")
Back to Journals » Journal of Inflammation Research » Volume 18
Alterations in Signaling Pathways and Therapeutic Strategies of Traditional Chinese Medicine in Granulomatous Lobular Mastitis
Authors Zhang M , Pu D, Meng F, Shi G, Li J
Received 17 April 2025
Accepted for publication 1 July 2025
Published 14 July 2025 Volume 2025:18 Pages 9185—9197
DOI https://doi.org/10.2147/JIR.S535195
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Mengdi Zhang,1 Dongqing Pu,2 Feifei Meng,1 Guangxi Shi,2 Jingwei Li2
1First Clinical Medical College, Shandong University of Traditional Chinese Medicine, Jinan City, People’s Republic of China; 2Department of Thyroid and Breast Diagnosis and Treatment Center, Affiliated Hospital of Shandong University of Traditional Chinese Medicine, Jinan City, People’s Republic of China
Correspondence: Jingwei Li, Department of Thyroid and Breast Surgery, Affiliated Hospital of Shandong University of Traditional Chinese Medicine, Jinan City, People’s Republic of China, Email [email protected]
Abstract: Granulomatous lobular mastitis (GLM), a clinically challenging inflammatory breast disease, is characterized by a high recurrence rate, strong invasiveness, and a trend of affecting younger individuals, posing significant challenges to patients and clinical practitioners. Its pathological processes involve the abnormal activation of multiple signaling pathways, including NF-κB, NLRP3, JAK/STAT, TLR, MAPK, PI3K/AKT/mTOR, and Nrf2/HO-1. These pathways contribute to the disease’s onset and progression by regulating inflammatory responses, immune reactions, and oxidative stress. Traditional Chinese Medicine (TCM) demonstrates unique therapeutic value in the treatment of GLM by virtue of its multi-component and multi-target mechanisms. Through modulation of the aforementioned signaling pathways, TCM can effectively inhibit inflammatory cascades, regulate immune imbalance, and ameliorate oxidative stress, thereby reducing lesion size, shortening disease course, and lowering recurrence rates. This article systematically reviews the research progress on GLM-related signaling pathways and integrates the latest evidence on the interventional mechanisms of Chinese medicine, aiming to provide new insights for precision clinical treatment.
Keywords: granulomatous lobular mastitis, signaling pathways, Chinese medicine, inflammation regulation, immune intervention
Introduction
Granulomatous lobular mastitis (GLM) is a rare chronic inflammation with breast lobule as the center. It is characterized by sudden breast mass and rapid progress, which is easy to form abscess, sinus and skin ulceration, seriously affecting the appearance of breast and the quality of life of patients, and may also be accompanied by distant symptoms such as erythema nodosum of lower limbs.1 GLM is similar to breast cancer and periductal mastitis in clinical manifestations, and can be differentiated by ultrasound, mammography, MRI and pathology.2 Epidemiology shows that the incidence of GLM is on the rise in recent years, and it mainly occurs in young women aged 30–40, especially in economically developed areas.3
 |
Figure 1 Alterations in signaling pathways in granulomatous lobular mastitis. |
Although the exact cause of GLM has not been fully clarified, it is considered that the disease is closely related to autoimmune disorder, hyperprolactinemia and oral contraceptives. Autoimmunity, genetic factors related to autoinflammation and genetic mutations related to innate immunity, especially macrophage function and phagocytosis, are important factors for GLM.4 Cystic neutrophil granulomatous mastitis is a rare subtype of GLM, which is usually related to Corynebacterium.5 The pathological feature of GLM is non-caseous necrotizing granuloma in lobules, accompanied by infiltration of immune cells such as macrophages, neutrophils and lymphocytes.6 This also suggests that a variety of immune cells and cell components may be involved in the occurrence and development of the disease. Because the recurrence rate is as high as 15.4%-24.8%,7 and there are some problems such as strong local tissue destruction and long treatment period,8 exploring effective pathogenesis and treatment strategies has become the focus of clinical research.
Progress in Research on Signaling Pathways Related to GLM
NF-κB Signaling Pathway
NF-κB was initially discovered by Sen and Baltimore in B lymphocytes and named for its specific binding to the enhancer of the immunoglobulin κ light chain gene.9 As a member of the Rel protein family, NF-κB consists of homodimers or heterodimers formed by five subunits: p105/p50 (NF-κB1), p100/p52 (NF-κB2), p65 (RelA), RelB, and RelC.10 In a resting state, NF-κB exists as a heterodimer bound to the inhibitory protein IκB, anchoring it in the cytoplasm in an inactive form.10 Upon activation of IκB kinase (IKK) by stimulatory factors such as TNF-α, IL-1, phorbol ester, or lipopolysaccharide, IKK catalyzes the phosphorylation and degradation of IκB, releasing activated NF-κB to translocate to the nucleus. There, it regulates inflammatory, stress, and immune responses by binding to response elements.11,12 The core components of NF-κB activation, including IKKγ, IKKα, IKKβ, and NF-κB subunits, are essential, and their deficiency can lead to severe inflammation in humans.13 Specifically, the p50/p65 heterodimer is the most prominent functional complex. It remains inactive in resting cells by binding to the IκBα inhibitory subunit, while phosphorylation of p65 activates the transcription of inflammatory factors such as TNF-α, IL-1β, IL-6, as well as chemokines and adhesion molecules, further exacerbating the inflammatory response.14
In physiological and pathological processes, NF-κB participates in immune and chronic inflammatory responses through complex molecular regulation. Its abnormal regulation is closely associated with cancer, autoimmune diseases, septic shock, and viral infections.15,16 Since the gene promoters/enhancers of numerous cytokines and inflammatory mediators contain κB binding sites, NF-κB can bind to these sites and accelerate the expression of related genes.15 Specifically, in the pathophysiology of GLM, the activation of Toll-like receptors (TLRs) triggers the NF-κB pathway, leading to the accumulation of proinflammatory factors such as TNF-α, IL-1β, and IL-6 in the mammary gland.17 Additionally, abnormalities in the NF-κB pathway are associated with hyperprolactinemia, which is one of the important pathogenic factors of GLM.18 The activation of NF-κB promotes the expression of inflammatory factors, which can in turn activate NF-κB, forming a positive feedback loop of “inflammatory amplification and continuous activation”.19 Studies have confirmed that proinflammatory cytokines such as IL-1β and TNF-α can directly activate NF-κB, and activated NF-κB further enhances their transcription levels, forming a typical positive feedback circuit.20,21 Coombe et al21 specifically pointed out that TNF-α, IL-1β, and NF-κB are key cytokines in the development and progression of GLM. Clinically, the mechanism of action of therapeutic drugs such as glucocorticoids is also closely related to the inhibition of NF-κB activity,22 suggesting that blocking the NF-κB pathway and reducing the levels of inflammatory mediators may become a new strategy for the treatment of GLM.
