")
Back to Journals » Journal of Inflammation Research » Volume 17
Analysis of a Familial IgAN Accompanied by COL4A3 Mutation
Authors Lin SQ, Deng JX, Jiang H, Xiang SH, Lin WJ, Qian FQ, Wu SC, Wang FZ
Received 13 August 2024
Accepted for publication 12 November 2024
Published 20 November 2024 Volume 2024:17 Pages 9269—9283
DOI https://doi.org/10.2147/JIR.S480279
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Sen-Qing Lin,* Jin-Xiu Deng,* Hui Jiang, Shi-Hong Xiang, Wen-Jing Lin, Feng-Qi Qian, Sen-Chao Wu, Fu-Zhen Wang
Department of Nephrology, Longyan First Affiliated Hospital of Fujian Medical University, Longyan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Fu-Zhen Wang; Sen-Chao Wu, Department of Nephrology, Longyan First Affiliated Hospital of Fujian Medical University, No. 105 Jiuyi North Road, Longyan, Fujian, People’s Republic of China, Tel +86 13599330411 ; +86 13860217030, Email [email protected]; [email protected]
Objective: IgA nephropathy (IgAN) is the prevailing primary glomerulonephritis globally and is the key factor contributing to the onset of chronic kidney disease and eventual progression to end-stage renal disease. This study aims to explore the mutated gene in a familial case of IgAN, especially COL4A3.
Methods: Family lineages diagnosed with familial IgAN at the Longyan First Hospital of Fujian Medical University were selected for this study, followed by comprehensive whole exome sequencing. After obtaining the sequencing data, bioinformatics analyses were conducted to discern potential mutated genes. These findings within the familial lineages were validated using Sanger sequencing to identify IgAN-associated mutated genes, based on literature references and in accordance with the genetic variation classification criteria determined by the American College of Medical Genetics and Genomics.
Results: Whole exome sequencing analysis of familial IgAN family lineages led to the identification of a total of 212,187 single nucleotide variant/insertion-deletion mutation sites, annotated using ANNOVAR. These sites were screened targeting four mutated genes, revealing three mutations of undetermined significance along with a single disease-causing mutation: a heterozygous disease-causing mutation within COL4A3 (p.G1167R). This mutation manifested across seven family members within the group, encompassing both family members diagnosed with kidney disease and those serving as normal carriers. Notably, one additional family member with IgAN within the familial lineage exhibited an absence of the pathogenic mutation.
Conclusion: This study identified four mutated genes that may be involved in the onset and progression of IgAN, further revealing the complex multigenic inheritance characteristics of IgAN. The underlying mechanisms of the disease require further investigation. Additionally, we discovered potential mutations associated with known genetic kidney diseases, such as COL4A3 mutations. Therefore, we recommend comprehensive genetic screening in familial cases of IgAN to improve disease diagnosis and facilitate genetic counseling.
Keywords: IgA nephropathy, COL4A3, bioinformatics analysis, familial, whole exome sequencing
Introduction
Primary IgA nephropathy (IgAN) represents a clinically significant immunopathological syndrome characterized by the primary manifestation of IgA or IgA-based immunoglobulins within the glomerular mesangial region. It is the most prevalent primary glomerular disorder globally.1 Despite the typically slow progression observed in the most cases, a notable subset of patients—approximately 25% to 30%—experience renal failure within a span of 20 to 25 years following disease onset, a consequence of the yet incompletely elucidated pathogenesis.2,3 This lack of understanding, diagnostic precision, and effective therapeutic methods constitutes the principal driver behind the chronic kidney disease and end-stage renal disease (ESRD) burden, imposing substantial socioeconomic strains on both healthcare systems and patients. It is worth noting that the incidence of IgAN demonstrates marked familial clustering and racial differences. Notably, first-degree relatives of individuals with IgAN exhibit an increased susceptibility to the disorder.
Predominantly prevalent among East Asians, followed by Europeans, IgAN exhibits a low incidence rate among individuals of African descent, thereby underscoring the profound influence of genetic factors in disease pathogenesis.4–10 With the widespread integration of second-generation sequencing technology, the research of hereditary kidney disorders has witnessed remarkable advancement. Presently, extensive studies have implicated nearly 30 susceptibility genes in sporadic IgAN.11–18 However, research relating to familial IgAN remains notably scarce, necessitating further assessment of the genetic underpinnings of IgAN.
Familial IgAN presents an increased risk of progression to ESRD when compared to sporadic cases. Recent chain analyses have pinpointed several loci of significant relevance, including 2q36, 3p23-24, 4q26-31, 6q22-23, and 17q12-22.19–21 yet they fail to definitively delineate the causal genes.7 Data from studies have indicated that changes in type IV collagen are present in approximately 20% of familial IgAN cases, encompassing variants within COL4A3/COL4A4 genes, and to a lesser extent, COL4A5 variants. Conversely, the likelihood of concurrent mutations in type IV collagen genes in sporadic IgAN is less than 10%. Situated within the chromosome 2q36 region, the COL4A3/COL4A4 genes exhibit significant association with IgAN. However, the precise relationship between IgAN and mutations in type IV collagen genes remains ambiguous, as IgAN has not been historically classified among diseases linked to mutations in type IV collagen genes as per existing literature.
In the current study, comprehensive whole exome sequencing analysis of a familial IgAN lineage in China identified four susceptibility genes, comprising three notably unidentified mutant genes along with one pathogenic mutation within the type IV collagen gene, COL4A3 (p.G1167R). This underscores the genetic complexity inherent to IgAN, with a broad spectrum of mutation phenotypes observed within type IV collagen genes. Consequently, there is a pressing need to broaden the spectrum of mutation phenotypes targeted in screening efforts and to contemplate diseases concomitant with mutations in type IV collagen genes within the Alport syndrome (AS).
Materials and Methods
Study Materials
Study Participants
Participants for the study were selected from a familial IgAN lineage diagnosed at Longyan First Affiliated Hospital of Fujian Medical University. The family, comprising 18 members, encompassed 10 males and 8 females, with ages spanning from 8 to 71 years. Ethical principles outlined in the Declaration of Helsinki were adhered to in the study, which received approval from the Ethics Committee of the hospital under acceptance number [2022] Lun Audit Scientific Research No. (168). Informed consent forms were signed by all participants.
