, GM-CSF, CCL3, CCL5) is also impaired under hypoxic conditions (Figure 1A).15
 |
Box 1 Imbalanced Receptors of NK Cells Under Hypoxia |
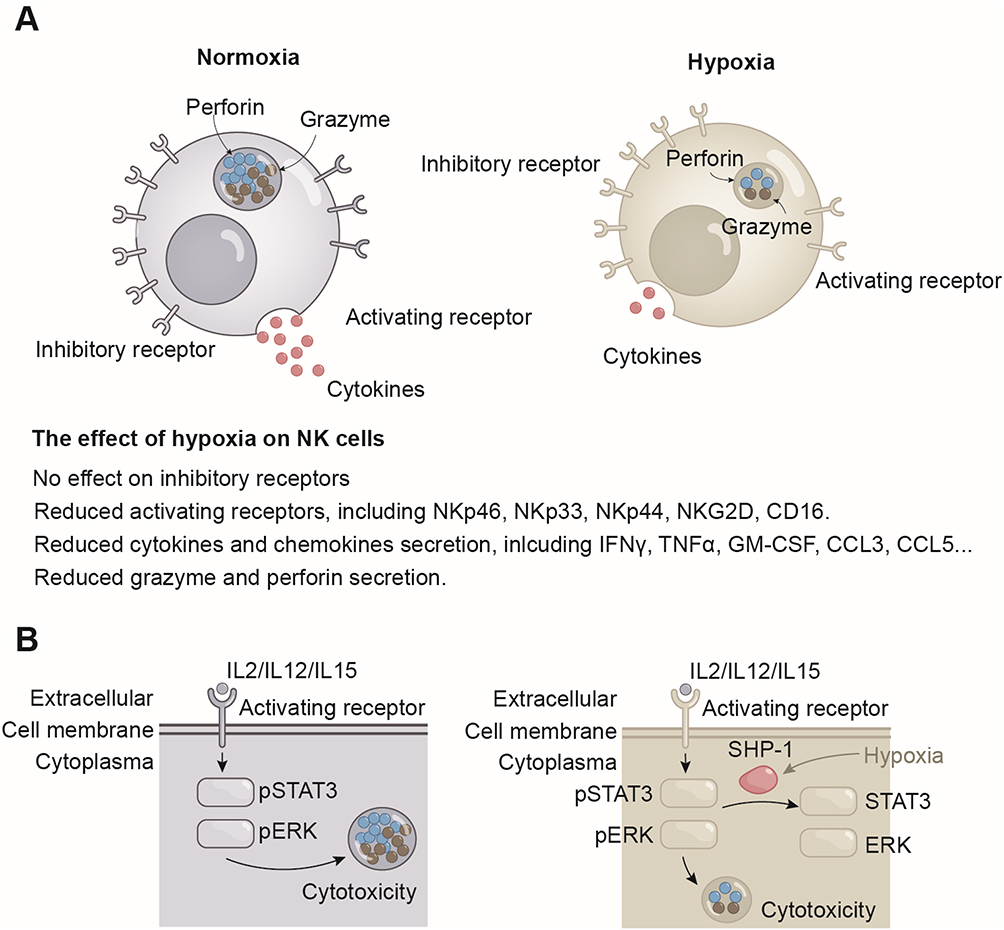 |
Figure 1 Effects of hypoxia on NK cells. (A) The expression of NK cell activating receptors is decreased under hypoxic conditions, whereas inhibitory receptors remain unaffected. Inhibition of activating receptors is associated with a reduction in the secretion of cytokines and chemokines, as well as impaired cytotoxicity in hypoxic NK cells. (B) Hypoxia impairs the cytotoxicity of NK cells by inducing the expression of SHP-1, which dephosphorylates STAT3 and ERK. Abbreviations: IFNγ, Interferon gamma; TNFα, Tumor necrosis factor alpha; GM-CSF, Granulocyte-macrophage colony-stimulating factor; CCL, Chemokine (C-C motif) ligands; STAT3, Signal transducer and activator of transcription 3; ERK, Extracellular signal-regulated kinase; SHP-1, Src homology region 2 domain-containing phosphatase-1; IL, Interleukin. |
A multitude of factors contribute to the reduction of NK cell function in hypoxia (Figure 1B). Many researchers hypothesize that this is a self-protective mechanism of NK cells after oxygen deprivation, whereby energy-expensive processes, including the production and secretion of cytokines, perforin and granzyme, are curtailed.21 Although the effects of hypoxic conditions in shaping the functions of NK cells have been increasingly recognized and investigated, reviews have been scanty, and have largely focused on the tumor microenvironment.22,23 This review provides a comprehensive analysis of the mechanisms by which NK cells adapt to hypoxia. It presents a summary of the relevant transcriptional, translational, and metabolic processes of hypoxic NK cells. Ultimately, it suggests a basis for the pathophysiological mechanisms that may be employed to overcome NK cell inhibition under hypoxic conditions. The methodology of the literature search is partially delineated in Box 2.
 |
Box 2 The Methodology of the Literature Search |
Transcriptional Adaptation to Hypoxia
HIF-1 Mediates NK Cell Adaptation Under Hypoxia
Hypoxia-inducible factors (HIF) are heterodimeric transcription factors that are stabilized under conditions of hypoxia.24 The level of HIF-1α is regulated by oxygen levels, whereas HIF-1β is constitutively expressed in cells.25 Under normoxia, prolyl hydroxylase (PHD) hydroxylates the proline residues (P402 and P564) in the HIF-1α, in conjunction with oxygen (O2), iron (Fe2+) and α-ketoglutarate.26 Hydroxylated HIF-1α is recognized and polyubiquitylated by the pVHL–elongin BC–CUL2 complex (referred to as the VHL complex), which then targeted it for proteasomal degradation.27 In the context of hypoxia, the process of hydroxylation mediated by PHD is inhibited, resulting in the failure of HIF-1α to undergo degradation.26 Stabilized HIF-1α binds to HIF-1β to form the heterodimer HIF-1. HIF-1 translocates into the nucleus and binds to hypoxia-responsive elements (HRE), thereby initiating transcription of a range of genes, including those involved in cell survival, angiogenesis, glycolysis, and invasion/metastasis (Figure 2A).21 In NK cells, the stabilization of HIF-1α is not solely contingent on the dysfunction of PHD due to hypoxia. It is also dependent on cytokine induced HIF-1α expression.28,29 The IL-2 and IL-15 signaling pathways promote the translation of HIF-1α through the phosphoinositide 3-kinases / mechanistic target of rapamycin complex 1 (PI3K/mTORC1) signaling pathway.28,29 Cytokines initiate the synthesis of HIF-1α, whereas hypoxia stabilizes the HIF-1α protein, ultimately allowing for the accumulation of HIF-1α in NK cells.
 |
Figure 2 Transcriptional alterations in NK cells under hypoxic conditions. (A) PHD hydroxylates HIF-1α in the presence of oxygen, iron ions, and α-ketoglutarate. Hydroxylated HIF-1α is polyubiquitylated by pVHL. Ubiquitinated HIF-1α is degraded by the proteasome. In contrast, under hypoxic conditions, the deprivation of oxygen results in the inhibition of the hydroxylation of HIF1α. HIF-1α is stabilized and translocates to the nucleus, where it forms a heterodimer with HIF-1β. (B) Under normoxia, NK cells promote the expression of cMyc and HIF-1α following cytokine activation. However, because HIF-1α is degraded by the proteasome, NK cells effector functions, primarily through cMyc. Conversely, under hypoxic conditions, stabilized HIF-1α competes with cMyc for binding to target genes. Specifically, in the context of normoxia, the cMyc/Max complex binds to Sp1, thereby promoting the expression of CDC25A, MSH2, and NBS1. In hypoxic conditions, HIF-1α competitively binds to Sp1, thereby inhibiting the expression of these genes. The cMyc/Max complex is able to repress transcription of the CDK1A gene via Mzi. Nevertheless, this inhibition can be overcome by the competitive binding of HIF-1α to Mzi. Abbreviations: HIF, Hypoxia inducible factor; PHD, Prolyl hydroxylases; pVHL, Hippel-Lindau tumor suppressor protein. CDC25, Cell division cycle 25; MSH2, MutS Homolog 2; NBS1, Nibrin; CDKIA, Cyclin-dependent kinase inhibitor gene. |
cMyc Mediate NK Cell Function Under Normoxia
Among the numerous transcription factors that influence NK cells, cMyc is the most intricately and closely associated with HIF-1. The two transcription factors share common target genes. cMyc is an important transcription factor whose function in normoxia is very similar to that of HIF-1 in hypoxia. Both transcription factors ultimately modulate NK cell function by regulating NK cell metabolism and protein synthesis.30 The cMyc to HIF1a transition reprograms NK cell metabolism, protein synthesis, and cell cycle progression at the transcriptional level, thereby fine-tuning the adaptive response of NK cells to hypoxic environments.31
cMyc is a pivotal transcription factor that plays a crucial role in regulating a multitude of cellular processes, including cell cycle progression, cell proliferation, apoptosis, cell metabolism, and cell transformation.32,33 cMyc inhibits the expression of genes that suppress cell proliferation, thereby facilitating the proliferation of cells.34,35 For example, cyclin-dependent kinase inhibitor gene (CDKN1A), as a pivotal cell cycle inhibitory gene, encodes the expression of cell-cycle inhibitor p21.cip1 CDKN1A is repressed by cMyc through the formation of a ternary repressive complex with Max and Mzi, which binds to the initiator element (INR) of the growth-suppressor gene.34,35 In addition, cMyc plays a role in promoting cell proliferation by increasing the expression of genes involved in DNA damage repair and cell cycling. In the absence of Mzi, the cMyc/Max complex binds directly to Sp1 to form a heterotrimer. This heterotrimer relies on Sp1 subunit to bind to the GC-rich regions of DNA damage repair and cell cycling genes, thereby promoting their expression.36 Furthermore, cMyc/Max directly binds to the E-box region of these genes, thereby promoting their expression.36 The transcription factor cMyc promote cell proliferation by inhibiting the expression of genes that inhibit cell cycle progression and by facilitating the transcription of genes that involved in cellular proliferation, mitochondrial biosynthesis, rRNA and protein biosynthesis, glycolytic and oxidative phosphorylation (Figure 2B).32
In the context of normoxia, cMyc, but not HIF-1α, is essential for the activation of NK cells induced by cytokines. The expression of cMyc promotes the secretion of IFNγ, perforin and granzyme by NK cells.37,38 cMyc is a pivotal transcription factor in the cytokine-induced metabolic reprogramming of NK cells.38 The level of cMyc is found to increase during the stimulation of NK cells with IL2/IL12.38 Elevated cMyc levels stimulate glycolysis and oxidative phosphorylation in NK cells, thereby orchestrating metabolic reprogramming of NK cells under normoxia.38 This metabolic reprogramming would prompt NK cells to exert effective cytotoxic effects and IFNγ secretion.38 In contrast, the absence of HIF-1α under normoxia did not affect the expression of IFNγ and the secretion of perforin and granzyme B in NK cells.38 This indicates that cMyc is the dominant factor under normoxic conditions. However, under hypoxic conditions, HIF-1α interacts with cMyc, gradually gaining dominance over NK cells under hypoxia.31,39
Transcriptional Adaptation of NK Cells to Hypoxia is Manifested in the Interaction of HIF-1 with cMyc
Although cMyc is a transcriptional center that controls NK cell proliferation and development under normoxic conditions, its activity is progressively replaced by HIF-1α under hypoxic conditions.31,39 HIF-1α has been demonstrated to play a pivotal role in the regulation of NK cell survival, metabolism, and adaptation.31,39 HIF-1α exerts its inhibitory effect on cMyc through various distinct pathways.31,39 For instance, HIF-1α inhibits the transcription of cMyc target genes by competitively binding to Sp1, thereby displacing the cMyc/Max complex from Sp1.40 HIF-1α is also capable of binding to Mzi1, which results in the displacement of the cMyc/Max complex and the removal of the inhibition of cell cycle inhibitory proteins (CDKN1A). This shift ultimately contributes to the slowed proliferation of NK cells and cell cycle arrest under hypoxic conditions. The HIF-1α target gene Mix also inhibits cMyc.41 In response to prolonged hypoxia, HIF-1 induces the production of Mix, which forms a complex with Max and binds to the E-Box, the binding site of cMyc on DNA. This blocks the binding of cMyc to the E-box, inhibiting the transcriptional activity of cMyc.41–43 Notably, this inhibitory effect of HIF-1α on cMyc is reversible. Upon restoration of oxygen levels to normal, cMyc function will also gradually recover.31 The interaction between cMyc and HIF that contributes to transcriptional adaptation of NK cells under hypoxic conditions (Figure 2B).