NLR Signaling Pathway
Martinon et al23 first introduced the concept of the inflammasome in 2002. As a core component of the innate immune system, the inflammasome constitutes the first defensive barrier against pathogen invasion and plays a fundamental role in maintaining immune homeostasis. Upon recognition of endogenous damage-associated molecular patterns (DAMPs) or exogenous pathogen-associated molecular patterns (PAMPs), pattern recognition receptors (PRRs) mediate the activation of signaling pathways, inducing the assembly and release of inflammatory corpuscles. However, abnormally persistent activation of these corpuscles can trigger inflammatory tissue damage. The innate immune system recognizes conserved molecular patterns through PRRs, and certain cytoplasmic PRRs, such as the NLR family, can recruit the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD) to form a multi-protein complex known as the inflammasome.24,25 Among these, the NLRP3 inflammasome is the most thoroughly studied inflammatory complex, consisting of NOD-like receptor thermal protein domain associated protein 3 (NLRP3), ASC, and pro-caspase-1. Stimulated by danger signals like ATP and reactive oxygen species (ROS), NLRP3 undergoes a conformational change upon recognizing DAMPs/PAMPs, recruiting ASC to form speck-like aggregates and activating caspase-1 protease. Activated caspase-1, on the one hand, cleaves pro-IL-1β and pro-IL-18, promoting their maturation and secretion, inducing Th1 cell differentiation and natural killer (NK) cell activation, and initiating adaptive immune responses.26 On the other hand, it mediates the cleavage of GasderminD protein, inducing pyroptosis and leading to the release of large amounts of inflammatory mediators into the extracellular space, exacerbating local inflammatory responses.27
In inflammatory diseases such as GLM, the NLRP3 inflammasome is abnormally activated by “danger signals” like ATP and ROS. This drives inflammatory cascade reactions by regulating the maturation and release of proinflammatory factors like IL-1β and IL-18.28,29 It’s worth noting that there is a bidirectional regulatory relationship between inflammatory responses and oxidative stress: excessive inflammatory states induce overproduction of ROS, and oxidative damage further activates the NLRP3 inflammasome, forming a positive feedback loop of “inflammation-oxidative stress” that intensifies tissue damage.30 As downstream target genes of the NF-κB signaling pathway, the transcription of NLRP3, pro-IL-1β, and pro-IL-18 is regulated by NF-κB.31 When TLRs recognize pathogens and initiate innate immune responses, the NF-κB pathway is activated and upregulates the expression of these genes, providing a material basis for NLRP3 inflammasome activation. IL-1β directly amplifies local inflammatory responses by promoting the chemotaxis and activation of neutrophils and macrophages, while IL-18 mediates Th1-type immune responses by inducing IFN-γ secretion and NK cell activation. The levels of both are significantly correlated with the degree of inflammatory damage.32 The overactivation of the NLRP3/IL-1β signaling pathway triggers immune-inflammatory imbalance, which is closely related to systemic symptoms such as erythema nodosum and arthritis that occur during the acute phase of GLM, suggesting that abnormal systemic immune responses play a key role in disease progression.33 The NLRP3 inflammasome, with its clear molecular structure and signal transduction mechanism, serves as a critical hub connecting innate and adaptive immunity. This complex not only participates in the pathological processes of various diseases such as type 2 diabetes, ulcerative colitis, and rheumatic immune diseases, but its central regulatory role in inflammatory responses also provides potential targets for the treatment of inflammatory-related diseases like GLM.34
JAK/STAT Signaling Pathway
In recent years, numerous studies14 have reported a close association between the IL-6/STAT3 signaling pathway and plasma cell mastitis in non-lactational mastitis.35 During immune regulation, the JAK1/STAT6 pathway exhibits a highly activated state. JAK1, a crucial member of the human tyrosine kinase protein family, can rapidly initiate the phosphorylation process of JAK1 when interleukin-4 (IL-4) specifically binds to its corresponding receptor. The phosphorylated and activated JAK1 drives the release of cytokines, which in turn promotes the phosphorylation of Signal Transducer and Activator of Transcription 6 (STAT6). This ultimately leads to the polarization of macrophages towards the M2 phenotype, playing a key role in maintaining immune homeostasis.36
Interleukin-6 (IL-6), an essential activator of the JAK2/STAT3 signaling pathway, has been conclusively identified as the core signaling pathway regulating increased prolactin levels in women.20 Generally, the IL-6/JAK2/STAT3 signaling pathway serves as a critical hub for intracellular gene transcription regulation.37 Under inflammatory stress conditions, IL-6 acts as an initiating factor, specifically activating JAK2 protein. The activated JAK2 protein further stimulates intracellular STAT3 protein, promoting prolactin secretion. Simultaneously, activation of this signaling pathway triggers the accumulation of proinflammatory factors in breast tissue. When STAT3 protein is activated, its phosphorylation level significantly increases, leading to conformational changes and nuclear translocation. Once in the nucleus, STAT3 can bind to specific DNA sequences, initiating the expression of apoptosis-related genes such as Caspase-9 and BAX. Caspase-9, a member of the cysteine-aspartic acid protease family, initiates the cascade reaction of cell apoptosis, accelerating the process. Meanwhile, BAX, as a pro-apoptotic protein, promotes changes in mitochondrial membrane permeability, releasing cytochrome C and further promoting cell apoptosis. Additionally, STAT3 enhances the expression of IL-6 protein, forming a positive feedback loop. On one hand, the activation of apoptosis genes like Caspase-9 and BAX triggers cell apoptosis, releasing inflammatory substances from within the cells and promoting inflammatory responses. On the other hand, the intensification of inflammatory reactions further stimulates IL-6 secretion, activating the JAK2/STAT3 signaling pathway. This leads to increased expression of apoptotic genes and release of inflammatory factors, creating a vicious cycle. Ultimately, this results in the release of large amounts of inflammatory substances into the extracellular space, triggering a strong inflammatory response.38,39
The activation mechanism of the IL-6/JAK2/STAT3 signaling pathway exhibits typical characteristics of a cascade reaction. Excessive activation of this signaling pathway disrupts the body’s immune balance, promotes local inflammatory responses, and enhances autoimmune resistance. This subsequently triggers excessive immune and inflammatory reactions, leading to local tissue damage.40 In clinical practice, patients with GLM often present with local breast lesions accompanied by non-breast manifestations such as erythematous nodules and arthritis. This phenomenon strongly suggests that inflammatory responses mediated by the IL-6/JAK2/STAT3 signaling pathway and autoimmune responses are two key factors in the development and progression of GLM. Further investigation into the regulatory mechanisms of this signaling pathway is expected to provide new targets and strategies for GLM treatment.
