")
Back to Journals » Journal of Inflammation Research » Volume 18
Anthrahydroquinone-2,6-Disulfonate Restores Lung Function in COPD Through Keap1/Nrf2 Pathway Activation
Authors Li Z , Chen J, Zhang J, Li N, Yi Y , Lu H, Li M, Liu R, Chen Y, Liu X
Received 7 March 2025
Accepted for publication 1 July 2025
Published 18 July 2025 Volume 2025:18 Pages 9559—9579
DOI https://doi.org/10.2147/JIR.S524429
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Tara Strutt
Zhao Li,1– 4,* Jun Chen,5,* Jiadong Zhang,1– 3,* Nan Li,1– 3 Yang Yi,1– 3 Hanjing Lu,1– 3 Min Li,1– 3 Rui Liu,1– 3 Yuanpeng Chen,1– 3 Xiaoran Liu1
1College of Emergency Trauma, Hainan Medical University, Haikou, Hainan, People’s Republic of China; 2Key Laboratory of Hainan Trauma and Disaster Rescue, the First Affiliated Hospital of Hainan Medical University, Haikou, Hainan, People’s Republic of China; 3Key Laboratory of Emergency and Trauma Ministry of Education, Hainan Medical University, Haikou, Hainan, People’s Republic of China; 4Haikou Affiliated Hospital of Central South University Xiangya School of Medicine, Haikou, Hainan, People’s Republic of China; 5Danzhou People’s Hospital, Danzhou, Hainan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaoran Liu, College of Emergency Trauma, Hainan Medical University, No. 3 Xueyuan Road, Haikou, Hainan, 571199, People’s Republic of China, Tel +8613118901829, Email [email protected]
Objective: Chronic obstructive pulmonary disease (COPD), the third leading cause of death globally, is strongly driven by oxidative damage, contributing to lung function decline and high mortality. Despite advances in understanding oxidative stress in COPD pathogenesis, effective strategies to mitigate oxidative damage and normalize lung function remain a challenge. Anthrahydroquinone-2,6-disulfonate (AH2QDS) has potent antioxidant properties, but its ability to alleviate persistent airway oxidative damage and normalize lung function in COPD patients is not fully elucidated. This study aimed to investigate the therapeutic effects of AH2QDS on COPD by exploring its mechanisms involving the Keap1-Nrf2 pathway via in vivo and in vitro models.
Methods: This study induced COPD-like pathology by using LPS in combination with CS / CSE, establishing both in vivo and in vitro models to evaluate the effects of AH2QDS on lung function, histopathology, oxidative stress markers, levels of inflammatory cytokines, and apoptosis. Molecular docking predicted the interaction between AH2QDS and Keap1-Nrf2 proteins, which was confirmed by immunoblotting.
Results: AH2QDS treatment significantly mitigated lung tissue injury and restored lung function parameters (MMEF, PEF) in COPD model rats. This treatment upregulated the expression of antioxidant enzymes (SOD, CAT, and GSH-PX) and reduced the levels of inflammatory cytokines (TNF-α, IL-6, IL-1β, and IL-33), ROS, and the total number of apoptotic cells (Annexin V+). Following AH2QDS treatment, the expression of the Nrf2 protein was increased, and the expression of its downstream antioxidant proteins, HO-1 and NQO1, was also upregulated. Molecular docking analysis revealed a high binding affinity between AH2QDS and Keap1-Nrf2 protein (binding energy: − 8.0 kcal/mol).
Conclusion: AH2QDS promotes the body’s capacity to counteract oxidative damage and reduces inflammatory cytokine levels by activating the Keap1‒Nrf2 pathway, thereby normalizing lung function in COPD.