Inclusion and Exclusion Criteria
This study included family lines meeting the diagnostic criteria for familial IgAN. Diagnostic criteria included confirmation of IgAN through renal biopsy tissue examination in two or more related family members within the same family.3,5,22,23 Exclusion criteria encompassed secondary IgAN resulting from allergic purpura, systemic lupus erythematosus, Sjögren’s syndrome, ankylosing spondylitis, malignant tumors, psoriasis, chronic liver disease, infectious diseases such as tuberculosis, HIV infection, and other conditions.
Renal Pathology Experimental Methods
Percutaneous Renal Biopsy
Renal biopsy specimens include more than 10 glomeruli. All specimens undergo staining with hematoxylin-eosin (HE), periodic acid-Schiff (PAS), periodic acid-methenamine silver (PAM), and Masson’s trichrome. Immunofluorescence staining is performed for IgG, IgA, IgM, C3, C4, and C1q.
Light Microscopy
Renal tissues are fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned continuously at a thickness of 2 μm. Sections are stained with HE, PAS, PASM, and Masson stains according to the following protocols:
PASM Staining
1: Dewaxing and hydration; 2: Periodic acid oxidation; 3: Methenamine silver staining; 4:2% sodium thiosulfate treatment; 5: Hematoxylin counterstaining; 6: Dehydration and clearing; 7: Mounting; 8: Microscopic observation.1.3.3 Immunopathology: Direct immunofluorescence is performed using frozen sections (3–5 µm thick). The sections are air-dried at room temperature and fixed in acetone at 4°C. After washing with PBS, fluorochrome-labeled antibodies are added. The slides are incubated at 37°C in an incubator. Unbound antibodies are washed off with PBS, and the sections are observed under a fluorescence microscope.
Electron Microscopy
The specimens are cut into small pieces measuring 1 mm × 1 mm × 1 mm. The tissue, pre-fixed in 3% glutaraldehyde, is further sectioned into ultrathin slices about 50 nm thick. The sections are double-stained with uranyl acetate and lead citrate and observed using a transmission electron microscope.
Research Method
General Data Collection
Detailed medical histories were obtained from the family members. Review of hospitalization data and outpatient records was conducted, along with auxiliary examinations, encompassing blood routine, biochemistry, urine sediment analysis, urinary CT, skin type IV collagen biopsy for certain members, and immunofluorescence to detect type IV collagen expression. Furthermore, ocular examination, pure tone audiometry, and other relevant tests were conducted.
Whole Exome Sequencing and Bioinformatics Analysis
Following informed consent from participants with pre-existing conditions and their family members, peripheral blood samples ranging from 2 mL to 5 mL were extracted and stored at −80 °C for an extended period. Genomic DNA extraction was performed based on the instructions provided by the manufacturer in the kit manufactured by Sangon Biotech (Shanghai) Co., Ltd. Peripheral blood samples were collected from a total of 10 family members, including the core participants and control members (I:1/I:2/I:3/I:4/II:2/II:1/II:4/II:5), for whole exome sequencing.
The Agilent 12g high-depth kit was uniformly used to generate raw data. Quality assessment of the sequencing data was conducted visually using FastQC and analyzed in comparison with the reference genome (hg38). This analysis involved assessing the ratio of Q20 and Q30 sequences, average depth of coverage, and ratio of exon regions covered by 1X~100X. After ensuring data quality and reliability, the GATK HaplotypeCaller method was used to detect single nucleotide variant (SNV)/insertion-deletion (InDel). Subsequently, all known information regarding SNV/InDel loci was compared and analyzed using ANNOVAR to assess variant frequency, functional characteristics, conservation, pathogenicity and so on. Screening for mutated genes among all SNV/InDel variants was performed through case and control analysis using human population databases (Alt(1000g), ExAC, gnomAD), and computerized prediction software (SIFT, PolyPhen-2, MutationTaster, CADD).
In this study, 5 participants (I:1/I:4/II:1/II:2/II:5) were considered as family cases, with at least 4 cases carrying the mutated gene, and 2 outgroup members (I:2/II:7) were used as controls to identify IgAN-related mutated genes. The detailed process is illustrated in Figure 1. Finally, mutated genes were screened based on previous literature reports and American College of Medical Genetics and Genomics (ACMG) criteria.
 |
Figure 1 Screening Process. Notes: Alt(1000g), ExAC, gnomAD represent population databases, SIFT: D (damaging); Polyphen2_HDIV: D (probably damaging); MutationTaster: (A = disease causing automatically, D = disease causing); Cadd ≥ 20: pathogenic. |
The Alt(1000g) database (http://www.internationalgenome.org/) represents the first database based on extensive data from numerous individual genomes, providing a comprehensive resource of human genetic and mutation data. ExAc (http://exac.broadinstitute.org/) is a collaboration among international researchers compiling complete exon sequences from 60,706 individuals and related genetic information into a mutation database. GnomAD (https://gnomad.broadinstitute.org/) is a resource developed by an international consortium of researchers, housing a dataset containing 123,136 exon sequences and 15,496 whole genome sequences from unrelated individuals.
PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org) are tools used for predicting whether missense mutations (mutations resulting in changes in amino acids) lead to changes in protein structure or function. MutationTaster is another commonly used tool for this purpose, accessible at http://www.mutationtaster.org/, used to predict whether a mutation affects transcriptional shearing, among other factors. Category III (CADD) is a comprehensive prediction tool facilitating quantitative ranking of functional, deleterious, and disease-causing variants across various functional categories, effect sizes, and genetic structures.24 Suggested pathogenicity thresholds for selection include: Polyphen2_HDIV (official annotation, B = benign, P = possibly damaging, D = probably damaging), SIFT (official annotation, D = damaging, T = tolerated), MutationTaster (official Note, A = disease causing automatic, D = disease causing, N = polymorphism, P = polymorphism_automatic), and CAdd ≥ 20.24 The ACMG Genetic Variation Classification Criteria and Guidelines for Interpretation of Screened Variant Loci categorize pathogenicity as follows: pathogenic, likely pathogenic, benign, likely benign, and of uncertain significance.