TLR Signaling Pathway
TLRs, serving as a critical bridge between the innate and adaptive immune systems, constitute a class of evolutionarily shared PRRs that play a pivotal role in pathogen recognition and immune signal transduction.41,42 These transmembrane proteins are widely expressed in immune-related cells such as monocytes, macrophages, eosinophils, and epithelial cells. By recognizing PAMPs like lipopolysaccharides and peptidoglycans, as well as endogenous DAMPs, TLRs trigger downstream signaling cascades. This leads to the release of chemokines to recruit inflammatory cells or the elimination of invading pathogens through the complement system.43 However, abnormal persistent activation of TLR signaling can break the body’s immune resistance to self-antigens, inducing autoimmune cascades. This becomes an important pathogenic mechanism for various inflammatory diseases, including GLM.44
Currently, 10 functional members of the TLR family have been identified, categorized into two groups based on their subcellular localization. TLRs located on the cell membrane surface (TLR1, 2, 4, 5, 6, 10) primarily recognize PAMPs such as bacterial cell wall components and lipoproteins. In contrast, TLRs found in organelles like the endoplasmic reticulum, endosomes, and lysosomes (TLR3, 7, 8, 9) specifically recognize viral or bacterial nucleic acids.45 In the pathological study of GLM, multiple clinical evidences suggest a significant overexpression of TLR2 and TLR4 in inflamed breast tissues of patients. Their expression levels decrease after hormone therapy, indicating a close correlation between abnormal activation of TLR2 and TLR4 and the onset of GLM.46 Similarly, upregulated expression of TLR2 and TLR4 on the surface of monocytes has been observed in patients with plasma cell mastitis, positively correlated with serum IL-6 concentration.47 High expression of TLR2 and TLR4 is also seen in acute non-lactational mastitis tissues.48 These findings collectively point to the critical involvement of the TLR2/4 signaling pathway in mammary inflammatory diseases.
Analyzing the signal transduction mechanism, TLR2 and TLR4 primarily activate nuclear factor κB (NF-κB) through the myeloid differentiation factor 88 (MyD88)-dependent pathway. This regulates the gene transcription of proinflammatory cytokines such as IL-1β, IL-6, and TNF-α.49 Recent studies further untangle that the TLR2/NF-κB and TLR4/NF-κB signaling axes form a proinflammatory signal amplification network through interactions with chemokines and adhesion molecules, driving disease progression.50 Notably, TLR activation not only directly induces NF-κB-mediated cytokine expression but can also activate the JAK/STAT signaling pathway by upregulating IL-6. This creates a synergistic activation of innate and adaptive immunity.51,52 The bidirectional regulatory relationship between the TLR pathway and IL-6 is significant in GLM pathogenesis. TLR2/4 recognizes endogenous danger signals to activate NF-κB, promoting IL-6 secretion. As a key inflammatory mediator, IL-6 can provide feedback to enhance TLR signal sensitivity, forming a positive feedback loop of “TLR2/4-NF-κB-IL-6.” This exacerbates local immune microenvironment disorders in the breast. Therefore, the NF-κB/IL-6 signaling axis mediated by TLR2/4 is speculated to be the core pathway in GLM’s pathophysiology. Its abnormal activation may promote disease occurrence and progression by regulating immune cell recruitment, cytokine network imbalance, and autoimmune responses. Deeply analyzing the spatiotemporal expression characteristics and interactions of TLR2, TLR4, and IL-6 in GLM inflammatory tissues will provide critical clues for elucidating the immunopathological mechanism of the disease.
MAPK Signaling Pathway
Mitogen-activated protein kinases (MAPKs) constitute a highly shared family of serine/threonine protein kinases that transduce extracellular signals through a cascade of dual phosphorylation reactions involving tyrosine and serine residues. These kinases play a pivotal role in regulating cellular proliferation, differentiation, apoptosis, and stress responses.53 The MAPK family primarily includes three subfamilies: p38 MAPK, c-Jun N-terminal kinase (JNK), and extracellular signal-regulated kinase (ERK). Upon stimulation by pathogen-associated molecular patterns such as lipopolysaccharide (LPS), MAPKs are activated via a “three-tiered kinase cascade” mechanism (MAP3K→MAP2K→MAPK). This activation leads to the phosphorylation of downstream transcription factors, inducing the synthesis and secretion of proinflammatory cytokines like IL-6 and TNF-α.54 It is noteworthy that MAPKs exhibit synergistic effects with the NF-κB signaling pathway. By sharing upstream regulators (eg, TLR2/4) and downstream target genes (eg, the IL-1β promoter region), these two pathways form a proinflammatory signal amplification network that jointly regulates the intensity and duration of immune-inflammatory responses.54
As a key member of the MAPK family, the p38 MAPK signaling pathway plays a unique role in transcriptional regulation, cytokine expression, and tissue repair. When cells are stimulated by inflammatory factors (such as TNF-α and IL-1β) or oxidative stress, p38 MAPK translocates from the cytoplasm to the nucleus. There, it activates transcription factors like ATF2 and CREB through phosphorylation, driving the gene expression of IL-1β, TNF-α, chemokines, and adhesion molecules. This process regulates immune cell recruitment and the formation of an inflammatory microenvironment.55 In a GLM pathological model, rat mammary tissues exhibited significant overexpression of miR-451a, MKK3 (an upstream kinase of p38 MAPK), and phosphorylated p38 MAPK (p-p38 MAPK) (P < 0.01), suggesting that hyperactivation of the p38 MAPK pathway is involved in disease progression.56
In experimental studies on mastitis, Jiang KF and colleagues found that polydatin could reduce LPS-induced inflammatory cell infiltration and proinflammatory factor release in mouse mammary tissue by inhibiting the TLR2-mediated p38 MAPK/NF-κB signaling axis.57 Multiple clinical studies have further confirmed that TCM compounds effectively improve Staphylococcus aureus-induced mastitis by targeting MAPK (p38, ERK, JNK) and NF-κB (p65, IκBα) pathways and inhibiting TLR2/4 receptor expression.58 Zahoor A’s in vitro and in vivo studies have untangled that Staphylococcus aureus inhibits hyperactivated innate immune responses by downregulating TLR-mediated phosphorylation of MAPK and NF-κB signaling proteins, providing a new perspective on the pathophysiology of bacterial mastitis.59
Given the upstream dominance of NF-κB and MAPK pathways in the transcriptional regulation of inflammatory factors,60 synergistic intervention strategies targeting both have emerged as important directions in the treatment of inflammatory diseases. By blocking p38 MAPK phosphorylation or inhibiting NF-κB nuclear translocation, not only can the cascade release of proinflammatory cytokines be reduced, but the “inflammation-oxidative stress” vicious cycle can also be broken. This approach provides experimental evidence and clinical insights for the targeted treatment of chronic inflammatory diseases such as GLM.