Keywords: COPD, AH2QDS, oxidative stress, lung function, Keap1-Nrf2
Introduction
Chronic obstructive pulmonary disease (COPD) is a persistent respiratory disease characterized by airway inflammation and progressive obstruction of airflow in the lungs and has become the third leading cause of death globally.1 COPD occurs and develops due to cigarette smoking and prolonged exposure to toxic gases and particulates,2 resulting in the apoptosis of airway epithelial cells and compromised cellular integrity and function; these changes lead to chronic inflammation and an impaired immune response, ultimately leading to respiratory diseases.3 Oxidative/antioxidant imbalance is an important factor in the development of this disease.4,5 A growing body of recent evidence has demonstrated that the Keap1/Nrf2 pathway plays a pivotal regulatory role in the pathogenesis of COPD.6 As a crucial component of the endogenous antioxidant defense system, this pathway not only participates in counteracting both exogenous and endogenous oxidative damage but also maintains redox homeostasis by regulating the expression of antioxidant proteins. When cells are exposed to oxidative stress (eg, cigarette smoke), reactive oxygen species (ROS) or electrophiles can directly modify specific cysteine residues in Keap1, inducing conformational changes that reduce its binding affinity for Nrf2. This modification enables Nrf2 to escape ubiquitin-mediated degradation. Following liberation from Keap1-mediated suppression, newly synthesized Nrf2 rapidly translocates into the nucleus, where it initiates transcription of downstream target genes.7 Clinical observations have revealed that COPD patients commonly exhibit downregulated Nrf2 expression or impaired Nrf2 function.8 This deficiency renders COPD patients more susceptible to oxidative injury, establishing a vicious cycle of “oxidative stress-inflammation-Nrf2 suppression-diminished antioxidant capacity” that perpetuates disease progression. Despite promising results from animal models and in vitro studies, the clinical efficacy of antioxidant therapies in COPD patients remains limited. Sulfhydryl-containing antioxidants, such as N-acetylcysteine (NAC), carbocisteine, and erdosteine, represent the most extensively investigated antioxidant agents to date. While NAC and carbocisteine have demonstrated moderate efficacy in reducing acute exacerbations, their effects on improving lung function and quality of life remain insignificant. Erdosteine shows some benefits for mild exacerbations but appears ineffective against moderate-to-severe exacerbations. Dietary antioxidants, including vitamin C, vitamin E, and resveratrol, exhibit potent antioxidant and anti-inflammatory effects in vitro but have failed to demonstrate significant clinical benefits in COPD patients. Although numerous antioxidant strategies show therapeutic potential in animal studies, only sulfhydryl-based antioxidants (eg, NAC and carbocisteine) have shown modest clinical benefits in COPD patients to date.9 Therefore, administering antioxidants, reducing the levels of oxygen free radicals and reactive oxygen species (ROS), and alleviating progressive impairment of lung function are key measures for preventing disease occurrence and ameliorating symptoms; however, effective drugs are lacking, and identification of novel compounds to restores lung function is urgently needed.
Anthrahydroquinone-2,6-disulfonate (AH2QDS), with the chemical formula C14H8O8S2.2Na and a molecular weight of 368.33, is characterized by its distinctive disulfonic acid structure. This compound demonstrates significant antioxidant properties. AH2QDS can increase the expression level of capillary endothelial cell adhesion proteins (ZO-1), and reduce pulmonary microvascular permeability through the PI3K/AKT pathways.10 This compound also reduces the level of oxidative stress within renal tissues, decreases endoplasmic reticulum stress and apoptosis and increases the cellular antioxidant capacity through Apelin/APJ pathway.11,12 However, whether AH2QDS can attenuate oxidative damage, restores lung function, and reduce inflammation in chronic airway inflammation in COPD patients is unclear.
In this study, we aimed to establish an animal model of COPD in Sprague–Dawley (SD) rats induced by lipopolysaccharide (LPS) + cigarette smoke (CS) and used a mixture of LPS + cigarette smoke extract (CSE) to intervene in HBE cells to establish a cellular model. Our results demonstrated that AH2QDS exerts potent antioxidant effects through specific targeting of the Keap1/Nrf2 pathway. Treatment with AH2QDS significantly enhanced nuclear translocation of Nrf2 while reducing Keap1 protein expression levels in HBE cells. This activation upregulated the expression of downstream antioxidant enzymes, including superoxide dismutase (SOD), catalase (CAT), and heme oxygenase-1 (HO-1), while decreasing oxidative damage markers such as malondialdehyde (MDA). Parallel improvements in pulmonary function parameters were observed in vivo, confirming the therapeutic potential of AH2QDS for COPD management.