Sanger Sequencing for Lineage Validation
Mutated genes were identified based on a bioinformatics analysis. Gene mutation results of all family members were acquired through the design of specific primers, PCR amplification, and subsequent sequencing. Following this, the results of Sanger sequencing validation were analyzed. By integrating these findings with genealogical charts, we determined the carrier status of the detected gene variants within the family lineage.
Results
General Results
After clinical investigation, inquiry about family history, and retrieval of outpatient and inpatient treatment data from our hospital and other hospitals, 18 family members were included in the study. Among them, IgAN was diagnosed in 3 cases (II:1/II:2/III:6) following renal puncture biopsy, while 2 cases (I:1/I:4) were diagnosed with ESRD and underwent hemodialysis for treatment, and 1 case (II:5) exhibited renal puncture biopsy pathology indicating thin basement membrane nephropathy. In total, renal disease was present in 6 patients. The remaining 3 cases (I:3/III:1/III:4) exhibited a single abnormal urinalysis with an unknown phenotype. Details of the family lineage chart are presented in Figure 2, and renal puncture pathology details are provided in Figure 3 Participant II:1 information is unavailable, and there are no high-definition pathology charts.
 |
Figure 2 Familial IgAN lineage map. Individual wild-type genes are represented by letters A, B, C, and D, while mutant genes are indicated by A*, B*, C*, D*. (A) COL4A3 gene; (B) ZNF107 gene; (C) ZIC5 gene; and (D) WDR76 gene. Members not labeled in the figure: no blood specimens were collected. |
 |
Figure 3 Renal pathology information. Notes: (A–C) Correspond to light microscopic PASM staining, immunofluorescence, and electron microscopy images of family member II:2, respectively. These images reveal partial glomerulosclerosis, with IgA deposition (2+) along the tethered membrane, along with segmental thinning of the basement membrane. The diagnosis rendered is IgA nephropathy, classified according to Lee’s grading as grade III. (D–F) Correspond to electron microscopic PASM staining, immunofluorescence, and electron microscopy images of family member II:5, respectively. While light microscopy findings were nonspecific, fluorescence revealed the presence of IgA, IgG, IgM, C3, with negative staining for C1q. Electron microscopy unveiled diffuse thinning of the basement membrane, measuring approximately 140 to 200 nm in thickness, with an intact dense layer devoid of tearing or layering changes. Notably, segmental fusion of the pedicle was observed. (G–I) Depict light microscopic PASM staining, immunofluorescence, and electron microscopy images of family member III:6 respectively. Notably, these images showcase focal proliferative IgA nephropathy characterized by diffuse, globular IgA deposition (++) along the tethered zone. Notably, no discernible thickening or thinning of the glomerular basement membrane is observed. The diagnosis provided is IgA nephropathy, with Lee’s grading indicating grade III. |
As described below, presenter (II:2), a 33-year-old male, with a history of “foamy urine for 1 year”, presented to our clinic in September 2022. No history of hypertension or diabetes mellitus was reported. Occult blood and proteinuria, with polymorphic erythrocytes, were observed in outpatient and inpatient urinalysis results. The 24-hour urinary protein was measured at 3961 mg/24 hours, urine volume at 23.38 dL, and creatinine levels were normal. Furthermore, he tested negative for anti-neutrophil cytoplasmic antibody (ANCA), anti-glomerular basement membrane IgM antibody, immune set, viral hepatitis-related antibody, infectious pathogenetic antibody, tumor index, immunofixation electrophoresis, complement C3, C4, IgA, IgM, IgG, etc. A renal puncture biopsy was performed, revealing IgAN (Figure 3). Following treatment with an angiotensin-converting enzyme inhibitor (ACEI) or angiotensin II receptor antagonist (ARB), the 24-hour urine protein was reduced to 770 mg/24h in May 2023, with the 24-hour urine volume measuring 27.5 dL.
Five additional cases of renal disease were identified within the family, each with distinct medical histories:
The mother (I:1) underwent a renal biopsy at the age of 40, revealing pathology indicative of nephritis (pathology report was lost). At the age of 59, she developed chronic kidney disease stage 5 and commenced regular hemodialysis at age 60.
The brother (II:1) was diagnosed with proteinuria at age 29, with a history of long-term hypertension. His 24-hour urine protein measured 3.088 g, and creatinine level was 78 μmol/L. Renal puncture biopsy pathology indicated IgAN. Treatment included 60 mg of prednisone, gradually tapered, along with ACEI/ARB therapy. Subsequent reviews of 24-hour urine protein indicated levels below 0.5 g, with normal creatinine levels.
The maternal aunt (I:4), aged 67, was hospitalized due to abdominal pain and nausea. With a history of hypertension for over 7 years, tests revealed creatinine levels of 414 μmol/L and urea nitrogen levels of 19.5 μmol/L. A CT scan indicated bilateral renal atrophy, indicating stage 5 chronic kidney disease. She was admitted for hemodialysis 2 years later; however, a renal biopsy was not performed.
A distant cousin (II:5), aged 49, underwent routine urine tests revealing proteinuria and hematuria across multiple medical facilities. The cousin was admitted to Longyan First Affiliated Hospital of Fujian Medical University in March 2024 due to persistent urinalysis abnormalities and a history of abdominal pain and nausea. 24-hour urine protein measured 2655.35 mg/24 h and creatinine 50 μmol/L. Renal puncture biopsy revealed IgA, IgG, IgM, C3, C1q (-) under fluorescence, along with diffuse thinning of the basement membrane observed under an electron microscope. Diagnosis: thin basement membrane nephropathy. Treatment included ARB therapy and renal preservation.
Another distant cousin (III:6), aged 18, presented with “foamy urine, occasionally with hematuria for six months”. On admission to the hospital, urinary protein measured 2+, occult blood 2+, erythrocyte count 288.40/ul, 24-hour urinary protein 756.6 mg, and urinary output 6.5 dL. Renal biopsy puncture indicated IgAN.