After dominance, HIF-1, which is primarily involved in transcriptional processes, is responsible for the induction of a multitude of hypoxic adaptive responses in NK cells. The impairment of NK cell cytotoxicity and antimicrobial efficacy induced by hypoxia is correlated with the activity of HIF-1. In response to IL-18 stimulation, NK cells are activated to exert cytotoxicity. However, hypoxia-induced accumulation of HIF-1 impairs the ability of NK cells to secrete perforin, granzyme, and IFNγ.44 HIF-1α-deficient NK cells unleash cytokine secretion function and cytotoxicity in response to IL-18, thereby limiting tumor volume.44 Nevertheless, the impact of HIF-1 on NK cells varied across different models. In the wound model, accumulation of HIF-1 under hypoxic conditions is a key driver of IFNγ secretion by NK cells.45 This strikingly disparate outcome may be linked to the pathophysiological processes underlying wound and tumorigenesis. This suggests the presence of an unknown factor that can reverse the inhibitory effect of HIF-1 on NK cells under hypoxic conditions.
In addition to their role in tumor cell cytotoxicity, NK cells affect tumor volume by interfering with angiogenesis in solid tumors. Research indicates that NK cells represent a significant source of secreted vascular endothelial growth factor receptor1 (sVEGFR1) in solid tumors.45,46 sVEGFR1 binds to VEGF, thereby exerting a negative regulatory effect on VEGF bioavailability.47–49 Such inhibition disrupts the balance between angiogenic and angiostatic factors during tumor cell induced angiogenesis.46 Imbalanced angiogenesis results in the formation of a large network of non-functional blood vessels within the tumor.46 Non-productive angiogenesis leads to an inadequate oxygen supply to tumor cells, which in turn limits tumor volume.46 HIF-1α promotes sVEGFR1 secretion by NK cells under hypoxic conditions.45,46 The absence of HIF-1α will impair the secretion function of NK cells, resulting in a reduction in sVEGFR1.46
In addition to its impact on NK cell functionality, HIF-1α severs as a vital transcription factor that protects NK cells from apoptosis during infection.50 HIF-1α is specifically elevated in NK cells during infection. The target genes of HIF-1α inhibit the expression of the pro-apoptotic protein BCL2/adenovirus E1B 19 kd-interacting protein (BNIP), thereby inhibiting apoptosis.50 Despite the substantial proliferation of NK cells observed in the presence of HIF-1α deficiency, the number of NK cells still declines.50 This decline is closely associated with unrestrained cell death.50
In summary, the transcriptional levels of NK cells under hypoxic conditions undergo adaptive changes. HIF-1 replaced cMyc as the major transcription factor for mobilizing metabolism and protein synthesis after NK cell activation. However, the contrasting results prompts the complexity of the effects of HIF-1α on NK cells. Further investigation of HIF-1α-mediated adaptation of NK cells to hypoxia becomes imperative.
Immunometabolism Reprogramming in Hypoxia
Immunometabolism in NK Cell
Immunometabolism describes changes in intracellular metabolic pathways during immune cell activation.51 NK cell functions are closely correlated with intrinsic cellular Immunometabolism.30 In resting NK cells, the levels of oxidative phosphorylation (OXPHOS) and glycolysis were maintained at baseline.52 This baseline rate of OXPHOS and glycolysis was adequate for the acute phase response of NK cells.30 Rates of OXPHOS or glycolysis were not elevated in NK cells after 4 hours of cytokine stimulation (IL15/IL15+IL12/IL15+IL18) or after 6 hours of receptor ligation (anti-NK1.1/anti-Ly49D).52 However, although stimulation did not increase metabolic activity, inhibition of glucose/glycolysis or overall OXPHOS inhibition resulted in almost complete elimination of receptor-stimulated IFN-γ production.52 This demonstrates the importance of the baseline metabolic levels in sustaining the acute response of NK cells. Growing evidence indicates that NK cells not only function in the acute phase, but also play an essential role in the prolonged immune response. In the overnight cytokine stimulation assay in vitro and the overnight pathogen infection assay in vivo, basal OXPHOS and glycolysis were significantly increased in NK cells.53 In addition, overnight activated NK cells exhibited higher expression of nutrient transporter receptors (amino acid transporter CD98 and transferrin receptor CD71) and higher glucose uptake than resting NK cells.53,54 Overall, the immune metabolism of NK cells is intricately associated with their function.
Suppression of OXPHOS by Hypoxia Impairs NK Cell Function
Given the irreplaceable position of oxygen in the production of intracellular ATP and as an electron acceptor in various biochemical reactions serving, it is not surprising that the metabolic reprogramming of NK cells is intricately related to the oxygen supply. The main metabolic approaches of NK cells are OXPHOS and glycolysis, both of which are closely related to hypoxia.53 OXPHOS represents the most efficient metabolic approach to producing ATP. There are two distinct patterns of metabolism that differ from glycolysis: the tricarboxylic acid cycle (TAC) and the electron transport chain,55 both of which are dependent on the normal function of mitochondria.
Hypoxia impairs OXPHOS through the inhibition of the TAC. The conversion of pyruvate to acetyl coenzyme A (acetyl-CoA) by pyruvate dehydrogenase (PDH) represents the initiation of TAC.21,56 HIF-1-induced pyruvate dehydrogenase kinase 1 (PDK1) impairs the pyruvate-acetyl-CoA transition by phosphorylating and inactivating the catalytic subunit of PDH.21,56 Furthermore, the HIF-1 protein was found to induce an increase in the activity of lactate dehydrogenase A.21,57–60 LDHA catalyzes the conversion of pyruvate to lactate, which subsequently impairs the conversion of pyruvate to acetyl-CoA. HIF-1 induced the expression of PDK1 and LDHA, which have ability to inhibit OXPHOS and promote glycolysis. Glycolysis is another important metabolic pathway for energy replenishment in the event of an imbalance between cellular oxygen supplies and metabolic demands.21,58–60 In the hypoxic environment, the inhibited OXPHOS will result in a reduction in the secretion of cytokines and cytotoxicity (Figure 3A).52
 |
Figure 3 The effects of hypoxia on the metabolism and mitochondrial morphology of NK cells. (A and B) Hypoxia inhibits the OXPHOS process from both the TAC cycle (A) and the ETC (B). Hypoxia stabilizes HIF-1, which in turn transcribes PDK1 and LDHA. PDK1 dephosphorylates PDH, thereby reducing the conversion of pyruvate to Acetyl-CoA. The expression of LDHA facilitates the conversion of pyruvate to lactate, which subsequently impairs the production of Acetyl-CoA. In essence, hypoxia impairs the TAC cycle by reducing the production of Acetyl-CoA (A). Oxygen is a crucial substrate for complex IV. In the context of hypoxia, electrons transferred to complex IV lack acceptor, resulting in an increased production of ROS. Hypoxia also induces the production of miR-210, which represses the transcription of complex I, II, and III. The inhibition of these three complexes has the effect of reducing the efficiency of electron transfer through the ETC (B). (C and D) Hypoxia-induced mitochondrial fragmentation inhibits OXPHOS in NK cells. NK cells possess large, tubular and densely packed mitochondrial. In contrast, the mitochondrial of NK cells under hypoxia become smaller, fragmented and distinct. The fragmentation of mitochondrial not only reduces the level of OXPHOS but also releases pro-apoptotic proteins that promote apoptosis in NK cells (C). The mechanism by which hypoxia regulates mitochondrial fragmentation is related to the activity of the DRP1 and FIS1. The inhibition of DRP1 by mTORC1 was found to be deregulated under hypoxic conditions. Activated DRP1 is recruited to the mitochondrial periphery and binds to FIS1, thereby inducing mitochondrial fragmentation (D). (E) Hypoxia inhibits mTORC1 through the protein Rhed. The lysosomal membrane translocation is a crucial step in the activation of mTORC1 and Rhed is a crucial protein that facilitates the translocation of mTORC1 to the surface of the lysosomal membrane. Inhibition of mTORC1 has a profound impact on multiple signaling pathways in NK cells, resulting in a reduction in their cytotoxicity and cytokine secretion. Abbreviations: LDHA, lactate dehydrogenase A; PDH, pyruvate dehydrogenase; PDK1, pyruvate dehydrogenase kinase 1; Acetyl-CoA, acetyl coenzyme A; TAC, tricarboxylic acid; OXPHOS, oxidative phosphorylation; HIF, hypoxia inducible factor; NDUFA4L2, NADH dehydrogenase (ubiquinone) 1 α subcomplex, 4-like 2; CoQ, coenzyme Q; NADH, Nicotinamide adenine dinucleotide; FADH2, flavin adenine dinucleotide; ATP, Adenosine triphosphate; AMP, Adenosine monophosphate; Cyt C, cytochrome C; OCR, Oxygen consume rate; Cas3, Caspase 3; DRP1, dynamin-related protein 1; FIS1, fission 1; SIAH2, seven in absentia homolog 2; mTORC1, mammalian target of rapamycin complex 1; AKAP121, A kinase anchoring protein 121; PKA, Protein kinase A; TSC, Tuberous sclerosis; PDK1, pyruvate dehydrogenase kinase 1; PIP3, Phosphatidylinositol (3,4,5)-trisphosphate; PIP2, Phosphatidylinositol 4.5-bisphosphate; PI3K, Phosphoinositide 3-kinases; mLST8, mammalian lethal with sec-13 protein 8; RAPTOR, regulatory-associated protein of mTOR; AMPK, AMP-activated protein kinase; REDD1, Regulated in development and DNA damage-response 1; Rheb, Ras homolog enriched in brain; BNIP3, Bcl-2/adenovirus E1B 19 kDa-interacting protein 3. |
At an intracellular oxygen level of 0.3%, ETC activity begins to be impaired.61 Complex 4 (also known as cytochrome c oxidase or COX) is most intimately associated with oxygen within ETC. It contributes 4 electrons to oxygen and produces 2 molecules of water.62 It is noteworthy that the suppressive impact of hypoxia on ETC did not result from a decrease in the level of oxygen as a substrate. Rather, it was mediated by a HIF-1-dependent mechanism.63 Specifically, hypoxia induces the up-regulation of the mitochondrial NADH dehydrogenase (ubiquinone) 1α subcomplex, 4-like 2 (NDUFA4L2) gene expression via HIF1. It has been demonstrated that NDUFA4L2 suppresses complex I activity.64 In addition, hypoxia induces miRNA (mir-210) expression through HIF-1, which represses iron-sulfur cluster assembly enzyme 1 (ISCU1) and ISCU2. These factors are pivotal in the assembly of iron-sulfur clusters within Complex I, II and III.65 Additionally, mir-210 has been demonstrated to suppress the expression of NDUFA4, succinate dehydrogenase complex subunit D (SDHD), and COX10, which are crucial components in the formation and assembly of ETC-associated complexes (Figure 3B).66,67
Hypoxia-Induced Mitochondrial Fragmentation Limits NK Cell Function
Mitochondria, as the primary organelles involved in metabolic processes, play a pivotal role in the functionality of NK cells. Normoxic NK cells exhibit large, tubular, and densely packed mitochondrial, whereas hypoxic NK cells display small, fragmented, and distinct mitochondrial within the cytoplasm.68 The morphology of mitochondrial fragmentation has been demonstrated to limit the secretion of granzyme B by NK cells, as well as anti-tumor function and survival.68 The oxygen consumption rate (OCR) of mitochondrial respiration is standard method for assessing the level of OXPHOS.69 In hypoxic NK cells with fragmented mitochondria, the basal OCR level is observed to be lower than that of normal NK cells.68 The basal and maximal OCR of hypoxic NK cells can be restored by the use of the mitochondrial fragmentation inhibitors, such as mdivi-1 or Dyn.68 Furthermore, unbiased metabolomics profiling of purified hypoxic and normoxic cultured NK cells revealed that the inhibition of TAC-related metabolic intermediates under hypoxia could be alleviated by mitochondrial fragmentation inhibitors (Figure 3C).68
Hypoxia-induced mitochondrial fragmentation represents a critical initiating event in the apoptotic process of NK cell. Mitochondrial release of sequestered pro-apoptotic proteins, which are stored in the intermembrane space, results in their translocation into the cytoplasm.70 The release of pro-apoptotic proteins, including cytochrome c, Smac/DIABLO, and serine protease HtrA2/Omi, initiates the apoptotic process, ultimately leading to cell death.70 Caspase 3 is the most significant executor of apoptosis. The intrinsic pathway, the extrinsic cellular pathway, and the perforin/granzyme pathway collectively induce apoptosis via caspase 3.70 The number of NK cells was found to be significantly reduced in the hypoxic microenvironment of the tumor. The level of cleaved caspase 3 in hypoxic NK cells has been observed to increase.68 In contrast, the number of NK cells in the hypoxic microenvironment of the tumor increased following the use of the mitochondrial fragmentation inhibitor mdivi-1.68
Hypoxia-induced mitochondrial fragmentation is associated with the mitochondrial protein fission 1 (FIS1) signaling pathway.71 FIS1, which localizes to the outer mitochondrial membrane, is a pivotal receptor that promotes mitochondrial fragmentation.72 The GTPase dynamin-related protein 1 (DRP1) functions as a ligand for FIS1, thereby activating FIS1 to facilitate mitochondrial fragmentation.72 In the context of hypoxia, DRP1 is recruited to the periphery of the outer mitochondrial membrane by FIS1.71 DRP1’s activating site, Ser616, is phosphorylated in hypoxic NK cells.72 The phosphorylation of Ser616 in DRP1 has been demonstrated to be associated with the activation of mechanistic target of rapamycin (mTOR) signaling. Inhibition of the mTOR signaling pathway results in a significant reduction in the level of phosphorylated DRP1Ser616 in NK cells.21,68,72
In addition to the facilitated activation of mTOR, the deregulation of the inhibitory effect on DRP1 is also a critical step in facilitating the binding of DRP1 to FIS1. In the presence of sufficient oxygen, Scaffolding protein A kinase anchoring protein 121 (AKAP121) inhibits DRP1 through a cAMP-regulated protein kinase A73 -mediated phosphorylation process.72 The inhibition is alleviated in hypoxic environments by seven in absentia homolog 2 (SIAH2).74,75 Hypoxia-induced SIAH2, an E3 ubiquitin ligase, mediates the ubiquitination and proteasome-mediated degradation of AKAP121.74,75 The degradation of AKAP121 results in the de-repression of DRP1, which in turn promotes the DRP1-FIS1-mediated mitochondrial fragmentation (Figure 3D).