Methods and Materials
Reagents
LPS was purchased from Sigma (USA) and was derived from Escherichia coli 0111:B4 (cat#L2630). The antibodies used were as follows: polyclonal antibody against Nrf2 (cat#16396-1-AP), polyclonal antibody against Keap1 (cat#10503-2-AP), polyclonal antibody against HO-1 (cat#10701-1-AP), polyclonal antibody against NQO1 (cat#11451-1-AP), and PCNA polyclonal antibody (cat#10205-2-AP) were purchased from Wuhan Three Eagles Biotechnology Co. GAPDH recombinant antibody (cat#GB15004-100) was purchased from Wuhan Sevier Biotechnology Co. HRP-labelled goat anti-rabbit (cat#BL052A) IgG (H+L) was purchased from Lanjieke Technology Co. An enhanced ECL chemiluminescence detection kit (cat#36222ES60) and a BCA protein assay kit (cat#20201ES76) were purchased from Next Sage Biotechnology (Shanghai) Co. Rat interleukin-6 (IL-6) ELISA kit (cat#ml064292), rat tumor necrosis factor-α (TNF-α) ELISA kit (cat#ml002859), rat interleukin-1β (IL-1β) ELISA kit (cat# ml037361), and rat interleukin-33 (IL-33) ELISA kit (cat#ml037355); Human IL-6 ELISA kit (cat# ml058097), Human TNF-α ELISA kit (cat#ml064303), Human IL-1β ELISA kit (cat#ml028592), and Human IL-33 ELISA kit (cat# ml063084) were purchased from Shanghai Enzyme-linked Biotechnology Co., Ltd.
CSE Production
The Dulbecco’s Modified Eagle Medium (DMEM, cat#11965118, Thermo Fisher) was introduced into a gas washing bottle to prepare CSE. For each 10 mL aliquot of the medium, one commercially available cigarette (Huangshan brand: tar content 10 mg, nicotine 0.8 mg, carbon monoxide 10 mg, length 84 mm) was combusted over a period of approximately 5 minutes. During the combustion process, the generated smoke was drawn through the medium using a 20 mL syringe to ensure thorough saturation. After the complete combustion of the cigarette, the flask was carefully rinsed to remove any residual particulate matter and then allowed to stand undisturbed for 5 minutes to facilitate equilibration. The OD of the resulting medium was measured at a wavelength of 320 nm using a microplate reader, with the recorded values falling within the range of 0.77 ± 0.08. The pH of the CS-containing medium was meticulously adjusted to 7.4 using a calibrated pH meter to ensure physiological relevance. Finally, the medium was sterilized by filtration through a 0.22 µm membrane filter, and the concentration of CSE was standardized to 100% for subsequent experimental use.13
Cell Culture, Intervention and Cell Viability Assay
The human airway epithelial (HBE) cell line (cat#HTX2109) was purchased from Shenzhen Haodi Huatuo Biotechnology Co. Cell viability was determined via a CCK-8 kit (cat#BS350A, Biosharp, China) after coculturing LPS+CSE and HBE cells for 24, 48, and 72 h. An LPS concentration of 100 μg/mL (Details can be found in the supplementary files Figure S1A-D) and a CSE concentration of 10% were chosen to treat the cells for 24 h (Details can be found in the supplementary files S1E-H). More than 50% of the cells were still viable, and the results were better than those of the intervention at 48 h and 72 h (Details can be found in the supplementary files Figure S1I). Pretreatment with 400 μM AH2QDS for 30 min was selected (Details can be found in the supplementary files Figure S2C), and the results were better than the cell viability of the 200 μM and 600 μM pretreatment groups (Details can be found in the supplementary files Figure S2A-B). Please refer to the supplementary data for specific details on the dosage and timing of LPS, CSE, and AH2QDS treatment. The cells used for the experiments were as follows: (1) Control; (2) LPS + CSE; (3) LPS + CSE after AH2QDS pretreatment for 30 min; and (4) AH2QDS.