Routine blood tests, biochemistry analysis, urinalysis with sediment analysis, urinary CT scans, and ultrasound examinations of the urinary system were conducted for all family members. Abnormalities in urinalysis were identified in 4 members (II:1/II:2/II:5/III:6), and 2 cases (I:1/I:4) progressed to ESRD with renal atrophy, totaling 6 family members with renal disorders. This observation aligned with the family history concerning the diagnosis and management of renal conditions. Additionally, 3 other cases (I:3/III:1/III:5/III:4) exhibited abnormal urinalysis results. Recommendations were made to conduct multiple reviews of urinalysis and urine sediment. However, three members declined, hindering the clarification of the clinical phenotype. Specific results are detailed in Table 1.
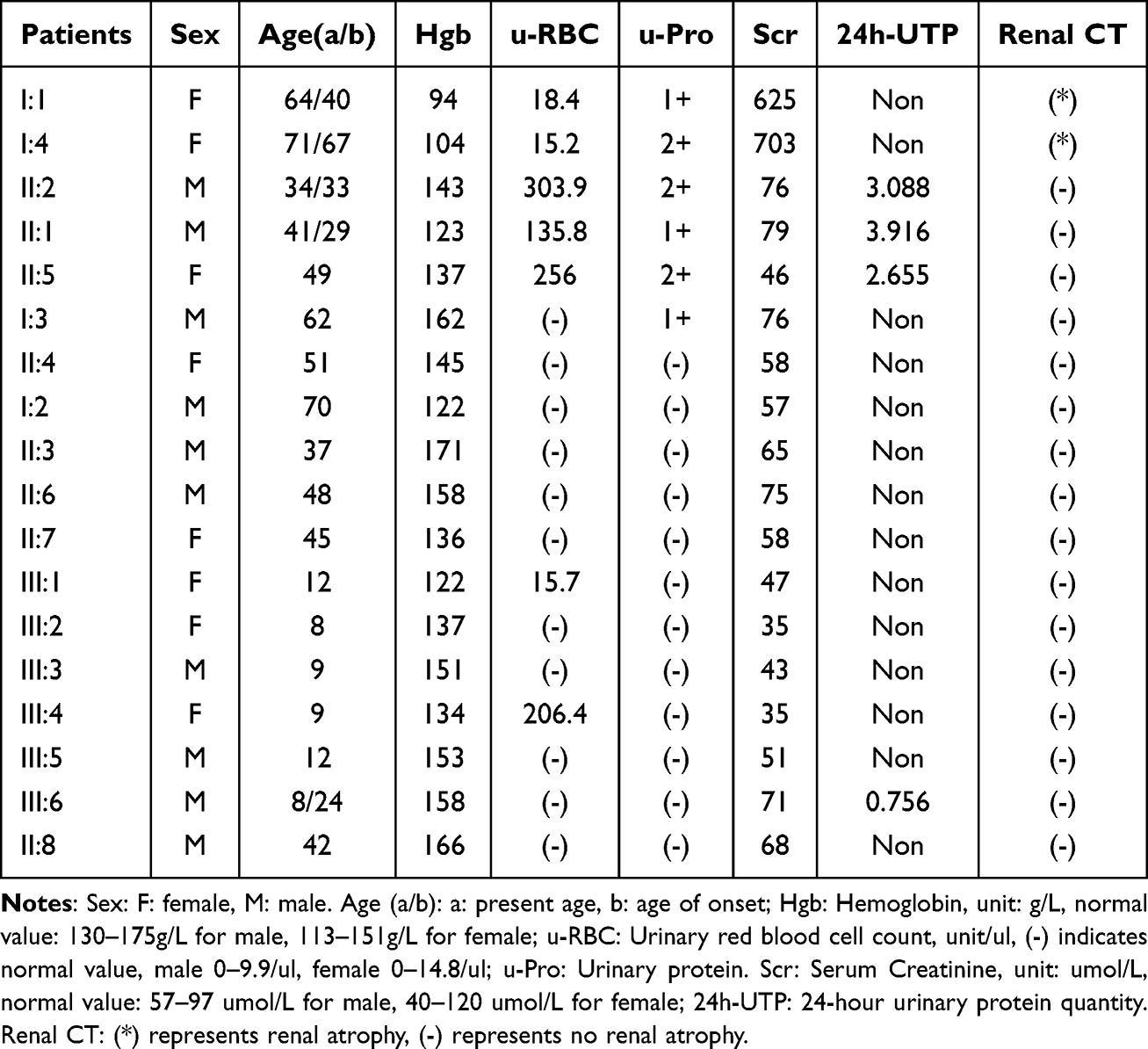 |
Table 1 Clinical Information of Family Members |
Furthermore, skin type IV collagen biopsies were performed on three members (II:1, II:2, and II:3), revealing the following: absence of anti-α5(IV) monoclonal antibody, anterior cone lens, peripapillary macular dots, speckled retinas, and other eye lesions. Pure tone audiometry results were within normal limits, as depicted in Figure 4.
 |
Figure 4 Eye examination and pure tone audiometry for II:1, II:2 and II:3. Notes: (A–C) Represent the results of eye examination and pure tone audiometry conducted on family member II:2. (D–F) Depict the findings of eye examination and pure tone audiometry for family member II:3. Finally, (G–I) Correspond to the outcomes of eye examination and pure tone audiometry of family member II:1. |
Results of Whole Exome Sequencing and Bioinformatics Analysis
Raw Data Quality Assessment and Comparison Analysis
Peripheral blood samples were collected from 10 core and control members (I:1/I:2/I:3/I:4/II:2/II:1/II:4/II:5/II:7/III:3). Initial confirmation of kinship relationships was conducted, as depicted in Figure 5. The markers demonstrated accuracy and consistency within the specimens and among the members. Whole exome sequencing was executed to analyze and extract raw data. FastQC software was used to assess the raw data quality. The proportion of sequencing base quality above Q30 exceeded 95%, indicating adherence to quality standards. During comparison analysis, approximately 99% of filtered reads were identified in relation to the total number of genomes read. Furthermore, the exon region coverage by comparison sequences at 20X exceeded 96%, affirming data reliability. Additional details are provided in Table 2.
 |
Table 2 Data Quality Control and Comparative Analysis Table |
 |
Figure 5 Genetic Diagram. Note: The larger the value, the closer the genetic relationship. |
Results of Bioinformatics Analysis
SNP/InDel mutations totaled 212,187. Figure 6 reveals the distribution of these mutations across all regional loci. Following bioinformatics analysis and screening, as described in Figure 1 in the Materials and Methods section, 5 mutation genes were identified: ZNF107 (p.C707R), ZIC5 (p.R76C), WDR76 (p.S614G), COL4A3 (p.G1167R), and KYAT3 (p.R44Q). Table 3 contains data on specific mutations. These genes, located in exons, have non-synonymous mutations with a frequency of less than 1% in common population databases, indicating that they are uncommon variants. The results of various computer prediction software tools, including SIFT, Polyphen2_HDIV, MutationTaster, and Cadd, indicate that these mutations are harmful.