PI3K/AKT/mTOR Signaling Pathway
The phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling pathway serves as a core pathway regulating cell proliferation, differentiation, metabolism, and survival. Its abnormal activation has been implicated in the pathological processes of various breast diseases, including breast cancer, breast hyperplasia, and GLM.61 In studies exploring the pathophysiology of GLM, a comparative analysis of breast tissue and peripheral blood samples from patients and healthy controls revealed significantly elevated phosphorylation levels of PI3K, AKT, and mTOR proteins in the observation group (GLM patients). This elevation was accompanied by a concurrent increase in the concentrations of proinflammatory cytokines such as IL-2 and IL-6, as well as the anti-inflammatory cytokine IL-10. Conversely, serum levels of immunoglobulins IgA, IgM, and IgG showed a significant decreasing trend. These differences between groups were statistically significant (P < 0.05).61
Further analysis indicated a clear correlation between the activation of the PI3K/AKT/mTOR pathway and the inflammatory response and immune dysfunction in GLM. As a hub for proinflammatory signals, this pathway regulates downstream transcription factors such as NF-κB and signal transducer and activator of transcription (STATs) through the phosphorylation and activation of AKT. This, in turn, promotes the gene transcription of cytokines like IL-6 and IL-10, leading to an imbalance in the local inflammatory microenvironment of the breast.61 It is noteworthy that IL-6, as a key mediator linking innate and adaptive immunity, can suppress B-cell differentiation and immunoglobulin class switching through the JAK/STAT pathway when abnormally elevated. This explains the observed decrease in IgA, IgM, and IgG levels.61
Mechanistically, it is speculated that the continuous activation of the PI3K/AKT/mTOR pathway drives the release of proinflammatory factors such as IL-2 and IL-6 by enhancing NF-κB activity, resulting in hyperactivation of macrophages, T-cells, and inflammatory cell infiltration. On the other hand, it impairs humoral immune responses by inhibiting B-cell receptor signaling and antibody production, forming a dual pathological state of “pro-inflammatory and immunosuppressive”.62 This association between pathway abnormalities and changes in immune molecule levels not only untangles the driving role of the PI3K/AKT/mTOR pathway in the initiation of GLM inflammation but also provides a new perspective for elucidating the molecular mechanisms underlying the reduction of immunoglobulins in the disease. It suggests that targeting this pathway could be a potential strategy to modulate immune imbalances in GLM.
Nrf2/HO-1 Signaling Pathway
The nuclear factor erythroid 2-related factor 2/Heme oxygenase-1 (Nrf2/HO-1) signaling pathway, as a core regulatory mechanism for endogenous antioxidant stress, plays a pivotal role in anti-inflammatory responses, oxidative defense, and the regulation of apoptosis.63 Abnormalities in this pathway are closely associated with the occurrence and development of various inflammatory diseases, making it an important target for current disease prevention and treatment.64
The transcription factor Nrf2 serves as a central regulatory molecule maintaining cellular redox homeostasis. It regulates the expression of over 200 downstream target genes, including key antioxidant enzymes such as HO-1 and NQO1, by recognizing antioxidant response elements (ARE).65,66 Under resting conditions, Nrf2 is bound to Kelch-like ECH-associated protein 1 (Keap1) and anchored in the cytoplasm in an inactive form. When cells are subjected to oxidative stress (such as reactive oxygen species, ROS, or electrophiles) or inflammatory stimuli, the cysteine residues of Keap1 undergo modification, leading to the dissociation of the Keap1-Nrf2 complex. The freed Nrf2 then transfers to the nucleus via a nuclear localization signal, where it binds to ARE and initiates the transcription program of antioxidant genes such as HO-1.67
As a key target gene product of Nrf2, HO-1 belongs to the heat shock protein 32 (HSP32) family. It exerts anti-inflammatory, antioxidant, and cytoprotective effects by catalyzing the degradation of heme into bilirubin, carbon monoxide, and iron ions. Bilirubin acts as a potent antioxidant to scavenge free radicals, carbon monoxide inhibits neutrophil chemotaxis and regulates macrophage polarization, and iron ions are sequestered by ferritin to avoid oxidative damage.68 Studies have confirmed that activation of the Nrf2/HO-1 pathway can inhibit proinflammatory signaling pathways such as NF-κB and MAPK, reducing the release of cytokines like TNF-α and IL-1β, thus forming a synergistic “antioxidant-anti-inflammatory” protective effect.
In experimental models of mastitis, Zhao et al69 found that dimethyl itaconate upregulates Nrf2 nuclear translocation and HO-1 protein expression while enhancing the stress response mediated by p38 and ERK phosphorylation. This inhibits the activation of TLR4 receptors and the phosphorylation of the NF-κB p65 subunit, thereby reducing LPS-induced pathological damage in mammary tissue and lowering levels of inflammatory factors such as IL-6 and TNF-α. This study reveals that the Nrf2/HO-1 pathway not only mitigates direct damage through antioxidant stress but also blocks inflammatory cascade reactions by inhibiting the TLR4/NF-κB signaling axis. This provides a new dual-pathway regulatory strategy for the treatment of inflammatory diseases. The elucidation of this mechanistic network highlights the bridging role of the Nrf2/HO-1 pathway in connecting oxidative defense and immune regulation, providing an important theoretical basis for the selection of therapeutic targets in chronic inflammatory diseases such as GLM.
Regulatory Mechanisms of Signaling Pathways in GLM Intervention by TCM
TCM exhibits multi-target advantages in the treatment of GLM, exerting anti-inflammatory, immunomodulatory, and antioxidant effects through the modulation of the aforementioned signaling pathways (Table 1).