Flow Cytometry and Intracellular ROS Detection
Apoptosis rates in each experimental group of cells were determined using an Annexin V-FITC/PI Apoptosis Detection Kit (cat#40302, Yeasen, Shanghai, China), a widely recognized method for distinguishing apoptotic cells based on phosphatidylserine externalization and membrane integrity. Flow cytometry (BD Accuri™ C6 Plus) was employed to assess the apoptosis rates accurately, enabling precise quantification of early and late apoptotic cell populations. For the measurement of intracellular ROS levels, a commercially available ROS detection kit (cat#BL714A; Anhui Biosharp Technology Co., Ltd.) that relies on a fluorescent probe to detect oxidative stress within cells was utilized. The cells were plated at an optimal density of 1 × 105 cells/mL in sterile Petri dishes and allowed to incubate under standard culture conditions (37°C, 5% CO2) for 24 h to ensure proper adherence and stabilization. After this initial incubation period, the cells were exposed to LPS and CSE for an additional 24 h to induce oxidative stress and inflammatory responses. Detection of fluorescence signals, indicative of ROS levels and apoptotic activity, was carried out using a laser-equipped detection system with excitation wavelengths of 488 nm and 515 nm, ensuring optimal sensitivity and specificity. The fluorescence intensity for each experimental group was subsequently quantified and analysed using ImageJ (version ×64; National Institutes of Health, USA) and FlowJo (version 10.8.1; BD Biosciences) software, which provide robust tools for data processing and statistical evaluation.
Enzyme-Linked Immunosorbent Assays (ELISAs), Measurement of the Mitochondrial Membrane Potential (MMP) and Transwell Assays
Reagents were added to the reaction mixtures following the manufacturer’s instructions for the MDA (cat#BL904A, White Shark, China), SOD (Cat# A001-3-2, Nanjing Jiancheng Bioengineering Institute, China), CAT (cat#E-BC-K031-M, Elabscience Biotechnology), and GSH-PX assay kits (cat#A005-1-2, Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The absorbance (OD) was measured at 532 nm and 450 nm using a microplate reader. The MMP was assessed via JC-1 (cat#BL711A, Biosharp) staining, with J-aggregates (high MMP) showing bright red fluorescence and J monomers (low MMP) displaying green fluorescence. Fluorescence images were captured using an inverted fluorescence microscope (Leica, Germany) and analysed with ImageJ software. Inflammatory cytokines (TNF-α, IL-1β, IL-33, and IL-6) were quantified via ELISA kits (Vancovi, Ltd). For migration assays, HBE cells (1 × 104 cells/well) were seeded in the upper chamber of Transwell plates with serum-free medium, while the lower chamber contained complete medium supplemented with 20% FBS. After 24 h (37°C, 5% CO2), the migrated cells on the lower membrane were stained with 0.1% crystal violet, and the nonmigrating cells were removed. The migrated cells were counted under an inverted light microscope.
Western Blotting
Protein extraction was performed following standard protocols, and protein concentrations were determined using a bicinchoninic acid (BCA) assay kit. Equal quantities of protein samples were resolved by electrophoresis on 10% SDS‒PAGE gels and subsequently transferred onto polyvinylidene difluoride (PVDF) membranes at a constant current of 400 mA. For minimization of nonspecific binding, the membranes were treated with a blocking solution containing 5% skim milk in TBST (0.05% Tween-20) for 1 h at room temperature. The membranes were then subjected to overnight incubation with the primary antibody at 4°C under gentle agitation. The membranes were washed three times with TBST and then incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody diluted at 1:10,000 for 1 h at room temperature. After further washing, the protein bands were visualized via enhanced chemiluminescence (ECL) reagent. For reprobing, the membranes were stripped with a stripping solution and reblocked. Finally, the protein band intensities were analysed and quantified using ImageJ software.
Establishment of an Animal COPD Model
A total of 24 male SD rats, aged 7–8 weeks and weighing 130–150 g, were obtained from Changsha Tianqin Co. The rats were maintained in a controlled environment at the Experimental Animal Centre of Hainan Medical University. After a 7-day acclimatization period, the rats were randomly divided into four groups (n = 6 per group): (1) Normal control (control group), (2) LPS intranasal administration combined with CS exposure (the LPS + CS group), (3) LPS intranasal administration combined with CS exposure plus intraperitoneal injection of AH2QDS 100 mg/kg (the LPS + CS + 100 mg/kg AH2QDS group), and (4) LPS intranasal administration combined with CS exposure plus intraperitoneal injection of erdosteine 30 mg/kg (the LPS + CS + 30 mg/kg erdosteine group). The modelling process involved daily CS exposure, with 20 cigarettes burned for 20 min per session, twice daily. LPS was administered via intranasal drops every 15 days, with no CS exposure on LPS administration days, for a total duration of 100 days. Beginning on day 60, PBS, AH2QDS, or erdosteine was administered via intraperitoneal injection every two days. All experimental procedures were approved by the Ethics Committee of Hainan Medical University (Approval No. HYLL-2024-063).