Overall, hypoxia activates DRP1 through two distinct mechanisms: mTOR and SIAH2. The activation of DRP1 results in the fragmentation of mitochondria through binding of FIS1, which ultimately leads to a reduction in the functionality and an increase in the death of NK cells.
mTOR is an Important Molecule in Hypoxia-Regulated NK Cell Metabolism
The mammalian target of rapamycin (mTOR) is a vital cellular metabolic checkpoint kinase that primarily promotes anabolic metabolism, including ribosome, RNA, and protein synthesis. Conversely, it inhibits catabolic metabolism, such as autophagy.76 mTOR is a component of two complexes, each with a unique structure and functions.76 mTOR complex 1 (mTORC1) is a heterotrimeric complex comprising regulatory-associated protein of mTOR (RAPTOR), mammalian lethal with sec-13 protein 8 (mLST8), and mTOR. The mTOR complex 2 (mTORC2) is a heterotetramer comprising the mLST8 and mTOR subunits, as well as the rapamycin insensitive companion of mTOR (RICTOR) and the mammalian stress activated MAPK-interacting protein 1 (mSIN1).77 mTORC1 rather than mTORC2, is responsible for regulating the development and activation-induced metabolic reprogramming of NK cells.
Coincidentally, hypoxia play a crucial role in the inhibition of mTORC1. mTORC1 activation is lysosome-dependent. Facilitating the translocation of mTORC1 to the lysosome necessitates the involvement of two classes of Ras-associated small G proteins: amino-acid-induced Rag GTPase and Rheb GTPase.78 Hypoxia exerts its inhibitory effect on mTORC1 primarily through the inhibition of Rheb activity.21 Rheb were found to be inhibited by the Tuberous sclerosis (TSC) complex in quiescent cells.78 The TSC complex is comprised of TSC1, TSC2, and TBC1 domain family member 7 (TBC1D7).78 In activated cells, the phosphoinositide 3-kinases (PI3K) signaling pathway is activated, resulting in the phosphorylation of downstream AKT.78 p-AKT then phosphorylates the TSC complex, thereby depriving the TSC complex’s inhibition of Rheb.78 In the absence of any inhibitory factors, Rheb will recruit mTORC1 for transfer to the lysosome, which ultimately leads to the activation of mTORC1.78 AMP-activated protein kinase (AMPK) is activated under hypoxia.79 The activation of AMPK will result in the inhibition of Rheb, rather than TSC.79 In addition, hypoxia has been demonstrated to induce the up-regulation of REDD1.80 Up-regulated REDD1 results in the release of TSC2 from its chaperone proteins 14-3-3.80 TSC2 then recombines with TSC1 and TBC1D7 to form the TSC complex, which inhibits Rheb activity.80 Bcl-2/adenovirus E1B 19 kDa-interacting protein 3 (BNIP3), a protein induced by HIF-1, was also found to bind directly to Rheb, thereby sequestering Rheb away from mTORC1.81 In hypoxic cells, the increase in glycolytic metabolism results in an elevation of lactate levels and the formation of an acidic environment. In such conditions, perinuclear lysosomes undergo a redistribution.82,83 Perinuclear lysosomes move away from perinuclear Rheb, thereby rendering lysosome-bound mTORC1 inactivation.21,82,83 Collectively, hypoxia affected the activity of mTORC1 by inhibiting Rheb. Inhibition of mTORC1 will compromise NK cell function (Figure 3E).
Translation Adaptation to Hypoxia
Translation represents a pivotal stage in the process by which NK cells accumulate effector proteins and receptors. mRNA is responsible for assembling genetic information into proteins through translation, thereby enabling NK cells to perform immune activity. The overall translation process is divided into three distinct phases: initiation, elongation and termination. In the context of hypoxic stress, cells experience a lack of nutrients and disrupted signaling, which in turn leads to increase in the production of unfolded and misfolded proteins. This, in turn, triggers endoplasmic reticulum stress. Hypoxia has been demonstrated to disrupt translation, resulting in imbalanced expression of NK cell receptors and inhibition of cytokine secretion.
Inhibition of Translation Initiation in NK Cells Under Hypoxic Conditions
The initiation of translation involves recruitment of the ribosomal 40S subunit, followed by the recognition of the mRNA’s AUG promoter by tRNAs. Among them, Eukaryotic initiation factor 2 (eIF2) is a crucial factor that mediates tRNA-mRNA binding and is one of the targets of hypoxia-regulated translation initiation. eIF2 has been shown to correlate with the expression of NKG2D, an activating receptor for NK cells.84 It has been demonstrated that the expression of NKG2D on the membrane of NK cells is upregulated following the activation of eIF2.84 However, eIF2 is subject to extensive inhibition under hypoxic conditions. The main receptor of the unfolded protein response, protein kinase RNA-like endoplasmic reticulum kinase (PERK), acts as a kinase and phosphorylates eIF2 to prevent transcription initiation.85,86 In the context of hypoxia, the accumulation of unfolded proteins leads to the activation of PERK, which in turn phosphorylates and inhibits eIF2 activity.85,86
In addition to the eIF2-mediated assembly of the 43S subunit binding complex, the recruitment of mRNA by the eukaryotic translation initiation factor 4F (eIF4F) complex is equally important for the initiation of protein translation.87 The eIF4F complex plays a crucial role in cap-dependent translation.87 eIF4F consists of the cap-binding protein eIF4E, the DEAD-box RNA helicase eIF4A, and the large scaffold protein eIF4G.88 It has been demonstrated that the production of IFNγ in NK cells is correlated with eIF4F.89 Hypoxia inhibits eIF4E by down-regulating the mTORC1 pathway, which ultimately affects eIF4F formation and inhibits the transcriptional initiation process.88 The mTORC1 signaling pathway plays a pivotal role in the formation of the eIF4F complex. mTORC1 inactivates 4E-BPs by phosphorylating them, thereby deregulating the inhibitory effect of 4E-BPs on eIF4E.90 In hypoxic conditions, mTORC1 is inhibited, which prevent eIF4E from dissociating from 4E-BPs.90 This sustained inhibition of eIF4E results in the failure of the eIF4F complex assembly, which ultimately leads to the inhibition of translation initiation.90 During prolonged hypoxia, the transporter 4-ET blocks eIF4E in the nucleus, which impedes the formation of eIF4F.90 In essence, the failure of eIF4F formation impedes the secretion of IFNγ by NK cells, as evidenced by a reduction in IFNγ translation under hypoxic conditions (Figure 4A).
 |
Figure 4 Effect of hypoxia on the translation process in NK cells. (A) During the initiation of translation, the inhibitory effect of hypoxia on mTORC1 prevents the release of eIF4E from its binding protein, 4E-BP. This results in the impaired formation of the eIF4F, which is important in translation initiation. In the context of hypoxia, the eIF2, which binds to tRNA, is inhibited by PERK. (B) During the transcription elongation phase, hypoxia invalids the inhibition of eEF2K by PHD2 and mTORC1. eEF2K inhibits eEF2, which is an important factor in transcription elongation. (C) eRF1 plays a pivotal role in the termination of transcription. It is inhibited under hypoxic conditions. (D) The production of large quantities of misfold proteins in response to hypoxia initiates the UPR pathway within the ER. The UPR comprises three receptors: The PERK, IRE1α, and ATF6. All three types of receptors bind to BIP and prevent its signaling activity. BIP binds to misfold proteins when misfold protein loading is increased. This results in the release of three receptors. Activated PERK represses eEF2 and promotes ATF4 transcription factor expression. Upon activation, IRE1α will cleave XBP1 to form XBP1s, which possesses transcriptional activity. Following its activation, ATF6 is transported to the Golgi apparatus. ATF6 in the Golgi is cleaved to form transcriptionally active ATF6f. The target genes of these transcription factors ultimately inhibit the translation process, thereby reducing the levels of endoplasmic reticulum misfold proteins. Abbreviations: eIF, Eukaryotic initiation factor 2; eEF, Eukaryotic translation elongation factor; RF, Release factor; A, Aminoacyl site; P, Peptidyl site; E, Exit site; eEF2K, Eukaryotic elongation factor 2 kinase; mTORC1, Mammalian target of rapamycin complex 1; PDH2, pyruvate dehydrogenase 2; JMJD4, Jumonji domain-containing 4; PERK, Protein Kinase RNA (PKR) -like Endoplasmic Reticulum Kinase; IRE1α, Inositol Requiring Enzyme 1; ATF, Activating Transcription Factor 6; UPR, Unfolded protein responses. ER, Endoplasmic reticulum. |
Inhibition of Translation Elongation in NK Cells Under Hypoxic Conditions
The elongation phase of translation is dependent upon the eukaryotic elongation factor 2 (eEF2). eEF2 plays an important role in the translocation of elongating ribosomal.91,92 The elevated expression of eEF2 in NK cells during the early stages of endotoxemia suggests a correlation between eEF2 and the functional capabilities of NK cells.93 eEF2 kinase (eEF2K), an important kinase negatively regulating eEF2, phosphorylates Thr56 of eEF2 and inhibits the translation elongation process.91,92,94,95 eEF2K is known to be normally subject to two factors, mTORC1 and PHD2, which are themselves subject to hypoxia. mTORC1 is responsible for the degradation of eEF2K via the proteasomal pathway. However, in the context of hypoxia, mTORC1 is inhibited.96 It has been demonstrated that PHD2 is capable of degrading eEF2K via a proteasomal degradation mechanism, in conjunction with the presence of Fe2+, O2, and α-ketoglutarate.97,98 This process is analogous to the degradation of HIF-1α. In the context of hypoxia, eEF2K is stabilized by the loss of PDH2-mediated proteasomal degradation, which occurs due to oxygen deprivation.26 The stabilization of eEF2K results in the inactivation of eEF2, which ultimately impedes the elongation process of translation.