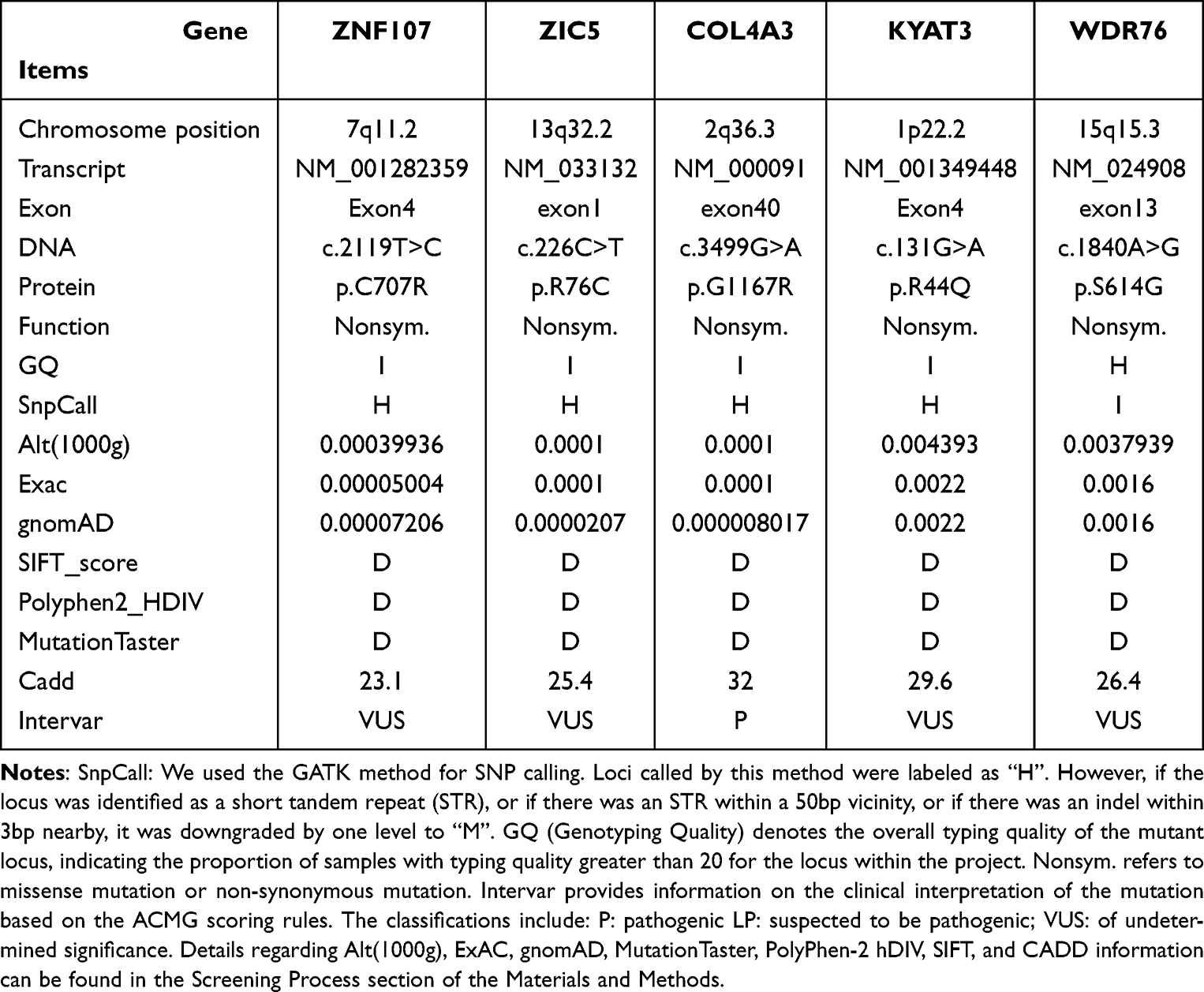 |
Table 3 Results of Susceptibility Gene Screening |
 |
Figure 6 Proportional distribution of SNV/InDel loci on different regions. |
Results of Sanger Validation Analysis
In this study, 5 mutated genes were screened for family lineage validation, and Sanger sequencing was conducted on 17 family members. Results revealed that carriers of the KYAT3 (p.R44Q) mutation genes within the family lineage encompassed I:1/I:4/II:1/II:2/II:6/III:1/III:5/III:6, which were prevalent among control members and thus excluded. Four IgAN-associated mutated genes were identified, with details of family validation presented in Figure. Among these, the COL4A3 (p.G1167R) pathogenic mutation had been previously reported in the literature and is classified as a pathogenic variant based on the ACMG criteria. This mutation was carried by 7 family members, specifically I:1, I:3, I:4, II:1, II:2, II:4, and II:5. It was present in all 7 members who were normal carriers of IgAN, ESRD, and thin basement membrane nephropathy (TBMN). The remaining 10 members exhibited normal COL4A3, among which III:6 (IgAN) did not carry the COL4A3 (p.G1167R) disease-causing mutation. The status of the remaining 4 members has not been reported and is classified as of undetermined significance based on the ACMG criteria.