Alveolar Lavage Fluid, Tissue Collection and Lung Histopathology
After 100 days of modelling, the SD rats were anaesthetised and sacrificed by dislocation of the neck. For bronchoalveolar lavage fluid (BALF), the lungs were removed, the left main bronchus was ligated, the left lung was slowly injected with 0.5 mL of ice-cold PBS, and BALF was subsequently collected from the lungs for subsequent experiments. The right lung tissue was fixed with 4% paraformaldehyde and dehydrated through a gradient in alcohol before being embedded in paraffin. After the paraffin blocks of the samples were cut into 4 μm sections, HE staining was performed. All analyses were performed by two pathologists who were blinded to group allocation. The mean linear intercept (MLI) and mean alveolar area (MAA) were determined to assess lung injury according to methods reported in the literature.14
Analysis of Lung Function
The SD rat model was anaesthetised with 2% sodium pentobarbital, and after endotracheal intubation, the rats were placed into the chamber of a lung function tester connected to a spirometer (Tawang, Shanghai, China), and the maximal mid-expiratory flow (MMEF), peak expiratory flow (PEF) and FEV0.3/FVC were measured. Pulmonary function tests were performed at three time points: pre-modeling, day 50, and day 100 post-modeling, with random selection of rats from each group. The respiratory parameters were set as follows: Respiratory frequency: 100 breaths/min. Breathing cycles: 5 cycles/measurement. Maximum inspiratory pressure: 28 cm H2O. Inspiration duration: 0.1 sec. Expiration maintenance time: 1 sec (rapid phase). Spontaneous breathing interval: 5 sec. Repeated measurements: 5 trials/rat.
RT‒qPCR
Total RNA was extracted using a TaKaRa kit, and DNA was obtained through reverse transcription with the Next Sage Kit. Quantitative polymerase chain reaction (PCR) amplification was performed using a real-time fluorescence quantitative PCR system (Thermo ABI QuantStudio3). In this study, human-specific primers were employed for in vitro experiments using COPD cellular models, while rat-specific primers were utilized for in vivo animal studies of COPD. The PCR primers used were designed with GAPDH as the endogenous control.
The specific primers utilized in this study are detailed in Table 1
 |
Table 1 The Specific Primers Utilized |
Molecular Docking Experiments
The crystal structure of the Keap1 (entry 7K2A) protein was downloaded from the RCSB Protein Data Bank (http://www.rcsb.org). The crystal structure of the protein molecule was processed with the software PyMOL to remove the water molecules, add the hydrogen ions and balance the charge. The 3D structure files of AH2QDS were drawn with ChemDraw and Chem3D software. Open AutoDock Tools 1.5.6 software was used for molecular docking. The Keap1 protein was hydrogenated, small molecules were hydrogenated, and the flexible bonds were determined. The molecular docking parameters were set with a grid plate as follows: Keap1: centre (X,Y,Z) = (−42.1, 23.5, 60.8), size (X×Y×Z) = (20.0×20.0×25.0). The docking method was set as semiflexible docking, the docking accuracy exhaustiveness = 25, and the docking algorithm was the Lamarckian genetic algorithm. AutoDock Vina 1.2.0 software was used for molecular docking.
Immunohistochemistry and Immunofluorescence
Paraffin-embedded tissue sections were initially deparaffinized using an ecofriendly deparaffinizing solution (item number G1128; Wuhan Kewei Biotechnology Co., Ltd), which effectively removes paraffin while preserving tissue integrity. The sections were then rehydrated through a descending alcohol series (100%, 95%, 80%, and 70% ethanol) to prepare them for subsequent staining procedures. To minimize nonspecific binding and increase antigen retrieval, the sections were immersed in 3% bovine serum albumin (BSA) for 30 min at room temperature. Next, the sections were incubated overnight at 4°C with primary antibodies targeting the Keap1 and Nrf2 proteins, which are key regulators of oxidative stress responses. After thorough washing to remove unbound primary antibodies, the sections were incubated with a horseradish peroxidase (HRP)-labelled goat anti-rabbit IgG secondary antibody at room temperature for 1 h. Finally, the sections were stained with diaminobenzidine (DAB) to visualize the antibody‒antigen complexes, counterstained with haematoxylin for 3 min at room temperature to highlight the nuclear structures, and observed under a light microscope for detailed analysis.