Intriguingly, eEF2K functions as a translation elongation repressor. However, for PD-L1, eEF2K emerged as a pivotal activator of its translation.99 The discrepancy in the translation results in the overexpression of PD-L1 on the surface of tumor cells under hypoxic conditions, thereby rendering it challenging for NK cells to recognize and eliminate tumor cells (Figure 4B).
Inhibition of Translation Termination in NK Cells Under Hypoxic Conditions
The translation termination phase requires the involvement of eukaryotic translation termination factor 1 (eRF1).100 eRF1 is the only class 1 eRF that recognizes all three stop codons.101 eRF1 recognizes stop codons and facilitates the hydrolysis of ester bonds in polypeptide chains with peptidyl-site tRNAs.101 This process of hydrolysis releases newly synthesized proteins from the ribosome and dissociates the ribosome and mRNA complex.101 Hydroxylation of eRF1 is a crucial step in the process of translation termination.102 Jumonji domain-containing 4 (JMJD4) is a crucial oxidase responsible for hydroxylating eRF1.102 Under hypoxic conditions, JMJD4 is inhibited, and eRF1 fails to recognize the stop codon, resulting in protein termination failure and increased production of mismatched proteins.102 Despite the absence of definitive evidence regarding the impact of hypoxia on the translation termination process in NK cells, it cannot be ruled out that hypoxia may inhibit the translation termination process in NK cells by affecting eRF1, which in turn affects NK cell protein synthesis (Figure 4C).
Hypoxia Induced Endoplasmic Reticulum Stress in NK Cells
Endoplasmic reticulum (ER) is of critical importance for the regulation of multitude of intracellular physiological process including protein folding and calcium homeostasis.103,104 Hypoxia and other cell stress results in a multitude of protein modifications, including protein oxidation, nitrosylation, and carbonylation.105 This exceeds the capacity of the ER for protein synthesis, resulting in an accumulation of unfolded proteins.106 In hypoxic conditions, the accumulation of misfolded and unfolded proteins can trigger the unfolded protein responses (UPR), a cellular mechanism that alleviates ER stress.105 The activation of the UPR has been demonstrated to alleviate ER stress through the attenuation of overall protein synthesis, ER-associated degradation pathway and autophagy mediated by PERK (Protein Kinase RNA (PKR) -like Endoplasmic Reticulum Kinase), IRE1α (Inositol Requiring Enzyme 1), and ATF6 (Activating Transcription Factor 6) (Figure 4D).107
The activation of IRE1 is a hallmark of NK cells that must constitutively deal with a high demand for protein synthesis, folding and secretion.37,108 The substrate transcription factor for IRE1 is XBP1.109,110 Upon activation of IRE1, XBP1 is cleaved to transcriptionally active XBP1s.109,110 XBP1s translocates to the nucleus and promotes NK cell proliferation and expansion.109,110 The proliferation of NK cells induced by XBP1s appears to be partially correlated with cMyc.37 However, as previously described, cMyc is antagonized by HIF1 under hypoxic conditions. This antagonism may have partially impaired the proliferation of NK cells induced by XBP1s. In addition to its role in NK cell proliferation, the IRE1α and its substrate XBP1 signaling pathway is also crucial for maintaining the migration, immune synapse formation and cytokine release of NK cells (Figure 4D).111
Upon an increase in the concentration of misfolded proteins, PERK is activated by oligomerization and auto-phosphorylation, which is due to the dissociation of immunoglobulin binding protein (BiP).112 Activated PERK phosphorylates eIF2α, which results in inhibition of translation.113 In the context of translational repression, the level of PERK-induced activating transcription factor 4 (ATF4) remains elevated.114 This phenomenon can be attributed to the presence of an internal ribosome entry site (IRES) within the ATF4 gene.115,116 Genes containing IRES are subject to selective enhancement upon eIF2α phosphorylation.117
The activation of ATF6 is initiated by the accumulation of unfolded peptide fragments within the cell, which subsequently leads to the translocation of ATF6 to the Golgi apparatus.118 Within the Golgi apparatus, ATF6 is cleaved by proteases to form the active ATF6f form.112 The N-terminal structural domain of ATF6f contains an alkaline leucine zipper structural domain that binds to DNA and promotes the expression of target genes in response to unfolded proteins.119
Although the direct effects of ER stress on NK cells remain poorly understood, it is clear that ER stress plays an important role in regulating NK cell responsiveness.120 NK cells exhibited a selective recognition of infected cells that had induced ER stress, while ignoring infected cells without ER stress.120,121 The cytolytic activity of NK cells is driven through ER stress and is particularly dependent on the activation of ATF6 and PERK downstream of ER stress.121 ATF6 is responsible for the up-regulation of key hub proteins in the ER proteostasis network, including BiP and calreticulin.122 Calreticulin, an ER lectin chaperone, has been demonstrated to bind to NKp46 and to promote cytotoxicity in NK cells.120 In order to make contact with NK cells, calreticulin needs to be externalized to the cell surface. The stimulation of ER chaperone externalization is correlated with PERK.123 In conclusion, the activation of ATF6 and PERK is responsible for the presentation of calreticulin at the cell surface. The presentation of calreticulin on the cell surfaces activates NKp46 and promotes NK cytotoxicity.
Hypoxic Metabolites Promote NK Cell Adaptation
In addition to the effects of hypoxia on NK cells, it is important to consider the impact of hypoxic metabolites. Lactic acid, adenosine, ROS, and other by-products of cellular metabolism in hypoxia can significantly affect NK cells and promote their adaptation to hypoxia (Figure 5).
 |
Figure 5 Effects of hypoxic metabolites on NK cells. In the context of hypoxia, there is a shift in cellular metabolism from oxidative phosphorylation to glycolysis. (A) This is accompanied by an increase in intracellular lactate levels. Normal lactate efflux from NK cells is inhibited by elevated lactate concentrations in the intracellular compartment. The accumulation of lactate in the cytosol results in the inhibition of glycolysis in NK cells. Inhibition of glycolysis results in an elevation of NADPH, which can bind to IFNγ mRNA, thereby preventing IFNγ transcription. Additionally, elevated lactic acid levels result in cellular acidification. The acidification of cell inhibits the calcineurin, which impairs the activity of NFAT, a transcription factor that regulates IFNγ. (B) Adenosine is another hypoxia induced immunosuppressive metabolite. HIF1 has been demonstrated to promote the expression of CD73. The enzymes CD73 and CD39 work in concert to convert ATP into adenosine. Adenosine binds to NK cell receptors, thereby inhibiting NK cell function through the cAMP-PKA signaling. (C) In the context of hypoxia, tumor cells are known to produce elevated levels of reactive oxygen species (ROS) due to oxygen deprivation. This phenomenon has been observed to elicit a similar response in myeloid immune cells, which are known to produce ROS in response to the enzyme NOX. A significant extracellular accumulation of reactive oxygen species (ROS) has been demonstrated to inhibit the expression of natural killer (NK) cell activation receptors, including NKp46 and NKG2D. Furthermore, the secretion of NK cell cytotoxicity mediators, including perforin, granzyme, and IFNγ, has also been shown to be impaired. In the meantime, NK cells have developed a defensive mechanism against ROS. Following IL-15 activation, the expression of the anti-ROS substance Trx1 was observed to increase. Conversely, the expression of the protein that inhibits Trx1 was found to decrease. Trx1 forms an antioxidant shield on the surface of NK cells, thereby protecting them from ROS. Moreover, the antioxidant protective shield afforded protection to the peripheral immune cells of NK cells, thereby maintaining optimal immune responsiveness. Abbreviations: OXPHOS, oxidative phosphorylation; LDHA, lactate dehydrogenase A; MCTs, monocarboxylate transporters; NFAT, Nuclear factor of activated T cells; IFNγ, Interferon gamma; HIF, Hypoxia-inducible factor; NT5E, ecto-5′-nucleotidase; ATP, Adenosine triphosphate; AMP, Adenosine monophosphate; PKA, Protein kinase A; CREB, cAMP response element-binding protein; TNFα, Tumor necrosis factor alpha; M-CSF, Macrophage colony-stimulating factor; MIP1α, Macrophage inflammatory protein-1 alpha; NADP, Nicotinamide adenine dinucleotide phosphate; ROS, Reactive oxygen species; IL, Interleukin; Trx1, Thioredoxin; TXN, Thioredoxin; TXNIP, Thioredoxin-interacting protein; TXNRD1, Thioredoxin Reductase 1. |
Lactate
In the context of hypoxia, the metabolic process of NK cells undergoes a shift from OXPHOS to anaerobic glycolysis.30 This shift is accompanied by the conversion of pyruvate to large quantities of lactate, a process that is catalyzed by the LDHA.30,51,57 Lactate is intimately associated with a reduction in the function of NK cells. In the tumor microenvironment, lactate accumulation is associated with a reduction in IFNγ production by NK cells.124 The apoptosis of NK cells is contingent upon the concentration of lactate exceeding 20 mM.124 This explains the observed reduction in the population of NK cells in tumors when high concentrations of lactate are present.124 The inhibition of lactate in tumor cells resulted in enhanced cytotoxicity and increased expression of activating receptors in NK cells.125
There are numerous subsets of NK cells, each exhibiting varying sensitivities to lactate. Two distinct populations of NK cells have been identified in the liver: tissue-resident NK (trNK) cells and conventional NK (cNK) cells.126–128 These NK cells exhibit disparate sensitivities to inflammation-induced lactate accumulation.129 Specifically, inflammation-induced lactate promoted apoptosis of trNK cells, resulting in a significant decrease in their number. Conversely, the number of cNK cells at the same lactate concentration was not significantly altered.129 With regard to the effector function of NK cells, it has been demonstrated that trNK cells are capable of producing IFNγ prior to cNK cells and that they play a protective role in the liver at the early stage of mouse cytomegalovirus (MCMV) infection.130 The disparity in lactate sensitivity is the underlying cause of the diminished functionality observed in trNK cells, which are expected to express IFNγ with greater rapidity.129 Nevertheless, impaired IFNγ expression of trNK cells permitted cNK cells to become the principal cells secreting IFNγ during MCMV infection.129 The different sensitivity of NK cells to lactate is inextricably linked to mitochondrial function. Sensitivity of trNK cells to lactate correlates with impairment of their mitochondrial function and the carbonic anhydrase 5B (Car5b) expression.129 The mitochondrial enzyme carbonic anhydrase is essential for the conversion of CO2 to bicarbonate.131 This is of critical important for NK cells in resisting intracellular pH changes in an acidic microenvironment.131
The inhibitory effect of lactate on NK cells is a topic of ongoing research. When NK cells are activated under normoxic conditions, they require a substantial amount of ATP for a brief period of time, which leads to aerobic glycolysis.132 This accelerated glucose metabolism in NK cells, so-called “Warburg effect”, is accompanied by the production of a considerable amount of lactate. In order to maintain the glycolysis, lactate needs to be exported via monocarboxylate transporters 1 (MCT1), which depends on a concentration gradient from cytoplasmic to extracellular lactate.133 Conversely, in the context of hypoxia, an increase in the extracellular lactate concentration results in a diminished concentration gradient, thereby impeding the transport of lactate from NK cells to extracellular compartment. The accumulation of intracellular lactate disrupts the energy metabolism of NK cell, resulting in a reduction in ATP levels.124 Furthermore, NADPH, which released by the lactate induced disruption of glycolysis, binds to IFNγ mRNA, thereby impeding the translation of IFNγ.134 The nuclear translocation of nuclear factor of activated T cells (NFAT), the transcription factor for IFNγ, is also disturbed by intracellular acidification due to lactate accumulation. The translocation of NFAT is mediated through calcineurin, a phosphatase that dephosphorylates NFAT. In low pH cells, calcineurin is suppressed, leading to the inhibition of NFAT translocation.135
Lactate has a profound effect on NK cells, but the exact mechanism is still a mystery. The extent to which lactate is important in hypoxia-induced NK cell inhibition and whether inhibition of lactate production can reverse the inhibitory effect of hypoxia on NK cells remains unexplored. The immunosuppressive effects of lactate make it an important clinical target for manipulation of NK cell function. The study of specific mechanisms for targeting lactate has profound implications for a wide range of diseases (Figure 5A).