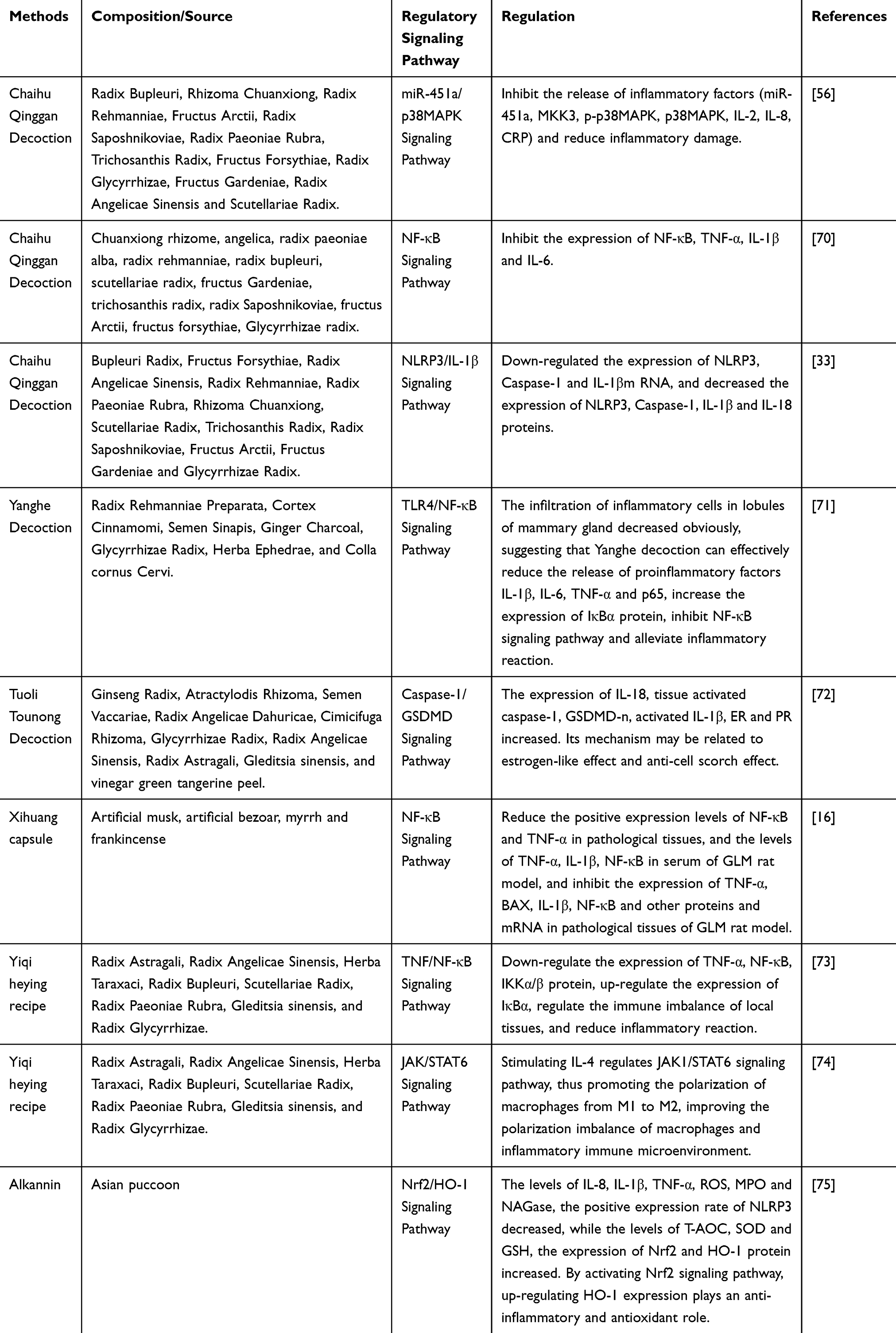 |
Table 1 Signaling Pathways and Mechanisms of Action in GLM Intervention by TCM |
Anti-Inflammatory Effects of Multi-Target Synergistic Pathway Regulation in TCM
TCM exerts pleiotropic anti-inflammatory effects in GLM by modulating interconnected signaling pathways. The classical formula Chaihu Qinggan Decoction (CHQD) suppresses inflammation through synergistic regulation of the NF-κB, NLRP3, and p38MAPK pathways. Its active components, such as baicalin and saikosaponin, inhibit NF-κB by blocking IκBα phosphorylation, reducing nuclear translocation of the p65 subunit and subsequent transcription of TNF-α, IL-1β, and IL-6.33,70 Simultaneously, CHQD downregulates NLRP3 inflammasome assembly, leading to a 30%-40% reduction in caspase-1 activation and mature IL-1β/IL-18 release in GLM rat models.33 In the p38MAPK pathway, inhibition of MKK3/p-p38MAPK decreases proinflammatory mediators (IL-8, CRP), alleviating neutrophil infiltration and tissue damage.56
Yanghe Decoction (YHD) targets the TLR4/NF-κB and MAPK pathways to inhibit inflammation. Cinnamic acid in YHD blocks TLR4-mediated MyD88 recruitment, inhibiting NF-κB activation and reducing IL-6/TNF-α expression by 45%.71 Additionally, inhibiting p38MAPK phosphorylation decreases COX-2/iNOS-dependent prostaglandin synthesis, mitigating oxidative tissue damage.57 These combined effects accelerate lesion resolution, with clinical studies showing a 25% increase in tumor reduction rate compared to monotherapy.71
Immunomodulation of the Inflammatory Microenvironment
TCM restores immune homeostasis in GLM by modulating macrophage polarization and the cytokine network. The Yiqi Heying Formula (YQHYF) promotes M2 macrophage polarization via the JAK/STAT6 pathway: IL-4 stimulation activates JAK1/STAT6, upregulates CD206 (an M2 marker), and downregulates iNOS (an M1 marker), shifting the M2/M1 ratio from 1:3 to 2:1.74 Simultaneously, YQHYF inhibits NF-κB-mediated hyperactivation of Th1 cells, reduces IFN-γ-driven granuloma formation, and enhances the Th2-type anti-inflammatory response (IL-10/TGF-β).73
The Tuoli Tounao Decoction (TTFD) suppresses cell pyroptosis and modulates sex hormone receptors. Saponins in TTFD block caspase-1/GSDMD-mediated pyroptotic pore formation, reducing IL-1β/IL-18 release by 60% and decreasing the pyroptosis rate from 25% to 8%.72 Ginsenoside Rg1 in TTFD, as an estrogen receptor modulator, increases Foxp3+ regulatory T cells (Tregs) by 60% and decreases IFN-γ+CD8+ T cells by 40%, thereby inhibiting autoreactive T cell responses.72 These mechanisms collectively correct immune imbalances, a key driver of GLM chronicity.
Protection Against Oxidative Stress
Oxidative stress exacerbates GLM inflammation, and TCM intervenes in this process by activating the Nrf2/HO-1 axis. The main component of purple gromwell, shikonin, dissociates Nrf2 from Keap1, promoting its nuclear translocation and upregulating HO-1 expression (a 2–3 fold increase). This enhances the activity of antioxidant enzymes (SOD, GSH), resulting in a 40% reduction in ROS levels.75 Additionally, shikonin disrupts the NLRP3-TXNIP interaction, decreasing the NLRP3-positive cell rate from 45% to 18% and alleviating pyroptotic inflammation.75 This dual effect on redox and inflammatory signaling creates a protective cycle: reduced ROS inhibits excessive activation of NF-κB, while decreased inflammation lowers the oxidative load. Experimental models demonstrate that shikonin treatment reduces MDA (a marker of lipid peroxidation) by 35% and MPO activity (a marker of neutrophil infiltration) by 28%, highlighting its potential in preventing tissue fibrosis.75
Conclusion
The pathophysiology of GLM involves the interaction of multiple signaling pathways, including NF-κB, NLRP3, JAK/STAT, and TLR, forming a complex inflammatory-immune regulatory network (Figure 1). Traditional Chinese medicine, through multi-component and multi-target interventions, has demonstrated clear efficacy in inhibiting inflammatory cascade reactions, regulating immune imbalances, and improving oxidative stress, providing diversified strategies for clinical treatment. In the clinical application and transformation of TCM prescriptions, there are some problems, such as unclear basis of effective substances and nonstandard quality standardization.Future research needs to further clarify the synergistic mechanisms of traditional Chinese medicine compounds and single components, combine metabolomics, proteomics, and other techniques to analyze precise targets, and promote the integration of traditional Chinese and Western medicine in the treatment of GLM towards precision and individualization.