Discussion
In this study, a Chinese familial lineage predisposed to IgAN, encompassing 16 immediate family members was analyzed. Among them, 6 family members exhibited renal disease, with 2 already progressing to ESRD, while the remaining 3 manifested abnormalities evident in a single urinalysis. The recurrent incidence of renal failure within this familial cluster, indicative of familial aggregation, is significant. Consequently, comprehensive whole exome sequencing coupled with bioinformatics screening was conducted on 10 members of this family lineage. The analysis revealed 4 mutated genes, comprising 1 pathogenic mutation and 3 mutations of indeterminate significance. The significant mutation sites of the 3 genes of indeterminate significance (ZNF107, ZIC5, and WDR76) have not previously been reported. These genes typically regulate transcription and maintain genome integrity.25–28 Despite these discoveries, our family validation results did not reveal a distinct phenotype attributable to individual genes nor a discernible gene co-segregation phenomenon. Therefore, subsequent analyses of the immune network are warranted. Further validation through basic experiments and ancillary tests will be pivotal in delineating their pathogenicity.
Notably, Cox et al29 documented the presence of 1–5 mutated genes in each of 8 IgAN families in their assessment of multiple rare genetic variants and co-segregation in IgAN. The singular gene within the familial lineages did not conform to the co-segregation pattern but demonstrated co-segregation when considered in conjunction with other genes. This observation resonates with our findings and previous literature reports. It underscores the notion that this complex and prevalent disease may not be ascribable to a solitary gene or variant; rather, the cumulative impact of rare variants necessitates consideration.
In this study, we identified the COL4A3 (p.G1167R) pathogenic mutation, characterized by a glycine missense mutation within COL4A3. This mutation has a key role in the formation of the triple helix structure of type IV collagen (α3α4α5). Notably, the (α3α4α5) heterotrimeric type IV collagen constitutes a significant component in the assembly of basement membranes across various tissues such as the glomerular basement membranes, eyes and ears. Upon mutation of the (α3α4α5) subtype of type IV collagen, the normal structural integrity of the glomerular basement membrane collagen network is compromised, resulting in glomerular filtration dysfunction and precipitating AS. The familial lineage under scrutiny harbors a pathogenic mutation in type IV collagen and exhibits a history of renal failure. Through analysis of the family tree, coupled with skin biopsy for type IV collagen, ocular examinations, and pure tone audiometry, a definitive diagnosis of autosomal dominant Alport syndrome (ADAS) was established.30,31 The glycine missense pathogenic mutation occurring at the 40th coding region of the COL4A3 gene has been documented in four papers.32–35 It has been associated with AS, focal segmental glomerular sclerosis (FSGS), and thin basement membrane nephropathy (TBMN). Notably, our study marks the first instance where this mutation has been linked to IgAN.
Within the family lineage, 3 family members (II:1/II:2/III:6) were diagnosed with IgA nephropathy (IgAN). Among them, II:1 and II:2 were carrying the COL4A3 (p.G1167R) mutation, while III:6 did not possess this pathogenic mutation. Notably, II:1 and II:2 were siblings, whose mother was diagnosed with ESRD and harbored the pathogenic mutation of this gene, consistent with autosomal dominant inheritance. Conversely, the parents of III:6—who was diagnosed with IgAN, served as normal controls.
Initial analysis of inheritance patterns suggested an autosomal recessive model; however, upon comprehensive assessment of the family tree, an autosomal dominant pattern of inheritance emerged as more plausible, although the possibility of sporadic IgAN could not be entirely dismissed. Renal pathology assessments revealed distinct findings: II:2 exhibited IgA immunofluorescence deposition at 2+ intensity, along with segmental thinning of a portion of the basement membrane observed under electron microscopy. Conversely, III:6 displayed IgA immunofluorescence deposition at 3+ intensity, without any discernible thickening or thinning of the basement membrane. Yuan et al36 reported that 31% of patients diagnosed with IgAN with thin basement membrane lesions exhibited mutations in the type IV collagen gene and displayed fewer typical IgAN characteristics, such as diminished IgA deposition, compared to those lacking thin basement membrane lesions, which corroborates our findings. Consequently, III:6 was classified as having sporadic IgAN, in contrast to II:1 and II:2, indicating a potential divergence in the pathogenesis of IgAN within the same family lineage. This observation underscores the complex nature of IgAN as a polygenic disorder, influenced by both environmental and genetic factors.
Within the family lineage, II:5 was identified as carrying a pathogenic mutation in the COL4A3 gene. Renal pathology studies revealed diffuse thinning of the basement membrane, measuring approximately 140 to 200 nm in thickness under electron microscopy, with no tearing or delamination changes. The diagnostic conclusion drawn was TBMN. Zhan et al32 conducted a study involving 22 children with AS, wherein they also identified a carrier of the p.G1167R mutation. Renal biopsy findings in this case revealed mild glomerular abnormalities under light microscopy, while electron microscopy demonstrated extensively thinned basement membranes without any IgA immunodeposition. TBMN, a benign familial hematuria condition attributed to mutations in the collagen type IV gene, typically presents with isolated hematuria. Some cases may feature mild proteinuria, with an overall favorable prognosis.37 However, in the case of II:5, clinically evident proteinuria was observed in this study, with a 24-hour urinary protein level measuring 2.655 g.
Given the family history of renal failure and the histopathological findings indicative of early-stage AS, vigilant screening for type IV collagen gene mutations is recommended in individuals diagnosed with TBMN, with emphasis placed on close follow-up to preempt the potential progression to AS. Furthermore, two other carriers of the causative mutation within the family lineage, II:4/I:3, currently do not exhibit overt signs of renal disease.
Furlano et al,38 in a group study examining ADAS, noted a broad spectrum of clinical manifestations among patients, ranging from asymptomatic presentation to renal failure. Significantly, they observed no discernible correlation between this clinical variability and the specific causative mutation or variant type, a finding that resonates with the results of our study. However, the diverse clinical presentations associated with ADAS pose diagnostic challenges in clinical practice.
In this study, we observed that heterozygous pathogenic mutations in COL4A3, known to precipitate AS, manifest significant individual variability even within the same family lineage. Notably, carriers of the pathogenic mutation within the family lineage encompassed patients exhibiting normal phenotypes, IgAN, and TBMN. Liu et al,34 in their study on AS, identified a patient harboring the COL4A3 (p.G1167R) mutation.