Adenosine
Tumors employ a series of subterfuges to evade immune escape, with the accumulation of extracellular adenosine being a common tactic.136 Adenosine, a ubiquitous immunosuppressive metabolite, impairs the infiltration, cytotoxicity and cytokines production of numerous immune cells, including NK cells and cytotoxic T cells.136–138 Extracellular adenosine triphosphate (ATP) is hydrolyzed to adenosine by nucleoside triphosphate dephosphorylase (CD39) and exto-5’-nucleotidase (CD73). Despite the identification of multiple mechanisms underlying ATP leakage to the extracellular compartment in various cell types, the exact mechanism remains elusive.139,140 In healthy tissues, the extracellular ATP concentration is typically low, in the nanomolar (nmol/l) range. In contrast, in diseased or stressed tissues, the extracellular ATP concentration increased to hundreds of micromolar (μmol/l).139,141,142 Extracellular ATP is initially hydrolyzed to adenosine monophosphate (AMP) by CD39, which is expressed on the cell membrane. Subsequently, AMP is recognized by CD73 on the cell membrane and hydrolyzed to adenosine.143 It is noteworthy that extracellular ATP signaling may have originally promoted antitumor immunity via P2 purinergic receptors.144 Following the catalysis by CD39 and CD73, the immunopromoting ATP was hydrolyzed to the immunosuppressive adenosine.145 As with ATP, extracellular adenosine concentrations elevate from nanomolar to micromolar range in the setting of disease and stress.146–148 The CD39-CD73-adenosine axis has emerged as a crucial regulatory pathway due to its potent immunosuppressive effects.
Tumor-induced hypoxic microenvironment is an important trigger for adenosine production. The deprivation of oxygen results in a reduction in the availability of energy and an increase in the concentration of extracellular ATP, which provides the material basis for the increase in adenosine.144 Moreover, hypoxia is a potent inducer of CD39 and CD73 on the cell membrane.136 HIF-1 has been demonstrated to promote the transcription of CD73 by binding to the promotor located in front of NT5E, the gene encoding CD73.149 Hypoxia is also associated with the inhibition of adenosine metabolism, which is accompanied by the accumulation of adenosine. Adenosine can be reconverted to AMP by adenosine kinase, but under hypoxic conditions, adenosine kinase is inhibited, which accelerates the accumulation of extracellular adenosine concentrations.150 The combined effects contribute to a significant increase in extracellular adenosine concentration in hypoxic micro-environments, which in turn leads to a profound suppression of immune cells, particularly NK cells.
The precise mechanism by which adenosine exerts an inhibitory effect on NK cells remains elusive. It has been demonstrated that the primary mode of inhibition exerted by adenosine is through the NK cell surface G-protein-coupled adenosine 2A receptors (A2AR).151 Adenosine inhibits IL-2 mediated TNFα release from NK cells.152 Activation of A2AR up-regulates the concentration of cAMP in NK cells, which in turn activates protein kinase A.73 Activated PKA inhibits cytotoxicity, cytokine secretion and immunometabolism in NK cells.153 A3R, another G-protein-coupled adenosine receptor expressed on NK cells, appears to antagonize A2AR signaling and promotes NK cell function by inhibiting cAMP activity.154 Nevertheless, this receptor is currently understudied. Activated PKA plays a pivotal role in adenosine-mediated inhibition of NK cells. In IL-2 primed NK cells, adenosine was found to inhibit cytotoxicity and cytokines production via PKA.151,155 Due to the metabolic instability of adenosine itself, some studies have employed an alternative, metabolically stable analog 2-chloroadenosine156 in lieu of adenosine to stimulate NK cells. As anticipated, CADO suppressed lymphocyte activation, Ly49D-crosslinking-stimulated NK cell cytotoxicity and cytokines secretion, including IFNγ and TNFα.157 In contrast to IL-2 stimulation, IL-12 and IL-15-primed NK cells exhibited the ability to overcome the inhibitory effects of adenosine, as evidenced by an increase in IFNγ expression in primed NK cells.158 Despite the observed increase in IFNγ expression in IL-12 and IL-15 primed NK cells, the observed enhancement in the lytic effect on tumor cells was not observed. This phenomenon indicates that adenosine may impede the exocytosis of IFNγ (Figure 5B).158
The substantial inhibitory of adenosine on NK cell function has prompted the development of therapeutic strategies that aim at inhibiting this pathway. One of the most interesting therapies is the application of engineered human NK cells.159,160 Engineered cell therapy is a process whereby the CD73 antibody gene is integrated into the NK cell specific genome. This results in the NK cell becoming activated upon encountering a tumor cell, and the NK cell itself secreting anti-CD73 antibodies.159,160 The antibody inhibits CD73, thereby reducing the accumulation of adenosine and restoring NK cell function.159,160 Hypoxia-induced adenosine has been demonstrated to exert potent immunosuppressive effects. Consequently, inhibition of adenosine production is expected to be the key to reversing the abnormal NK cell function caused by hypoxia.
ROS
Reactive oxidative species (ROS) exerts profound inhibitory effects on NK cells. In the hypoxic microenvironment, as the ETC is disrupted by oxygen deprivation, tumor cells and a variety of infiltrating myeloid immune cells, such as macrophages, myeloid derived suppressor cells (MDSC) and granulocytes, release large amounts for ROS into the intercellular space.161 NK cells that are exposed to ROS exhibit apoptosis at an earlier stage than T cells. Chronic myeloid leukemia cells contribute to the premature apoptosis of NK cells through the generation of ROS.162 This inhibitory effect can be alleviated by the use of ROS scavengers.162 Monocyte derived and macrophage derived hydrogen peroxide has been demonstrated to significantly inhibit effector function of NK cells, resulting in a decrease in CD3 and CD16 on the surface of NK cells. This decrease is one of the mechanisms by which monocytes and macrophages exhibit their immunosuppressive effects.163,164 Furthermore, ROS suppressed the expression of NK cell surface activation receptors NKG2D and NKp46, thereby impeding the cytotoxicity and cytokine secretion functions of NK cells.165–167
To resist the inhibitory effects of ROS, NK cells develop the antioxidant thioredoxin to counteract elevated extracellular ROS.168 Thioredoxin acts as a scavenger of ROS by reducing oxidized cysteine residues to the reduced state and cleaving disulfide bonds.169,170 In response to IL-15 stimulation, NK cells up-regulate the expression of thioredoxin genes (TXN and TXN2). In addition, IL-15 primed NK cells also inhibit the gene expression of thioredoxin interacting protein by up-regulating mTORC1. This protein acts as an inhibitory counterpart of thioredoxin and exerts negative regulation of thioredoxin.168 The up-regulation of thioredoxin results in an increased density of thiols on the surface of NK cells. This high density of thiols on the cell membrane serves to form an antioxidant protective shield against high extracellular concentrations of ROS.168 The antioxidant effect of NK cells allows NK cells located in the high ROS region of the tumor core to maintain higher cell cytotoxicity and IFNγ secretion than peripheral NK cells. Additionally, the level of ROS in NK cells in the tumor core region was found to be lower than that in peripheral NK cells.168 It is noteworthy that the antioxidant shield formed by NK cells not only preserves the functionality of themselves in high ROS environment but also protects bystander lymphocytes, such as T cells, from ROS.168
The precise mechanism by which ROS inhibit NK cells remains elusive, and further research is necessary to elucidate the manner in which ROS attack NK cells, resulting in a reduction in their cytotoxicity, cytokines secretion, and ultimately in their apoptosis (Figure 5C).
Targeting Hypoxic NK Cells Immunotherapy
The inhibitory effects of hypoxia on NK cells are complex, involving not only the adaptation of NK cells to hypoxia and its metabolites, but also interactions with other cells under hypoxic conditions. These alterations collectively act on the NK cells to cause a decrease in their cytotoxicity and cytokine secretion. For example, tumor cells undergo adaptive changes in response to hypoxia, which enables them to evade the surveillance of NK cells.171 The expression of MICA, the NK-specific recognition receptor on the surface of tumor cells under hypoxia, is decreased. A reduction in MHC class I-related chain molecules A17 results in the inability of NKG2D receptors to recognize tumor cells, which ultimately leads to their immune escape.171 Furthermore, hypoxia results in the upregulation of autophagy in tumor cells. Autophagy degrades perforin and granzyme released by NK cells into tumor cells, rendering tumor cells resistant to their cytotoxicity.172 The complexity and variability of hypoxia inhibition of NK cell function is further compounded by the cell-cell interactions involved.
The complexity of the hypoxia microenvironment has hindered the advancement of NK cell therapy targeting hypoxia. It can be postulated that enhancing the oxygen concentration within the microenvironment may assist in alleviating the impaired cytotoxicity function and decreased cytokine secretion of NK cells under hypoxia.123,173–175 One of the most significant challenges in tumor therapy has been the development of effective treatments for hypoxic microenvironment, particularly those found in solid tumors. It has been demonstrated that exercise can induce oxygenation in the tumor microenvironment, which in turn restores the functionality of NK cells.176 However, the efficacy of exercise remains to be demonstrated.177 Myo-inositol-trispyrophosphate (ITPP) enhances oxygen tension within solid tumors by stabilizing vascular normalization through the activation of endothelial Phosphatase-and-Tensin-homologue (PTEN).178,179 The ITPP-induced increase in intra-tumoral oxygen resulted in an increase in the number of NK cells and a decrease in the number of immunosuppressive cells.180 Similar to the ITPP mechanism, manganese dioxide nanoparticles, which were encapsulated into polylactic-co-glycolic, catalyze the degradation of tumor-produced hydrogen peroxide, resulting in the generation of oxygen.181 The therapeutic measures of manganese dioxide nanoparticles not only elevated oxygen in the tumor microenvironment, but also consumed ROS, adenosine and lactic acid.181 In addition to the tumor microenvironment, sepsis and severe trauma can result in the body undergoing hypoxia.182,183 The administration of oxygen therapy at this time will, in fact, serve to exacerbate the poor prognosis of septic and trauma patients through the mechanism of ischemia-reperfusion injury.184 Our recent research has also demonstrated that oxygen therapy following severe trauma is associated with a reduction in NK cell numbers. Further research is required to elucidate the correlation between oxygen therapy and the immune system in other diseases.
In addition to alleviating hypoxia in the microenvironment, an additional therapeutic approach is to enhance the tolerance of NK cells to hypoxia. It has been demonstrated that the stimulation of NK cells by cytokines enhances their tolerance to hypoxia. IL-2 primed NK cells prevent NKG2D down-regulation in hypoxic environment.13 A high-affinity NK cell that is capable of expressing the high-affinity CD16 receptor and internal IL-2 is able to maintain cytotoxicity under hypoxic conditions.185 Hypoxia has been demonstrated to reduce the phosphorylation levels of extracellular signal-regulated kinase (ERK) and signal transducer and activator of transcription 3 (STAT3) in an Src homology region 2 domain-containing phosphatase-1 (SHP-1) dependent manner. Pharmacological blockade of SHP-1 activity has been shown to partially restore the cytotoxicity caused by hypoxia.15 However, the promotion of STAT3 and JAK signaling pathways has the unintended consequence of inducing PD-L1 expression in tumor cells, which impairs the cytotoxicity of NK cells.186 As previously stated, tumor cells are able to resist the cytocidal effects of NK cells through autophagy. Inhibition of autophagy has been demonstrated to enhance the cytotoxicity of NK cells under hypoxic conditions.172 However, the development of NK cells is heavily dependent on autophagy, and selective inhibition of autophagy in tumor cells represents a key objective of this therapeutic approach.187 It can be posited that increasing the oxygen content of the microenvironment and increasing the tolerance of NK cells to hypoxia represent prospective potential treatments.
Conclusion and Perspective
In conclusion, the functional impairment of NK cells in a hypoxic environment is the result of a series of adaptive responses. These responses result in NK cells downregulating their own high-energy processes and avoiding potential damage caused by hypoxia. In the context of hypoxia, the transcription factor of NK cells undergoes a shift from cMyc to HIF-1, resulting in an altered mRNA profile at the transcriptional level. Furthermore, the energy metabolism of NK cells undergoes a transition from aerobic respiration to anaerobic glycolysis, thereby providing a rapid and continuous energy supply for NK cell function in the short term. Nevertheless, this metabolic shift is insufficient to maintain cytotoxicity and cellular function over an extended term. The long-term cytotoxicity and cellular function of NK cells are affected by mitochondrial fragmentation under conditions of hypoxia, which leads to an energy crisis. Hypoxia induced misfolding protein causes ER stress, resulting in the downregulating of activating receptors in NK cells. In addition to adapting to hypoxia, NK cells are affected by a variety of metabolites produced under such conditions, including lactate, adenosine, and ROS. These byproducts are immunosuppressive and collectively inhibit NK cell activity.