For analysis of the nuclear translocation of Nrf2 via immunofluorescence, groups of cells were incubated with 4% paraformaldehyde for 10 min and 0.1% Triton X-100 (cat#ST1723, Beyotime) for 15 min. The cells were incubated with a primary antibody (rabbit anti-Nrf2) overnight at 4°C, followed by incubation with a secondary anti-goat anti-mouse IgG (Alexa Fluor™ Plus 488/555) for 1 h at 37°C, and the nuclei were labelled with DAPI. Images were obtained with a laser confocal microscope (Olympus FV3000, Tokyo).
Statistical Analysis
Data were analyzed using Prism 10.0 software (GraphPad, USA), and results were presented as mean ± standard error of the mean (SEM). Shapiro–Wilk test was used to assess normality, and a two-tailed Student’s t-test was employed to evaluate the statistical significance of differences between two experimental groups. For experiments involving more than two groups, depending on the experimental design, either one-way or two-way analysis of variance (ANOVA) was applied, followed by Tukey’s test for post-hoc analysis. For samples not following a normal distribution, the Mann–Whitney U-test or Kruskal–Wallis test was used. A P value of less than 0.05 was considered statistically significant for all analyses.
Results
LPS+CS Generate a COPD Model in SD Rats
A COPD animal model was created to explore the effects of AH2QDS on COPD.15 As the pathomorphological index of the lung tissue is the most important parameter for evaluating the success of animal model establishment,16 we aimed to observe the pathological changes in lung tissue in an animal model. The rats were exposed to the CS for 100 days, and LPS was administered intranasally once every 15 days (Figure 1A). Figure 1B–D shows that, compared with those in the control group, lung histopathology revealed gradual widening of the alveolar septa, fusion and enlargement of the alveolar lumens and a gradual increase in the MLI and MAA of the lung tissue in the LPS+CS group from 50 days to 100 days. These alterations align with the characteristic pathological features of COPD. Lung function tests are used to diagnose and grade the severity of COPD;17 thus, an assessment model involving PEF, MMEF and FEV0.3/FVC was established. Figure 1E–G shows significant decreases in the PEF, MMEF, and FEV0.3/FVC in the rats in the LPS+CS group. These findings suggest that LPS+CS exposure caused airway obstruction and a progressive reduction in lung function.