Renal biopsy findings in this case revealed FSGS changes along with thinning of the glomerular basement membrane. This observation underscores the notable divergence in clinical presentations and renal pathology associated with mutations in type IV collagen genes. However, it is essential to recognize the inherent changes in pathogenesis, therapeutic approaches, and prognostic outcomes between AS and other renal diseases.36,37,39
In summary, our findings underscore the broad spectrum of phenotypic manifestations inherent to AS, each endowed with distinctive characteristics. Therefore, it is proposed that all conditions linked to mutations in type IV collagen genes be categorized under AS, thereby facilitating standardized diagnosis and treatment. This recommendation resonates with the propositions outlined in the International Society of Nephrology meeting report.39 Also, in line with the AS guidelines, broadening the spectrum of phenotypes under consideration for type IV collagen gene testing and advocating for screening in cases of persistent proteinuria, steroid-resistant nephrotic syndrome, FSGS, familial IgAN, and unexplained end-stage renal failure is deemed pivotal for enhancing disease diagnosis and management, genetic counseling, and informed decisions regarding renal transplantation.40
The present study demonstrates notable strengths along with certain limitations. Firstly, the inclusion of a large family lineage spanning three generations, encompassing both close and distant blood relations, highlights distinctive features of familial aggregation. Within this heterozygous pathogenic genetic family lineage linked to COL4A3, nearly all conceivable phenotypic presentations were observed, rendering the study highly representative. Secondly, four family members underwent renal puncture pathological biopsy, with some participants undergoing ophthalmologic examinations and pure tone audiometry, thereby enhancing the comprehensiveness of clinical data.
However, despite these strengths, the study also bears certain limitations. While whole exome sequencing technology enjoys widespread application across diverse medical domains, providing economic and efficient advantages, it harbors inherent limitations. Notably, it may overlook variants within the non-coding regions, potentially impacting the comprehensive understanding of genetic contributions in IgAN research.
Conclusion
This study identified four mutated genes that may be involved in the onset and progression of IgAN, further revealing the complex multigenic inheritance characteristics of IgAN. The underlying mechanisms of the disease require further investigation. Additionally, we discovered potential mutations associated with known genetic kidney diseases, such as COL4A3 mutations. Therefore, we recommend comprehensive genetic screening in familial cases of IgAN to improve disease diagnosis and facilitate genetic counseling.
Abbreviations
QC, quality control; IgAN, IgA nephropathy; ESRD, End-stage renal disease; CKD, Chronic kidney disease; WES, whole exome sequencing; ACEI, angiotensin-converting enzyme inhibitor; ARB, angiotensin II receptor antagonis; ACMG, the American College Medical Genetics Genomics; AS, Alport syndrome; ADAS, autosomal dominant Alport syndrome; ANCA, anti-neutrophil cytoplasmic antibody; TBMN, thin basement membrane nephropathy; FSGS, Focal segmental glomerular sclerosis.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
This study was conducted with approval from the Ethics Committee of Longyan First Affiliated Hospital of Fujian Medical University (No. 2022-168). This study was conducted in accordance with the declaration of Helsinki. Written informed consent was obtained from all participants or patient guardians.
Consent for Publication
All participants or patient guardians signed a document of consent to publish.
Acknowledgments
We would like to acknowledge the hard and dedicated work of all the staff who implemented the intervention and evaluation components of the study.
Funding
Longyan City Science and Technology Plan Project (No:2022LYF17097).
Disclosure
The authors declare that they have no competing interests.
References
1. Ding J, Nurko N, Faure F. Evidence-based guidelines for the diagnosis and treatment of primary IgA nephropathy (2016). Chin J Pediatr. 2017;55(9):4. doi:10.3760/cma.j.issn.0578-1310.2017.01.002
2. Schena FP. A retrospective analysis of the natural history of primary IgA nephropathy worldwide. Am J Med. 1990;89(2):209–215. doi:10.1016/0002-9343(90)90300-3
3. Rovin BH, Adler SG, Barratt J, et al. KDIGO 2021. clinical practice guideline for the management of glomerular diseases. Kidney Int. 2021;100(4):S1–S276.
4. Hsu SI, Ramirez SB, Winn MP, et al. Evidence for genetic factors in the development and progression of IgA nephropathy. Kidney Int. 2000;57(5):1818–1835. doi:10.1046/j.1523-1755.2000.00032.x
5. Julian BA, Quiggins PA, Thompson JS, et al. Familial IgA nephropathy. Evidence of an inherited mechanism of disease. N Engl J Med. 1985;312(4):202–208. doi:10.1056/NEJM198501243120403
6. Hall YN, Fuentes EF, Chertow GM, et al. Race/ethnicity and disease severity in IgA nephropathy. BMC Nephrol. 2004;5(10). doi:10.1186/1471-2369-5-10
7. Kiryluk K, Julian BA, Wyatt RJ, et al. Genetic studies of IgA nephropathy: past, present, and future. Pediatr Nephrol. 2010;25(11):2257–2268. doi:10.1007/s00467-010-1500-7
8. Gharavi AG, Moldoveanu Z, Wyatt RJ, et al. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J Am Soc Nephrol. 2008;19(5):1008–1014. doi:10.1681/ASN.2007091052
9. Magistroni R, D’agati VD, Appel GB, et al. New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney Int. 2015;88(5):974–989. doi:10.1038/ki.2015.252