Overall, it is now well established that hypoxia plays an important role in the effector function of NK cells. Despite the numerous outstanding questions (Box 3), future research in this area will undoubtedly reveal more exciting and intriguing mechanisms and therapeutic opportunities.
 |
Box 3 Important Outstanding Questions in Relation to the Effects of NK Cells Under Hypoxic Conditions |
Acknowledgments
This review was supported in part by National Natural Science Foundation of China 81873870 (ZhaoHui Tang) and Intramural Cultivation Fund of Tongji Hospital affiliated to Tongji Medical College of Huazhong University of Science & Technology 2023B01 (TeDing Chang).
Disclosure
The authors declare no competing interests.
References
1. Caligiuri MA. Human natural killer cells. Blood. 2008;112(3):461–469. doi:10.1182/blood-2007-09-077438
2. Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immunol. 2013;132(3):536–544. doi:10.1016/j.jaci.2013.07.006
3. Vivier E, Rebuffet L, Narni-Mancinelli E, Cornen S, Igarashi RY, Fantin VR. Natural killer cell therapies. Nature. 2024;626(8000):727–736. doi:10.1038/s41586-023-06945-1
4. Vivier E, Raulet DH, Moretta A. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331(6013):44–49. doi:10.1126/science.1198687
5. Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16(1):7–19. doi:10.1038/nrc.2015.5
6. Moretta L, Bottino C, Pende D, Mingari M, Biassoni R, Moretta A. Human natural killer cells: their origin, receptors and function. Eur J Immunol. 2002;32(5):1205–1211. doi:10.1002/1521-4141(200205)32:5<1205::AID-IMMU1205>3.0.CO;2-Y
7. Moretta A, Bottino C, Mingari MC, Biassoni R, Moretta L. What is a natural killer cell? Nat Immunol. 2002;3(1):6–8. doi:10.1038/ni0102-6
8. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity. 2017;47(5):820–833. doi:10.1016/j.immuni.2017.10.008
9. Sender R, Weiss Y, Navon Y. The total mass, number, and distribution of immune cells in the human body. Proc Natl Acad Sci U S A. 2023;120(44):e2308511120. doi:10.1073/pnas.2308511120
10. Hudspeth K, Silva-Santos B, Mavilio D. Natural cytotoxicity receptors: broader expression patterns and functions in innate and adaptive immune cells. Front Immunol. 2013;4:69. doi:10.3389/fimmu.2013.00069
11. Balsamo M, Manzini C, Pietra G. Hypoxia downregulates the expression of activating receptors involved in NK -cell-mediated target cell killing without affecting ADCC. Eur J Immunol. 2013;43(10):2756–2764. doi:10.1002/eji.201343448
12. Fink T, Ebbesen P, Koppelhus U, Zachar V. Natural Killer Cell-Mediated Basal and Interferon-Enhanced Cytotoxicity against Liver Cancer Cells is Significantly Impaired Under In Vivo Oxygen Conditions. Scand J Immunol. 2003;58(6):607–612. doi:10.1111/j.1365-3083.2003.01347.x
13. Sarkar S, Germeraad WTV, Rouschop KMA. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS One. 2013;8(5):e64835. doi:10.1371/journal.pone.0064835
14. Hasmim M, Messai Y, Ziani L. Critical Role of Tumor Microenvironment in Shaping NK Cell Functions: implication of Hypoxic Stress. Front Immunol. 2015;6:482. doi:10.3389/fimmu.2015.00482
15. Teng R, Wang Y, Lv N. Hypoxia Impairs NK Cell Cytotoxicity through SHP-1-Mediated Attenuation of STAT3 and ERK Signaling Pathways. J Immunol Res. 2020;2020:4598476. doi:10.1155/2020/4598476
16. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9(5):495–502. doi:10.1038/ni1581
17. Klionsky DJ. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2021;17:1–382.
18. Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol. 2015;15(4):243–254. doi:10.1038/nri3799
19. Bauer S, Groh V, Wu J. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285(5428):727–729. doi:10.1126/science.285.5428.727
20. Vivier E, Morin P, O’Brien C, Druker B, Schlossman SF, Anderson P. Tyrosine phosphorylation of the Fc gamma RIII(CD16): zeta complex in human natural killer cells. Induction by antibody-dependent cytotoxicity but not by natural killing. J Immunol. 1991;146(1):206–210. doi:10.4049/jimmunol.146.1.206
21. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev mol Cell Biol. 2020;21(5):268–283. doi:10.1038/s41580-020-0227-y
22. Zhang Y, Guo F, Wang Y. Hypoxic tumor microenvironment: destroyer of natural killer cell function. Chin J Cancer Res. 2024;36(2):138–150. doi:10.21147/j.issn.1000-9604.2024.02.04
23. Garces-Lazaro I, Kotzur R, Cerwenka A, Mandelboim O. NK Cells Under Hypoxia: the Two Faces of Vascularization in Tumor and Pregnancy. Front Immunol. 2022;13:924775. doi:10.3389/fimmu.2022.924775
24. Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12(1):9–22. doi:10.1038/nrc3183
25. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510–5514. doi:10.1073/pnas.92.12.5510
26. Markolovic S, Wilkins SE, Schofield CJ. Protein Hydroxylation Catalyzed by 2-Oxoglutarate-dependent Oxygenases. J Biol Chem. 2015;290(34):20712–20722. doi:10.1074/jbc.R115.662627
27. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. doi:10.1016/j.cell.2012.01.021
28. Coulibaly A, Velásquez SY, Kassner N, Schulte J, Barbarossa MV, Lindner HA. STAT3 governs the HIF-1alpha response in IL-15 primed human NK cells. Sci Rep. 2021;11(1):7023. doi:10.1038/s41598-021-84916-0
29. Cluff E, Magdaleno CC, Fernandez E. Hypoxia-inducible factor-1 alpha expression is induced by IL-2 via the PI3K/mTOR pathway in hypoxic NK cells and supports effector functions in NKL cells and ex vivo expanded NK cells. Cancer Immunol Immunother. 2022;71(8):1989–2005. doi:10.1007/s00262-021-03126-9
30. O’Brien KL, Finlay DK. Immunometabolism and natural killer cell responses. Nat Rev Immunol. 2019;19(5):282–290. doi:10.1038/s41577-019-0139-2
31. Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12(2):108–113. doi:10.1016/j.ccr.2007.07.006
32. Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22–35. doi:10.1016/j.cell.2012.03.003
33. Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A. 1982;79(24):7824–7827. doi:10.1073/pnas.79.24.7824
34. Seoane J, Le HV, Massague J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419(6908):729–734. doi:10.1038/nature01119
35. Herold S, Wanzel M, Beuger V. Negative regulation of the mammalian UV response by Myc through association with Miz-1. Mol Cell. 2002;10(3):509–521. doi:10.1016/S1097-2765(02)00633-0
36. Adhikary S, Marinoni F, Hock A. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell. 2005;123(3):409–421. doi:10.1016/j.cell.2005.08.016
37. Dong H, Adams NM, Xu Y. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat Immunol. 2019;20(7):865–878. doi:10.1038/s41590-019-0388-z
38. Loftus RM, Assmann N, Kedia-Mehta N. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat Commun. 2018;9(1):2341. doi:10.1038/s41467-018-04719-2
39. Huang LE. Carrot and stick: HIF-alpha engages c-Myc in hypoxic adaptation. Cell Death Differ. 2008;15(4):672–677. doi:10.1038/sj.cdd.4402302
40. Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004;23(9):1949–1956. doi:10.1038/sj.emboj.7600196
41. Corn PG, Ricci MS, Scata KA. Mxi1 is induced by hypoxia in a HIF-1-dependent manner and protects cells from c-Myc-induced apoptosis. Cancer Biol Ther. 2005;4(11):1285–1294. doi:10.4161/cbt.4.11.2299
42. Zhang H, Gao P, Fukuda R. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11(5):407–420. doi:10.1016/j.ccr.2007.04.001
43. Harper SE, Qiu Y, Sharp PA. Sin3 corepressor function in Myc-induced transcription and transformation. Proc Natl Acad Sci U S A. 1996;93(16):8536–8540. doi:10.1073/pnas.93.16.8536
44. Ni J, Wang X, Stojanovic A. Single-Cell RNA Sequencing of Tumor-Infiltrating NK Cells Reveals that Inhibition of Transcription Factor HIF-1alpha Unleashes NK Cell Activity. Immunity. 2020;52(6):1075–1087e1078. doi:10.1016/j.immuni.2020.05.001
45. Sobecki M, Krzywinska E, Nagarajan S. NK cells in hypoxic skin mediate a trade-off between wound healing and antibacterial defence. Nat Commun. 2021;12(1):4700. doi:10.1038/s41467-021-25065-w
46. Krzywinska E, Kantari-Mimoun C, Kerdiles Y. Loss of HIF-1alpha in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat Commun. 2017;8(1):1597. doi:10.1038/s41467-017-01599-w
47. Stockmann C, Doedens A, Weidemann A. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456(7223):814–818. doi:10.1038/nature07445
48. Mazzone M, Dettori D, Leite de Oliveira R. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136(5):839–851. doi:10.1016/j.cell.2009.01.020
49. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10(6):417–427. doi:10.1038/nrd3455