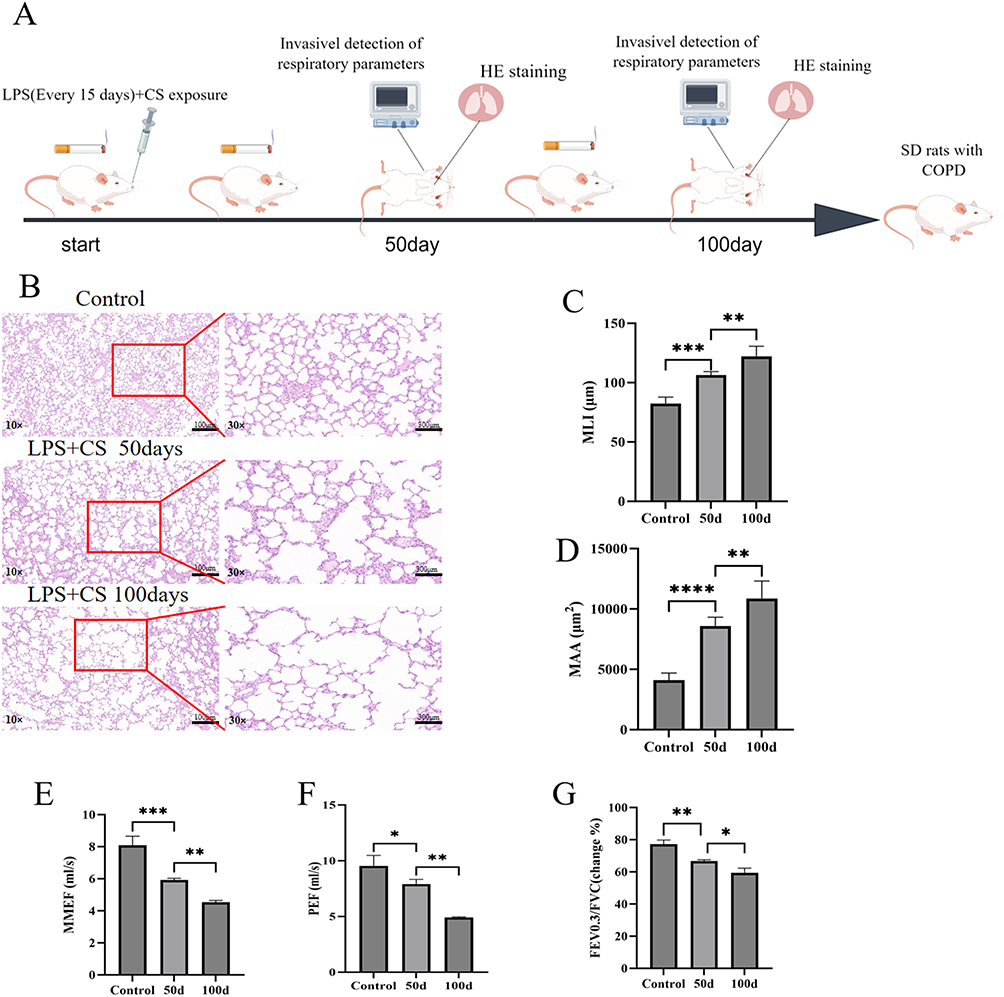 |
Figure 1 LPS + CS induced a COPD model in rats. (A) Schematic diagram of the model generated by Figdraw. (B) HE staining of lung tissues from each group: control, LPS + CS 50 days, and LPS + CS 100 days (lower power images10×, lower power images30×, scale bar=100μm). (C) MLI (µm) for each group. (D) MAA (µm²) for each group. (E–G) Lung function measurements: MMEF, PEF, and FEV0.3/FVC ratios. n ≥ 3, ns (not statistically significant), *P < 0.05, **P < 0.01, ***P< 0.001, ****P ˂ 0.0001. |
Oxidative stress status and lung inflammation in rats were evaluated by determining the levels of antioxidants and the expression of cellular inflammatory factors. As shown in Figure 2A–D, the data indicate a decrease in the levels of the key antioxidants SOD, GSH-PX, and CAT and an increase in the lipid peroxidation marker MDA in the rats exposed to LPS+CS at 50 and 100 days. These findings suggest that oxidative stress, indicated by reduced antioxidant levels and elevated MDA, is a critical pathological consequence of LPS+CS exposure. The progressive decrease in antioxidant levels and increase in MDA over time reflect the persistence and worsening of oxidative stress, which could contribute to lung tissue damage.
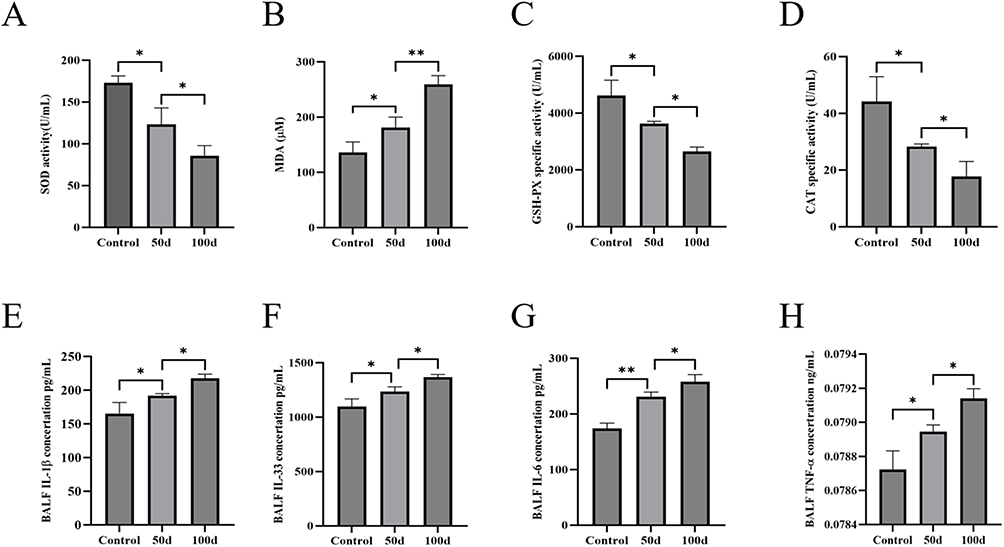 |
Figure 2 Levels of antioxidant and inflammatory factors in the plasma and BALF of rats with LPS+CS-induced COPD. (A) Serum SOD levels. (B) Serum MDA levels. (C) Serum GSH-PX levels. (D) Serum CAT levels. (E) BALF IL-1β levels. (F) BALF IL-33 levels. (G) BALF IL-6 levels. (H) BALF TNF-α levels. n ≥ 3; *P < 0.05, **P < 0.01. |
As demonstrated in Figure 2E–H, the levels of inflammatory markers, including IL-1β, TNF-α, IL-33, and IL-6, were substantially increased in the BALF of the rats in the LPS+CS group compared with those in the control group. These cytokines have essential roles in triggering and sustaining pulmonary inflammatory responses. The gradual increase in inflammatory factor levels over time (from 50 to 100 days) reflects the chronic nature of inflammation in response to continuous LPS+CS exposure, indicating that both oxidative stress and inflammation are exacerbated over prolonged exposure periods and that airway inflammation persists and gradually worsens. In summary, these findings highlight the sustained and progressive nature of both oxidative stress and lung inflammation during LPS+CS exposure.