Abbreviations
GLM, Granulomatous lobular mastitis; TCM, Traditional Chinese Medicine; IKK, IκB kinase; TLRs, Toll-like receptors; DAMPs, Damage-associated molecular patterns; PAMPs, Pathogen-associated molecular patterns; PRRs, Pattern recognition receptors; NLRP3, NOD-like receptor thermal protein domain associated protein 3; ROS, Reactive oxygen species; NK, Natural killer; IL-4, Interleukin-4; STAT6, Signal Transducer and Activator of Transcription 6; IL-6, Interleukin-6; NF-κB, Nuclear factor κB; MyD88, Myeloid differentiation factor 88; MAPKs, Mitogen-activated protein kinases; JNK, Jun N-terminal kinase; ERK, Extracellular signal-regulated kinase; LPS, Lipopolysaccharide; p-p38 MAPK, Phosphorylated p38 MAPK; STATs, Signal transducer and activator of transcription; Nrf2/HO-1, Nuclear factor erythroid 2-related factor 2/Heme oxygenase-1; ARE, Antioxidant response elements; Keap1, Kelch-like ECH-associated protein 1; HSP32, Heat shock protein 32.
Data Sharing Statement
All the original data of this study belong to the author.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Shandong Natural Science Foundation (No.ZR2022LZY019), Shandong Province Traditional Chinese Medicine Science and Technology Project (No.Z-2023002T) and Quality - improvement and Innovation Project for Doctoral Students of Shandong University of Traditional Chinese Medicine (No. 33[2024], Documents of the University’s Research Affairs).
Disclosure
All authors declare no financial or non-financial competing interests in this work.
References
1. Xiaoyan L, Qianjun C. Expert Consensus on the Diagnosis and Treatment of Granulomatous Lobular Mastitis with Traditional Chinese Medicine (2021 Edition). Chinese Journal of Integrated Traditional and Western Medicine Surgery. 2022;28(05):597–602.
2. Jiao Y, Chang K, Jiang Y, Zhang J. Identification of periductal mastitis and granulomatous lobular mastitis: a literature review. Ann Transl Med. 2023;11(3):158. doi:10.21037/atm-22-6473
3. Zhang M, Pu D, Feng D, Shi G, Li J. Rare and Complicated Granulomatous Lobular Mastitis (2000–2023): a Bibliometrics Study and Visualization Analysis. J Inflamm Res. 2024;17:3709–3724. doi:10.2147/JIR.S465844
4. Ozer L, Koksal H. Whole exome sequencing for identifying rare genetic variants related to idiopathic granulomatous mastitis. Clin Rheumatol. 2025;44(4):1843–1850. doi:10.1007/s10067-025-07343-w
5. Houcine Y, Maaoui A, Riahi N, Sakhri S, Driss M. The histologic diagnosis and the management of cystic neutrophilic granulomatous mastitis: a case report. J Med Case Rep. 2024;18(1):541. doi:10.1186/s13256-024-04787-7
6. Tingting Z, Hao Y, Shengjia W, Yongzhong Y. The Pathophysiology of Granulomatous Lobular Mastitis and Its Association with Autoimmunity: a Review and Prospect. J Southeast Univ. 2024;43(04):650–654.
7. Yuan QQ, Xiao SY, Farouk O, et al. Management of granulomatous lobular mastitis: an international multidisciplinary consensus (2021 edition). Military Med Res. 2022;9(1):20. doi:10.1186/s40779-022-00380-5
8. Chan CW. The treatment conundrum that is idiopathic granulomatous mastitis. Ann Acad Med Singap. 2021;50(8):596–597. doi:10.47102/annals-acadmedsg.2021286
9. Dorrington MG, Fraser IDC. NF-κB Signaling in Macrophages: dynamics, Crosstalk, and Signal Integration. Front Immunol. 2019;10:705. doi:10.3389/fimmu.2019.00705
10. Wertz IE, Dixit VM. Signaling to NF-kappaB: regulation by ubiquitination. Cold Spring Harb Perspect Biol. 2010;2(3):a003350. doi:10.1101/cshperspect.a003350
11. Lu K, Zhao J, Liu W. Macrophage stimulating 1-induced inflammation response promotes aortic aneurysm formation through triggering endothelial cells death and activating the NF-κB signaling pathway. J Recept Signal Transduction Res. 2020;40(4):374–382. doi:10.1080/10799893.2020.1738484
12. Hoffmann AA, Weeks AR, Sgrò CM. Opportunities and challenges in assessing climate change vulnerability through genomics. Cell. 2021;184(6):1420–1425. doi:10.1016/j.cell.2021.02.006
13. Spehlmann ME, Eckmann L. Nuclear factor-kappa B in intestinal protection and destruction. Curr Opin Gastroenterol. 2009;25(2):92–99. doi:10.1097/MOG.0b013e328324f857
14. Zhang Z, Rigas B. NF-kappaB, inflammation and pancreatic carcinogenesis: NF-kappaB as a chemoprevention target (review). Int J Oncol. 2006;29(1):185–192.
15. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2(17023). doi:10.1038/sigtrans.2017.23
16. Dai X, Maoyang X. Exploring the Mechanism of Xihuang Capsule in the Treatment of Granulomatous Mastitis Based on Network Pharmacology and Experimental Validation. Chin J Hospital Pharm. 2022;42(09):889–895.