50. F Victorino. HIF1alpha is required for NK cell metabolic adaptation during virus infection. Elife. 2021;10:1.
51. O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–565. doi:10.1038/nri.2016.70
52. Keppel MP, Saucier N, Mah AY, Vogel TP, Cooper MA. Activation-specific metabolic requirements for NK Cell IFN-gamma production. J Immunol. 2015;194(4):1954–1962. doi:10.4049/jimmunol.1402099
53. Donnelly RP, Loftus RM, Keating SE. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol. 2014;193(9):4477–4484. doi:10.4049/jimmunol.1401558
54. Marçais A, Cherfils-Vicini J, Viant C. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. 2014;15(8):749–757. doi:10.1038/ni.2936
55. Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature. 1996;383(6597):263–266. doi:10.1038/383263a0
56. Garcia-Bermudez J, Baudrier L, La K. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol. 2018;20(7):775–781. doi:10.1038/s41556-018-0118-z
57. Welte S, Kuttruff S, Waldhauer I, Steinle A. Mutual activation of natural killer cells and monocytes mediated by NKp80-AICL interaction. Nat Immunol. 2006;7(12):1334–1342. doi:10.1038/ni1402
58. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3(3):187–197. doi:10.1016/j.cmet.2006.01.012
59. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–185. doi:10.1016/j.cmet.2006.02.002
60. Iyer NV, Kotch LE, Agani F. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998;12(2):149–162. doi:10.1101/gad.12.2.149
61. Wilson DF, Rumsey WL, Green TJ, Vanderkooi JM. The oxygen dependence of mitochondrial oxidative phosphorylation measured by a new optical method for measuring oxygen concentration. J Biol Chem. 1988;263(6):2712–2718. doi:10.1016/S0021-9258(18)69126-4
62. Cooper CE. The steady-state kinetics of cytochrome c oxidation by cytochrome oxidase. Biochim Biophys Acta. 1990;1017(3):187–203. doi:10.1016/0005-2728(90)90184-6
63. Wheaton WW, Chandel NS. Hypoxia. Hypoxia regulates cellular metabolism. Am J Physiol Cell Physiol. 2011;300(3):C385–393. doi:10.1152/ajpcell.00485.2010
64. Tello D, Balsa E, Acosta-Iborra B. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011;14(6):768–779. doi:10.1016/j.cmet.2011.10.008
65. Chan SY, Zhang -Y-Y, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009;10(4):273–284. doi:10.1016/j.cmet.2009.08.015
66. Puissegur MP, Mazure NM, Bertero T. iR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011;18(3):465–478. doi:10.1038/cdd.2010.119
67. Chen Z, Li Y, Zhang H, Huang P, Luthra R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene. 2010;29(30):4362–4368. doi:10.1038/onc.2010.193
68. Zheng X, Qian Y, Fu B. Mitochondrial fragmentation limits NK cell-based tumor immunosurveillance. Nat Immunol. 2019;20(12):1656–1667. doi:10.1038/s41590-019-0511-1
69. Plitzko B, Loesgen S. Measurement of Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) in Culture Cells for Assessment of the Energy Metabolism. Biol Protoc. 2018;8(10):e2850. doi:10.21769/BioProtoc.2850
70. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. doi:10.1080/01926230701320337
71. Fuhrmann DC, Brune B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017;12:208–215. doi:10.1016/j.redox.2017.02.012
72. Kim H, Scimia M, Wilkinson D. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell. 2011;44(4):532–544. doi:10.1016/j.molcel.2011.08.045
73. Lichtenegger FS, Rothe M, Schnorfeil FM. Targeting LAG-3 and PD-1 to Enhance T Cell Activation by Antigen-Presenting Cells. Front Immunol. 2018;9:385. doi:10.3389/fimmu.2018.00385
74. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi:10.1038/nature09663
75. Liu L, Feng D, Chen G. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177–185. doi:10.1038/ncb2422
76. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168(6):960–976. doi:10.1016/j.cell.2017.02.004
77. Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. 2018;18(12):744–757. doi:10.1038/s41568-018-0074-8
78. Menon S, Dibble C, Talbott G. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell. 2014;156(4):771–785. doi:10.1016/j.cell.2013.11.049
79. Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21(4):521–531. doi:10.1016/j.molcel.2006.01.010
80. Brugarolas J, Lei K, Hurley RL. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18(23):2893–2904. doi:10.1101/gad.1256804
81. Li Y, Wang Y, Kim E. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J Biol Chem. 2007;282(49):35803–35813. doi:10.1074/jbc.M705231200
82. Thomas GV, Tran C, Mellinghoff IK. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12(1):122–127. doi:10.1038/nm1337
83. Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin Jr WG. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell. 2003;4(2):147–158. doi:10.1016/S1535-6108(03)00187-9
84. Jin F, Zhaozhen W, Xiao H. The PI3K/Akt/GSK-3beta/ROS/eIF2B pathway promotes breast cancer growth and metastasis via suppression of NK cell cytotoxicity and tumor cell susceptibility. Cancer Biol Med. 2019;16(1):38–54. doi:10.20892/j.issn.2095-3941.2018.0253
85. Koumenis C, Naczki C, Koritzinsky M. Regulation of Protein Synthesis by Hypoxia via Activation of the Endoplasmic Reticulum Kinase PERK and Phosphorylation of the Translation Initiation Factor eIF2α. mol Cell Biol. 2002;22(21):7405–7416. doi:10.1128/MCB.22.21.7405-7416.2002
86. Blais JD, Addison CL, Edge R. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. mol Cell Biol. 2006;26(24):9517–9532. doi:10.1128/MCB.01145-06
87. Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat Rev mol Cell Biol. 2004;5(10):827–835. doi:10.1038/nrm1488
88. Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev mol Cell Biol. 2010;11(2):113–127. doi:10.1038/nrm2838
89. Piersma SJ, Pak-Wittel MA, Lin A, Plougastel-Douglas B, Yokoyama WM. Activation Receptor-Dependent IFN-gamma Production by NK Cells Is Controlled by Transcription, Translation, and the Proteasome. J Immunol. 2019;203(7):1981–1988. doi:10.4049/jimmunol.1900718
90. Koritzinsky M, Magagnin MG, van den Beucken T. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 2006;25(5):1114–1125. doi:10.1038/sj.emboj.7600998
91. Proud CG. Peptide-chain elongation in eukaryotes. Mol Biol Rep. 1994;19(3):161–170. doi:10.1007/BF00986958
92. Nygard O, Nilsson L. Translational dynamics. Interactions between the translational factors, tRNA and ribosomes during eukaryotic protein synthesis. Eur J Biochem. 1990;191(1):1–17. doi:10.1111/j.1432-1033.1990.tb19087.x
93. Kim EY, Ner-Gaon H, Varon J. Post-sepsis immunosuppression depends on NKT cell regulation of mTOR/IFN-gamma in NK cells. J Clin Invest. 2020;130(6):3238–3252. doi:10.1172/JCI128075
94. Ryazanov AG, Shestakova EA, Natapov PG. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature. 1988;334(6178):170–173. doi:10.1038/334170a0
95. Carlberg U, Nilsson A, Nygard O. Functional properties of phosphorylated elongation factor 2. Eur J Biochem. 1990;191(3):639–645. doi:10.1111/j.1432-1033.1990.tb19169.x
96. Connolly E, Braunstein S, Formenti S, Schneider RJ. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. mol Cell Biol. 2006;26(10):3955–3965. doi:10.1128/MCB.26.10.3955-3965.2006
97. Romero-Ruiz A, Bautista L, Navarro V. Prolyl hydroxylase-dependent modulation of eukaryotic elongation factor 2 activity and protein translation under acute hypoxia. J Biol Chem. 2012;287(12):9651–9658. doi:10.1074/jbc.M111.299180
98. Moore CE, Mikolajek H, Regufe da Mota S. Elongation Factor 2 Kinase Is Regulated by Proline Hydroxylation and Protects Cells during Hypoxia. mol Cell Biol. 2015;35(10):1788–1804. doi:10.1128/MCB.01457-14
99. Wu Y, Xie J, Jin X. eEF2K enhances expression of PD-L1 by promoting the translation of its mRNA. Biochem J. 2020;477(22):4367–4381. doi:10.1042/BCJ20200697
100. Isken O, Maquat LE. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21(15):1833–1856. doi:10.1101/gad.1566807
101. Bertram G, Bell HA, Ritchie DW, Fullerton G, Stansfield I. Terminating eukaryote translation: domain 1 of release factor eRF1 functions in stop codon recognition. RNA. 2000;6(9):1236–1247. doi:10.1017/S1355838200000777
102. Feng T, Yamamoto A, Wilkins S. Optimal translational termination requires C4 lysyl hydroxylation of eRF1. Mol Cell. 2014;53(4):645–654. doi:10.1016/j.molcel.2013.12.028
103. Martins AS, Alves I, Helguero L, Domingues MR, Neves BM. The Unfolded Protein Response in Homeostasis and Modulation of Mammalian Immune Cells. Int Rev Immunol. 2016;35(6):457–476. doi:10.3109/08830185.2015.1110151
104. Kaufman RJ, Scheuner D, Schröder M. The unfolded protein response in nutrient sensing and differentiation. Nat Rev mol Cell Biol. 2002;3(6):411–421. doi:10.1038/nrm829
105. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev mol Cell Biol. 2012;13(2):89–102. doi:10.1038/nrm3270
106. Li A, Song NJ, Riesenberg BP, Li Z. The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: mechanisms and Opportunities. Front Immunol. 2019;10:3154. doi:10.3389/fimmu.2019.03154
107. Almanza A, Carlesso A, Chintha C. Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J. 2019;286(2):241–278. doi:10.1111/febs.14608
108. Vetters J, van Helden M, De Nolf C. Canonical IRE1 function needed to sustain vigorous natural killer cell proliferation during viral infection. iScience. 2023;26(12):108570. doi:10.1016/j.isci.2023.108570
109. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi:10.1016/S0092-8674(01)00611-0
110. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. mol Cell Biol. 2003;23(21):7448–7459. doi:10.1128/MCB.23.21.7448-7459.2003
111. Bednarska K, Gunawardana J, Vari F. The IRE1-XBP1s Pathway Impairment Underpins NK Cell Dysfunction in Hodgkin Lymphoma, That Is Partly Restored by PD-1 Blockade. Blood. 2019;134(Supplement_1):2795. doi:10.1182/blood-2019-126372
112. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2(6):326–332. doi:10.1038/35014014
113. Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci U S A. 2009;106(39):16657–16662. doi:10.1073/pnas.0903775106
114. Shi M, Chai Y, Zhang J, Chen X. Endoplasmic Reticulum Stress-Associated Neuronal Death and Innate Immune Response in Neurological Diseases. Front Immunol. 2021;12:794580. doi:10.3389/fimmu.2021.794580
115. Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal. 2007;9(12):2357–2371. doi:10.1089/ars.2007.1764
116. Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A. 2004;101(31):11269–11274. doi:10.1073/pnas.0400541101
117. Locker N, Easton LE, Lukavsky PJ. HCV and CSFV IRES domain II mediate eIF2 release during 80S ribosome assembly. EMBO J. 2007;26(3):795–805. doi:10.1038/sj.emboj.7601549
118. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. mol Biol Cell. 1999;10:3787–3799. doi:10.1091/mbc.10.11.3787
119. Sovolyova N, Healy S, Samali A, Logue SE. Stressed to death - mechanisms of ER stress-induced cell death. Biol Chem. 2014;395(1):1–13. doi:10.1515/hsz-2013-0174
120. Sen Santara S, Lee D-J, Crespo Â. The NK cell receptor NKp46 recognizes ecto-calreticulin on ER-stressed cells. Nature. 2023;616(7956):348–356. doi:10.1038/s41586-023-05912-0
121. Castellanos JP, Genereux JC. Calreticulin surface presentation: a signal for natural killer cells to attack. Signal Transduct Target Ther. 2023;8(1):289. doi:10.1038/s41392-023-01551-z
122. Shoulders MD, Ryno L, Genereux J. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013;3(4):1279–1292. doi:10.1016/j.celrep.2013.03.024
123. Van Krieken R, Tsai YL, Carlos AJ, Ha DP, Lee AS. ER residential chaperone GRP78 unconventionally relocalizes to the cell surface via endosomal transport. Cell mol Life Sci. 2021;78:5179–5195. doi:10.1007/s00018-021-03849-z
124. Brand A, Singer K, Koehl G. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016;24(5):657–671. doi:10.1016/j.cmet.2016.08.011
125. Long Y, Gao Z, Hu X. Downregulation of MCT 4 for lactate exchange promotes the cytotoxicity of NK cells in breast carcinoma. Cancer Med. 2018;7(9):4690–4700. doi:10.1002/cam4.1713
126. Yokoyama WM, Sojka DK, Peng H, Tian Z. Tissue-resident natural killer cells. Cold Spring Harb Symp Quant Biol. 2013;78:149–156. doi:10.1101/sqb.2013.78.020354
127. Sojka DK, Plougastel-Douglas B, Yang L. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. Elife. 2014;3:e01659. doi:10.7554/eLife.01659
128. Daussy C, Faure F, Mayol K. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med. 2014;211(3):563–577. doi:10.1084/jem.20131560
129. Dodard G, Tata A, Erick TK. Inflammation-Induced Lactate Leads to Rapid Loss of Hepatic Tissue-Resident NK Cells. Cell Rep. 2020;32(1):107855. doi:10.1016/j.celrep.2020.107855
130. Weizman OE, Adams NM, Schuster IS. ILC1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell. 2017;171(4):795–808e712. doi:10.1016/j.cell.2017.09.052
131. Shah GN, Rubbelke TS, Hendin J. Targeted mutagenesis of mitochondrial carbonic anhydrases VA and VB implicates both enzymes in ammonia detoxification and glucose metabolism. Proc Natl Acad Sci U S A. 2013;110(18):7423–7428. doi:10.1073/pnas.1305805110
132. Sheppard S, Santosa EK, Lau CM. Lactate dehydrogenase A-dependent aerobic glycolysis promotes natural killer cell anti-viral and anti-tumor function. Cell Rep. 2021;35(9):109210. doi:10.1016/j.celrep.2021.109210
133. Fischer K, Hoffmann P, Voelkl S. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109(9):3812–3819. doi:10.1182/blood-2006-07-035972
134. Chang CH, Curtis J, Maggi L. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153(6):1239–1251. doi:10.1016/j.cell.2013.05.016
135. Hisamitsu T, Nakamura TY, Wakabayashi S. Na+ /H+ Exchanger 1 Directly Binds to Calcineurin A and Activates Downstream NFAT Signaling, Leading to Cardiomyocyte Hypertrophy. mol Cell Biol. 2012;32(16):3265–3280. doi:10.1128/MCB.00145-12
136. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17(12):709–724. doi:10.1038/nrc.2017.86
137. Hatfield SM, Kjaergaard J, Lukashev D. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med. 2015;7(277):277ra230. doi:10.1126/scitranslmed.aaa1260
138. Hatfield SM, Kjaergaard J, Lukashev D. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1alpha-dependent and extracellular adenosine-mediated tumor protection. J Mol Med. 2014;92(12):1283–1292. doi:10.1007/s00109-014-1189-3
139. Lazarowski ER. Vesicular and conductive mechanisms of nucleotide release. Purinergic Sig. 2012;8(3):359–373. doi:10.1007/s11302-012-9304-9
140. Di Virgilio F, Adinolfi E. Extracellular purines, purinergic receptors and tumor growth. Oncogene. 2017;36(3):293–303. doi:10.1038/onc.2016.206
141. Yegutkin GG. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: functional implications and measurement of activities. Crit Rev Biochem mol Biol. 2014;49(6):473–497. doi:10.3109/10409238.2014.953627
142. Falzoni S, Donvito G, Di Virgilio F. Detecting adenosine triphosphate in the pericellular space. Interface Focus. 2013;3(3):20120101. doi:10.1098/rsfs.2012.0101
143. Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol. 2001;41(1):775–787. doi:10.1146/annurev.pharmtox.41.1.775
144. Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. 2010;29(39):5346–5358. doi:10.1038/onc.2010.292
145. Ohta A. A Metabolic Immune Checkpoint: adenosine in Tumor Microenvironment. Front Immunol. 2016;7:109. doi:10.3389/fimmu.2016.00109
146. Ohta A, Gorelik E, Prasad SJ. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A. 2006;103(35):13132–13137. doi:10.1073/pnas.0605251103
147. Miller WL, Thomas RA, Berne RM, Rubio R. Adenosine production in the ischemic kidney. Circ Res. 1978;43(3):390–397. doi:10.1161/01.RES.43.3.390
148. Ballarin M, Fredholm BB, Ambrosio S, Mahy N. Extracellular levels of adenosine and its metabolites in the striatum of awake rats: inhibition of uptake and metabolism. Acta Physiol Scand. 1991;142(1):97–103. doi:10.1111/j.1748-1716.1991.tb09133.x
149. Synnestvedt K, Furuta GT, Comerford KM. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110(7):993–1002. doi:10.1172/JCI0215337
150. Decking UK, Schlieper G, Kroll K, Schrader J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res. 1997;81(2):154–164. doi:10.1161/01.RES.81.2.154
151. Raskovalova T, Huang X, Sitkovsky M, Zacharia LC, Jackson EK, Gorelik E. Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J Immunol. 2005;175(7):4383–4391. doi:10.4049/jimmunol.175.7.4383
152. Miller JS, Cervenka T, Lund J, Okazaki IJ, Moss J. Purine metabolites suppress proliferation of human NK cells through a lineage-specific purine receptor. J Immunol. 1999;162(12):7376–7382. doi:10.4049/jimmunol.162.12.7376
153. Chambers AM, Matosevic S. Immunometabolic Dysfunction of Natural Killer Cells Mediated by the Hypoxia-CD73 Axis in Solid Tumors. Front Mol Biosci. 2019;6:60. doi:10.3389/fmolb.2019.00060
154. Wang J, Matosevic S. Adenosinergic signaling as a target for natural killer cell immunotherapy. J Mol Med. 2018;96(9):903–913. doi:10.1007/s00109-018-1679-9
155. Raskovalova T, Lokshin A, Huang X, Jackson EK, Gorelik E. Adenosine-mediated inhibition of cytotoxic activity and cytokine production by IL-2/NKp46-activated NK cells: involvement of protein kinase A isozyme I (PKA I). Immunol Res. 2006;36(1–3):91–99. doi:10.1385/IR:36:1:91