10. Barbour SJ, Cattran DC, Kim SJ, et al. Individuals of pacific Asian origin with IgA nephropathy have an increased risk of progression to end-stage renal disease. Kidney Int. 2013;84(5):1017–1024. doi:10.1038/ki.2013.210
11. Kiryluk K, Li Y, Scolari F, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet. 2014;46(11):1187–1196. doi:10.1038/ng.3118
12. Feehally J, Farrall M, Boland A, et al. HLA has strongest association with IgA nephropathy in genome-wide analysis. J Am Soc Nephrol. 2010;21(10):1791–1797. doi:10.1681/ASN.2010010076
13. Gharavi AG, Kiryluk K, Choi M, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43(4):321–327. doi:10.1038/ng.787
14. Kiryluk K, Li Y, Sanna-Cherchi S, et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet. 2012;8(6):e1002765. doi:10.1371/journal.pgen.1002765
15. Yu X-Q, Li M, Zhang H, et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat Genet. 2011;44(2):178–182. doi:10.1038/ng.1047
16. Li M, Foo J-N, Wang J-Q, et al. Identification of new susceptibility loci for IgA nephropathy in Han Chinese. Nat Commun. 2015;6:7270. doi:10.1038/ncomms8270
17. Li M, Wang L, Shi D-C, et al. Genome-wide meta-analysis identifies three novel susceptibility loci and reveals ethnic heterogeneity of genetic susceptibility for IgA nephropathy. J Am Soc Nephrol. 2020;31(12):2949–2963. doi:10.1681/ASN.2019080799
18. Kiryluk K, Sanchez-Rodriguez E, Zhou X-J, et al. Genome-wide association analyses define pathogenic signaling pathways and prioritize drug targets for IgA nephropathy. Nat Genet. 2023;55(7):1091–1105. doi:10.1038/s41588-023-01422-x
19. Paterson AD, Liu X-Q, Wang K, et al. Genome-wide linkage scan of a large family with IgA nephropathy localizes a novel susceptibility locus to chromosome 2q36. J Am Soc Nephrol. 2007;18(8):2408–2415. doi:10.1681/ASN.2007020241
20. Gharavi AG, Yan Y, Scolari F, et al. IgA nephropathy, the most common cause of glomerulonephritis, is linked to 6q22-23. Nat Genet. 2000;26(3):354–357. doi:10.1038/81677
21. Bisceglia L, Cerullo G, Forabosco P, et al. Genetic heterogeneity in Italian families with IgA nephropathy: suggestive linkage for two novel IgA nephropathy loci. Am J Hum Genet. 2006;79(6):1130–1134. doi:10.1086/510135
22. D’amico G. Natural history of idiopathic IgA nephropathy: role of clinical and histological prognostic factors. Am J Kidney Dis. 2000;36(2):227–237. doi:10.1053/ajkd.2000.8966
23. Rodrigues JC, Haas M, Reich HN. IgA Nephropathy. Clin J Am Soc Nephrol. 2017;12(4):677–686. doi:10.2215/CJN.07420716
24. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi:10.1038/ng.2892
25. Gallina I, Colding C, Henriksen P, et al. Cmr1/WDR76 defines a nuclear genotoxic stress body linking genome integrity and protein quality control. Nat Commun. 2015;6:6533. doi:10.1038/ncomms7533
26. Ishiguro A, Inoue T, Mikoshiba K, et al. Molecular properties of Zic4 and Zic5 proteins: functional diversity within Zic family. Biochem Biophys Res Commun. 2004;324(1):302–307. doi:10.1016/j.bbrc.2004.09.052
27. Jeong W-J, Park J-C, Kim W-S, et al. WDR76 is a RAS binding protein that functions as a tumor suppressor via RAS degradation. Nat Commun. 2019;10(1):295. doi:10.1038/s41467-018-08230-6
28. Li XA, Kokame K, Okubo K, et al. Cloning and characterization of a novel human gene encoding a zinc finger protein with 25 fingers. Biochim Biophys Acta. 1999;1489(2–3):405–412. doi:10.1016/S0167-4781(99)00206-7
29. Cox SN, Pesce F, El-Sayed Moustafa JS, et al. Multiple rare genetic variants co-segregating with familial IgA nephropathy all act within a single immune-related network. J Intern Med. 2017;281(2):189–205. doi:10.1111/joim.12565
30. Kashtan CE, Michael AF. Alport syndrome. Kidney Int. 1996;50(5):1445–1463. doi:10.1038/ki.1996.459
31. Savige J, Ariani F, Mari F, et al. Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr Nephrol. 2019;34(7):1175–1189. doi:10.1007/s00467-018-3985-4
32. Zhang L, Sun B-C, Zhao B-G, et al. An overview of the multi-pronged approach in the diagnosis of Alport syndrome for 22 children in Northeast China. BMC Nephrol. 2020;21(1):294. doi:10.1186/s12882-020-01962-y
33. Heidet L, Arrondel C, Forestier L, et al. Structure of the human type IV collagen gene COL4A3 and mutations in autosomal Alport syndrome. J Am Soc Nephrol. 2001;12(1):97–106. doi:10.1681/ASN.V12197
34. Liu J-H, Wei -X-X, Li A, et al. Novel mutations in COL4A3, COL4A4, and COL4A5 in Chinese patients with Alport Syndrome. PLoS One. 2017;12(5):e0177685. doi:10.1371/journal.pone.0177685
35. Kamiyoshi N, Nozu K, Fu XJ, et al. Genetic, clinical, and pathologic backgrounds of patients with autosomal dominant Alport syndrome. Clin J Am Soc Nephrol. 2016;11(8):1441–1449. doi:10.2215/CJN.01000116
36. Yuan X, Su Q, Wang H, et al. Genetic Variants of the COL4A3, COL4A4, and COL4A5 genes contribute to thinned glomerular basement membrane lesions in sporadic IgA nephropathy patients. J Am Soc Nephrol. 2023;34(1):132–144. doi:10.1681/ASN.2021111447
37. Savige J, Rana K, Tonna S, et al. Thin basement membrane nephropathy. Kidney Int. 2003;64(4):1169–1178. doi:10.1046/j.1523-1755.2003.00234.x
38. Furlano M, Martínez V, Pybus M, et al. Clinical and genetic features of autosomal dominant Alport syndrome: a cohort study. Am J Kidney Dis. 2021;78(4):560–570.e1. doi:10.1053/j.ajkd.2021.02.326
39. Kashtan CE, Ding J, Garosi G, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV α345: a position paper of the Alport syndrome classification working group. Kidney Int. 2018;93(5):1045–1051. doi:10.1016/j.kint.2017.12.018
40. Savige J, Storey H, Watson E, et al. Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: refining the ACMG criteria. European J Human Genetics. 2021;29(8):1186–1197. doi:10.1038/s41431-021-00858-1
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.