17. Khan MZ, Khan A, Xiao J, et al. Overview of Research Development on the Role of NF-κB Signaling in Mastitis. Animals. 2020;10(9):1625. doi:10.3390/ani10091625
18. Friedrichsen S, Harper CV, Semprini S, et al. Tumor necrosis factor-alpha activates the human prolactin gene promoter via nuclear factor-kappaB signaling. Endocrinology. 2006;147(2):773–781. doi:10.1210/en.2005-0967
19. Li H, Lin X. Positive and negative signaling components involved in TNFalpha-induced NF-kappaB activation. Cytokine. 2008;41(1):1–8. doi:10.1016/j.cyto.2007.09.016
20. Kong C, Zhang C, Wu Y, et al. The expression and meaning of CD68, CD163, CD57, and IgG4 in granulomatous lobular mastitis. Gland Surg. 2020;9(4):936–949. doi:10.21037/gs-20-419
21. Coombe RF, Hamed H. An update on granulomatous mastitis: a rare and complex condition. Br J Hosp Med. 2021;82(5):1–7. doi:10.12968/hmed.2020.0718
22. King EM, Holden NS, Gong W, Rider CF, Newton R. Inhibition of NF-kappaB-dependent transcription by MKP-1: transcriptional repression by glucocorticoids occurring via p38 MAPK. J Biol Chem. 2009;284(39):26803–26815. doi:10.1074/jbc.M109.028381
23. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi:10.1016/s1097-2765(02)00599-3
24. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. doi:10.1016/j.cell.2010.01.040
25. Sharma D, Kanneganti TD. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213(6):617–629. doi:10.1083/jcb.201602089
26. Wang Z, Zhang S, Xiao Y, et al. NLRP3 Inflammasome and Inflammatory Diseases. Oxid Med Cell Longev. 2020;2020:4063562. doi:10.1155/2020/4063562
27. Zeng C, Wang R, Tan H. Role of Pyroptosis in Cardiovascular Diseases and its Therapeutic Implications. Int J Biol Sci. 2019;15(7):1345–1357. doi:10.7150/ijbs.33568
28. Ran X, Yan Z, Yang Y, et al. Dioscin Improves Pyroptosis in LPS-Induced Mice Mastitis by Activating AMPK/Nrf2 and Inhibiting the NF-κB Signaling Pathway. Oxid Med Cell Longev. 2020;2020:8845521. doi:10.1155/2020/8845521
29. Yu S, Liu X, Yu D, Changyong E, Yang J. Morin Protects LPS-Induced Mastitis via Inhibiting NLRP3 Inflammasome and NF-κB Signaling Pathways. Inflammation. 2020;43(4):1293–1303. doi:10.1007/s10753-020-01208-x
30. Guo W, Liu J, Sun J, et al. Butyrate alleviates oxidative stress by regulating NRF2 nuclear accumulation and H3K9/14 acetylation via GPR109A in bovine mammary epithelial cells and mammary glands. Free Radic Biol Med. 2020;152:728–742. doi:10.1016/j.freeradbiomed
31. Parys K, Colaianni NR, Lee HS, et al. Signatures of antagonistic pleiotropy in a bacterial flagellin epitope. Cell Host Microbe. 2021;29(4):620–634.e9. doi:10.1016/j.chom
32. Huixia H, Yan L. Research Progress on the Role of Interleukin-18 in Organ Transplantation. Chin J Microcirculation. 2018;28(04):72–74+80.
33. Yao Z, Lifang L, Jialu L, et al. Mechanism of Chaihu Qinggan Decoction in Treating Rat Model of Granulomatous Lobular Mastitis by Intervening in NLRP3/IL-1β Pathway. Chin J Experiment Tradit Med Formulas. 2022;28(15):1–7.
34. Wang Y, Zhang H, Xu Y, Peng T, Meng X, Zou F. NLRP3 induces the autocrine secretion of IL-1β to promote epithelial-mesenchymal transition and metastasis in breast cancer. Biochem Biophys Res Commun. 2021;560:72–79. doi:10.1016/j.bbrc.2021.04.122
35. Liu Y, Zhang J, Zhou YH, et al. IL-6/STAT3 signaling pathway is activated in plasma cell mastitis. Int J Clin Exp Pathol. 2015;8(10):12541–12548.
36. Daniel B, Nagy G, Horvath A, et al. The IL-4/STAT6/PPARγ signaling axis is driving the expansion of the RXR heterodimer cistrome, providing complex ligand responsiveness in macrophages. Nucleic Acids Res. 2018;46(9):4425–4439. doi:10.1093/nar/gky157
37. Liu Y, Sun Y, Zhou Y, et al. Sinomenine hydrochloride inhibits the progression of plasma cell mastitis by regulating IL-6/JAK2/STAT3 pathway. Int Immunopharmacol. 2020;81:106025. doi:10.1016/j.intimp.2019.106025
38. Back DB, Choi BR, Han JS, et al. Characterization of Tauopathy in a Rat Model of Post-Stroke Dementia Combining Acute Infarct and Chronic Cerebral Hypoperfusion. Int J Mol Sci. 2020;21(18):6929. doi:10.3390/ijms21186929
39. El-Sherbiny M, El-Sayed RM, Helal MA, et al. Nifuroxazide Mitigates Angiogenesis in Ehlrich’s Solid Carcinoma: molecular Docking, Bioinformatic and Experimental Studies on Inhibition of IL-6/Jak2/Stat3 Signaling. Molecules. 2021;26(22):6858. doi:10.3390/molecules26226858
40. Zhang F, Zhao P, Qian Z, Zhong M. Central Nervous System Inflammation Induced by Lipopolysaccharide Up-Regulates Hepatic Hepcidin Expression by Activating the IL-6/JAK2/STAT3 Pathway in Mice. Front Nutr. 2021;8:649640. doi:10.3389/fnut.2021.649640
41. Quach H, Wilson D, Laval G, et al. Different selective pressures shape the evolution of Toll-like receptors in human and African great ape populations. Hum Mol Genet. 2013;22(23):4829–4840. doi:10.1093/hmg/ddt335
42. El-Sherbiny M, Eisa NH, Nf AE-M, Elsherbiny NM, Said E, Khodir AE. Anti-inflammatory/anti-apoptotic impact of betulin attenuates experimentally induced ulcerative colitis: an insight into TLR4/NF-kB/caspase signalling modulation. Environ Toxicol Pharmacol. 2021;88:103750. doi:10.1016/j.etap.2021.103750
43. Lim KH, Staudt LM. Toll-like receptor signaling. Cold Spring Harb Perspect Biol. 2013;5(1):a011247. doi:10.1101/cshperspect.a011247
44. Zhang Y, Liu J, Wang C, Liu J, Lu W. Toll-Like Receptors Gene Polymorphisms in Autoimmune Disease. Front Immunol. 2021;12:672346. doi:10.3389/fimmu.2021.672346
45. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461. doi:10.3389/fimmu.2014.00461
46. Han L. Clinical Efficacy and Immune Mechanism of Jiawei Tounong Powder in the Treatment of Granulomatous Mastitis at the Abscess Stage Based on the Expression Levels of IL-6, TLR2, and TLR4. Shandong Univ Tradit Chin Med. 2023. doi:10.27282/d.cnki.gsdzu.2023.001276
47. Bao F, Jinfeng W, Xiaofei L, et al. Expression Significance of TLR2, TLR4, and HMGB1 in Peripheral Blood of Patients with Plasma Cell Mastitis. Chin J Curr Adv Gen Surg. 2021;24(04):316–318+322.
48. Daoyuan T, Linlin Z, Zhen L, et al. Expression and Significance of Toll-like Receptors 2/4 in Non-lactating Mastitis Tissues. Chin J Breast Dis. 2020;14(02):92–97.