156. Bastard P. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:423–+.
157. Lokshin A, Raskovalova T, Huang X, Zacharia LC, Jackson EK, Gorelik E. Adenosine-mediated inhibition of the cytotoxic activity and cytokine production by activated natural killer cells. Cancer Res. 2006;66(15):7758–7765. doi:10.1158/0008-5472.CAN-06-0478
158. Chambers AM, Wang J, Lupo KB, Yu H, Atallah Lanman NM, Matosevic S. Adenosinergic Signaling Alters Natural Killer Cell Functional Responses. Front Immunol. 2018;9:2533. doi:10.3389/fimmu.2018.02533
159. Wang J, Toregrosa-Allen S, Elzey BD. Multispecific targeting of glioblastoma with tumor microenvironment-responsive multifunctional engineered NK cells. Proc Natl Acad Sci U S A. 2021;118(45). doi:10.1073/pnas.2107507118
160. Chambers AM, Lupo KB, Wang J. Engineered natural killer cells impede the immunometabolic CD73-adenosine axis in solid tumors. Elife. 2022;11. doi:10.7554/eLife.73699
161. Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160(1):1–40. doi:10.1016/j.cbi.2005.12.009
162. Mellqvist UH, Hansson M, Brune M, Dahlgren C, Hermodsson S, Hellstrand K. Natural killer cell dysfunction and apoptosis induced by chronic myelogenous leukemia cells: role of reactive oxygen species and regulation by histamine. Blood. 2000;96(5):1961–1968. doi:10.1182/blood.V96.5.1961
163. Kono K, Salazar‐Onfray F, Petersson M. Hydrogen peroxide secreted by tumor-derived macrophages down-modulates signal-transducing zeta molecules and inhibits tumor-specific T cell-and natural killer cell-mediated cytotoxicity. Eur J Immunol. 1996;26(6):1308–1313. doi:10.1002/eji.1830260620
164. Aydin E, Johansson J, Nazir FH, Hellstrand K, Martner A. Role of NOX2-Derived Reactive Oxygen Species in NK Cell-Mediated Control of Murine Melanoma Metastasis. Cancer Immunol Res. 2017;5(9):804–811. doi:10.1158/2326-6066.CIR-16-0382
165. Romero AI, Thoren FB, Brune M, Hellstrand K. NKp46 and NKG2D receptor expression in NK cells with CD56 dim and CD56 bright phenotype: regulation by histamine and reactive oxygen species. Br J Haematol. 2006;132(1):91–98. doi:10.1111/j.1365-2141.2005.05842.x
166. Izawa S, Kono K, Mimura K. H(2)O(2) production within tumor microenvironment inversely correlated with infiltration of CD56(dim) NK cells in gastric and esophageal cancer: possible mechanisms of NK cell dysfunction. Cancer Immunol Immunother. 2011;60(12):1801–1810. doi:10.1007/s00262-011-1082-7
167. El-Hag A, Clark RA. Down-regulation of human natural killer activity against tumors by the neutrophil myeloperoxidase system and hydrogen peroxide. J Immunol. 1984;133(6):3291–3297. doi:10.4049/jimmunol.133.6.3291
168. Yang Y, Neo SY, Chen Z. Thioredoxin activity confers resistance against oxidative stress in tumor-infiltrating NK cells. J Clin Invest. 2020;130(10):5508–5522. doi:10.1172/JCI137585
169. Holmgren A. Thioredoxin and glutaredoxin systems. J Biol Chem. 1989;264(24):13963–13966. doi:10.1016/S0021-9258(18)71625-6
170. Collet JF, Messens J. Structure, function, and mechanism of thioredoxin proteins. Antioxid Redox Signal. 2010;13(8):1205–1216. doi:10.1089/ars.2010.3114
171. Yamada N, Yamanegi K, Ohyama H. Hypoxia downregulates the expression of cell surface MICA without increasing soluble MICA in osteosarcoma cells in a HIF-1alpha-dependent manner. Int J Oncol. 2012;41(6):2005–2012. doi:10.3892/ijo.2012.1630
172. Baginska J, Viry E, Berchem G. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A. 2013;110(43):17450–17455. doi:10.1073/pnas.1304790110
173. Yun S, Lee SH, Yoon SR, Myung PK, Choi I. Oxygen tension regulates NK cells differentiation from hematopoietic stem cells in vitro. Immunol Lett. 2011;137(1–2):70–77. doi:10.1016/j.imlet.2011.02.020
174. Arvindam US. 675 Oxygen concentration alters natural killer cell phenotype and function in the solid tumor microenvironment. Journal for ImmunoTherapy of Cancer. 2020;8:A405–A406.
175. Kennedy PR. 365 Oxygen levels found in bone marrow (5%) provide an optimal niche for the differentiation of induced-pluripotent stem cell-derived NK cells for the treatment of AML. Journal for ImmunoTherapy of Cancer. 2023;11:A414–A414.
176. Idorn M, Hojman P. Exercise-Dependent Regulation of NK Cells in Cancer Protection. Trends Mol Med. 2016;22(7):565–577. doi:10.1016/j.molmed.2016.05.007
177. Pal A, Zimmer P, Schmidt ME. No Evidence for Effect of Exercise on Transcriptome of NK Cells in Breast Cancer Patients Undergoing Adjuvant Therapy: results From a Pilot Study. Front Physiol. 2019;10:959. doi:10.3389/fphys.2019.00959
178. Kieda C, Greferath R, Crola Da Silva C, Fylaktakidou KC, Lehn J-M, Nicolau C. Suppression of hypoxia-induced HIF-1α and of angiogenesis in endothelial cells by myo-inositol trispyrophosphate-treated erythrocytes. Proc Natl Acad Sci U S A. 2006;103(42):15576–15581. doi:10.1073/pnas.0607109103
179. Kieda C, El Hafny-Rahbi B, Collet G. Stable tumor vessel normalization with pO(2) increase and endothelial PTEN activation by inositol trispyrophosphate brings novel tumor treatment. J Mol Med. 2013;91(7):883–899. doi:10.1007/s00109-013-0992-6
180. El Hafny‐Rahbi B, Brodaczewska K, Collet G. Tumour angiogenesis normalized by myo-inositol trispyrophosphate alleviates hypoxia in the microenvironment and promotes antitumor immune response. J Cell Mol Med. 2021;25(7):3284–3299. doi:10.1111/jcmm.16399
181. Murphy DA, Cheng H, Yang T, Yan X, Adjei IM. Reversing Hypoxia with PLGA-Encapsulated Manganese Dioxide Nanoparticles Improves Natural Killer Cell Response to Tumor Spheroids. Mol Pharm. 2021;18(8):2935–2946. doi:10.1021/acs.molpharmaceut.1c00085
182. Yan EB, Satgunaseelan L, Paul E. Post-traumatic hypoxia is associated with prolonged cerebral cytokine production, higher serum biomarker levels, and poor outcome in patients with severe traumatic brain injury. J Neurotrauma. 2014;31(7):618–629. doi:10.1089/neu.2013.3087
183. Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc. 2013;88(10):1127–1140. doi:10.1016/j.mayocp.2013.06.012
184. Bar-Or D, Carrick MM, Mains CW, Rael LT, Slone D, Brody EN. Sepsis, oxidative stress, and hypoxia: are there clues to better treatment? Redox Rep. 2015;20(5):193–197. doi:10.1179/1351000215Y.0000000005
185. Solocinski K, Padget MR, Fabian KP. Overcoming hypoxia-induced functional suppression of NK cells. J Immunother Cancer. 2020;8(1):e000246. doi:10.1136/jitc-2019-000246
186. Xu LJ, Ma Q, Zhu J. Combined inhibition of JAK1,2/Stat3-PD-L1 signaling pathway suppresses the immune escape of castration-resistant prostate cancer to NK cells in hypoxia. Mol Med Rep. 2018;17(6):8111–8120. doi:10.3892/mmr.2018.8905
187. Lopez-Soto A, Bravo-San Pedro JM, Kroemer G, Galluzzi L, Gonzalez S. Involvement of autophagy in NK cell development and function. Autophagy. 2017;13(3):633–636. doi:10.1080/15548627.2016.1274486
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Development and Validation of a Novel Nomogram Integrated with Hypoxic and Lactate Metabolic Characteristics for Prognosis Prediction in Hepatocellular Carcinoma
Qiu X, Dong L, Wang K, Zhong X, Xu H, Xu S, Guo H, Wei X, Chen W, Xu X
Journal of Hepatocellular Carcinoma 2024, 11:241-255
Published Date: 2 February 2024
Research Progress of NK Cells in Glioblastoma Treatment
Wu H, Liu Q, Wang F, Gao W, Zhou F, Zhao H
OncoTargets and Therapy 2025, 18:87-106
Published Date: 18 January 2025
TGF-β Decreases NK Cell Mobility and Cytotoxic Efficacy in Complex in vitro Models of the Leukemia Microenvironment
Švubová V, Janstová L, Jedlička M, Mašínová E, Szabová J, Feglarová T, Kuglerová K, Bosáková V, Brodská B, Boráková K, Kundrát D, Trsová I, Böhmová M, Kuželová K, Hrdý J, Gašová Z, Vydra J, Dostálová Merkerová M, Hortová-Kohoutková M, Frič J
ImmunoTargets and Therapy 2025, 14:589-604
Published Date: 18 June 2025