49. Dutta D, Jana M, Majumder M, Mondal S, Roy A, Pahan K. Selective targeting of the TLR2/MyD88/NF-κB pathway reduces α-synuclein spreading in vitro and in vivo. Nat Commun. 2021;12(1):5382. doi:10.1038/s41467-021-25767-1
50. Xiong T, Zheng X, Zhang K, et al. Ganluyin ameliorates DSS-induced ulcerative colitis by inhibiting the enteric-origin LPS/TLR4/NF-κB pathway. J Ethnopharmacol. 2022;289:115001. doi:10.1016/j.jep.2022.115001
51. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi:10.1101/cshperspect.a001651
52. Anthoney N, Foldi I, Hidalgo A. Toll and Toll-like receptor signalling in development. Development. 2018;145(9):dev156018. doi:10.1242/dev.156018
53. Canovas B, Nebreda AR. Diversity and versatility of p38 kinase signalling in health and disease. Nat Rev Mol Cell Biol. 2021;22(5):346–366. doi:10.1038/s41580-020-00322-w
54. Guma M, Stepniak D, Shaked H, et al. Constitutive intestinal NF-κB does not trigger destructive inflammation unless accompanied by MAPK activation. J Exp Med. 2011;208(9):1889–1900. doi:10.1084/jem.20110242
55. Song YF, Zhao L, Bc W, et al. Corrigendum to ‘The circular RNA TLK1 exacerbates myocardial ischemia/reperfusion injury via targeting miR-214/RIPK1 through TNF signaling pathway. Free Radic Biol Med. 2022;179(179):435–436. doi:10.1016/j.freeradbiomed.2021.03.012
56. Jingqun S, Jie L, Lian L, et al. Mechanism of Chaihu Qinggan Decoction in the Intervention of Granulomatous Lobular Mastitis in Rats Based on miR-451a/p38MAPK Pathway. Sichuan J Tradit Chin Med. 2025;43(02):105–111.
57. Jiang KF, Zhao G, Deng GZ, et al. Polydatin ameliorates Staphylococcus aureus-induced mastitis in mice via inhibiting TLR2-mediated activation of the p38 MAPK/NF-κB pathway. Acta Pharmacol Sin. 2017;38(2):211–222. doi:10.1038/aps.2016.123
58. Akhtar M, Shaukat A, Zahoor A, et al. Hederacoside-C Inhibition of Staphylococcus aureus-Induced Mastitis via TLR2 & TLR4 and Their Downstream Signaling NF-κB and MAPKs Pathways In Vivo and In Vitro. Inflammation. 2020;43(2):579–594. doi:10.1007/s10753-019-01139-2
59. Zahoor A, Yang Y, Yang C, et al. Gas6 negatively regulates the Staphylococcus aureus-induced inflammatory response via TLR signaling in the mouse mammary gland. J Cell Physiol. 2020;235(10):7081–7093. doi:10.1002/jcp.29604
60. Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92(2):689–737. doi:10.1152/physrev.00028.2011
61. Zhang HW, Hu JJ, Fu RQ, et al. Flavonoids inhibit cell proliferation and induce apoptosis and autophagy through downregulation of PI3Kγ mediated PI3K/AKT/mTOR/p70S6K/ULK signaling pathway in human breast cancer cells. Sci Rep. 2018;8(1):11255. doi:10.1038/s41598-018-29308-7
62. Yang Z, Xijing W, Shuqun Z, et al. Study on the Pathogenesis of Granulomatous Lobular Mastitis Involving PI3K/AKT/mTOR Pathway and Immunoglobulin. Chin Med Herald. 2018;15(30):8–10.
63. Zhang Q, Liu J, Duan H, Li R, Peng W, Wu C. Activation of Nrf2/HO-1 signaling: an important molecular mechanism of herbal medicine in the treatment of atherosclerosis via the protection of vascular endothelial cells from oxidative stress. J Adv Res. 2021;34:43–63. doi:10.1016/j.jare.2021.06.023
64. Wu D, Zhang X, Liu L, Guo Y. Key CMM Combinations in Prescriptions for Treating Mastitis and Working Mechanism Analysis Based on Network Pharmacology. Evid Based Complement Alternat Med. 2019;2019:8245071. doi:10.1155/2019/8245071
65. Pillai R, Hayashi M, Zavitsanou AM, Papagiannakopoulos T. NRF2: kEAPing Tumors Protected. Cancer Discov. 2022;12(3):625–643. doi:10.1158/2159-8290.CD-21-0922
66. Deng HF, Yue LX, Wang NN, et al. Mitochondrial Iron Overload-Mediated Inhibition of Nrf2-HO-1/GPX4 Assisted ALI-Induced Nephrotoxicity. Front Pharmacol. 2021;11:624529. doi:10.3389/fphar.2020.624529
67. Xu C, Song Y, Wang Z, et al. Pterostilbene suppresses oxidative stress and allergic airway inflammation through AMPK/Sirt1 and Nrf2/HO-1 pathways. Immun Inflamm Dis. 2021;9(4):1406–1417. doi:10.1002/iid3.490
68. Dang X, He B, Ning Q, et al. Alantolactone suppresses inflammation, apoptosis and oxidative stress in cigarette smoke-induced human bronchial epithelial cells through activation of Nrf2/HO-1 and inhibition of the NF-κB pathways. Respir Res. 2020;21(1):95. doi:10.1186/s12931-020-01358-4
69. Zhao C, Jiang P, He Z, et al. Dimethyl itaconate protects against lippolysacchride-induced mastitis in mice by activating MAPKs and Nrf2 and inhibiting NF-κB signaling pathways. Microb Pathog. 2019;133:103541. doi:10.1016/j.micpath.2019.05.024
70. Lina M, Jingjing W, Meina Y, et al. Exploring the Mechanism of Chaihu Qinggan Decoction in the Treatment of Granulomatous Mastitis Through the Combination of Network Pharmacology and Experimental Validation. J New Chin Med. 2024;56(23):16–24. doi:10.13457/j.cnki.jncm.2024.23.003
71. Haoyu L, Minmin Y, Mengdi Z, et al. Exploring the Mechanism of Yanghe Decoction in the Treatment of Granulomatous Lobular Mastitis Based on Network Pharmacology and Experimental Validation. Chin J Inf Traditional Chin Med. 2025;32(03):34–41. doi:10.19879/j.cnki.1005-5304.202407330
72. Zhao Z, Ximeng Z, Tangshun W, et al. Effect of Tuoli Tounong Decoction on Caspase-1/GSDMD Signaling Pathway in Granulomatous Lobular Mastitis. Global Traditional Chin Med. 2022;15(09):1537–1542.
73. Ziwei D, Qiqi S, Xiaofei L. Mechanism Study of Yiqi Heying Traditional Chinese Medicine in Regulating TNF-α-induced Immune Status and NF-κB Signaling Pathway Expression in Granulomatous Lobular Mastitis. Lishizhen Medicine and Materia Medica Res. 2024;35(11):2614–2618.
74. Yanjun C. Clinical Efficacy and Mechanism Study of Yiqi Heying Formula in the Treatment of Granulomatous Mastitis Based on Macrophage Phenotype Polarization from JAK/STAT6 Pathway. Shandong Univ Tradit Chin Med. 2023. doi:10.27282/d.cnki.gsdzu.2023.001275
75. Fanfan L, Yang X, Xiaoxu W. Therapeutic Effect of Shikonin on Experimental Rat Granulomatous Lobular Mastitis by Regulating Nrf2/HO-1 Signaling Pathway. Chin J Clin Anat. 2024;42(01):26–32. doi:10.13418/j.issn.1001-165x.2024.1.06
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.