AH2QDS Mitigates Histopathological Lung Damage and Restores Pulmonary Function
As shown in Figure 3A–C, lung histopathology revealed that alveolar wall thickening, alveolar lumen fusion and expansion, and inflammatory cell infiltration were significantly reduced. Additionally, the MLI and MAA were substantially restored in the AH2QDS treatment group, outperforming the erdosteine group. In terms of lung function, as shown in Figure 3D and E, the MMEF was approximately 3.73 mL/s and the PEF was approximately 5.75 mL/s in the LPS+CS group, while the MMEF was 6.35–8.99 mL/s and the PEF was 7.8–12.04 mL/s in the AH2QDS-treated group (Table 2); the PEF and MMEF values in the treatment group were superior and better than those of the erdosteine group. The results indicated that AH2QDS could restore lung function (PEF, MMEF) in rats with COPD.
 |
Table 2 Lung Function Parameters |
 |
Figure 3 Effects of AH2QDS and erdosteine on pathological lung injury and lung function in SD rats. (A) HE staining (lower power images 10×, lower power images 30×, scale bar=100μm). (B) MAA for each experimental group (µm2). (C) MLI measurement for each experimental group (µm). (D) MMEF values for each experimental group. (E) PEF values for each experimental group. n ≥ 3, *P ˂ 0.05, **P ˂ 0.01, ***P ˂ 0.001, ****P ˂ 0.0001. |
AH2QDS Alleviates Oxidative Stress Injury and Decreases Inflammatory Cytokine Levels in a Rat Model of COPD
The levels of antioxidant factors, including SOD, GSH-PX, and CAT, as well as the oxidative damage marker MDA, were assessed. As shown in Figure 4A–D, the LPS+CS group presented a marked reduction in SOD, CAT, and GSH-PX levels, along with a substantial increase in MDA levels. Treatment with AH2QDS or erdosteine notably elevated the expression of SOD, CAT, and GSH-PX while reducing the MDA level. Notably, the efficacy of AH2QDS was superior to that of erdosteine in increasing GSH-PX expression. These results suggest that AH2QDS effectively reduces oxidative damage in the lungs, restores lung function, and outperforms erdosteine.
 |
Figure 4 AH2QDS inhibits LPS+ CS-induced oxidative stress and inflammation. (A–D) Determination of SOD, MDA, GSH-PX and CAT levels in lung tissue homogenates. (E–H) IL-1β, IL-33, IL-6, and TNF-α levels in the BALF. n≥3, ns: not statistically significant, *P˂0.05, **P ˂0.01, ***P ˂0.001, ****P˂0.0001. |
Small airway inflammation is a key characteristic of COPD.18 The effect of AH2QDS on airway inflammation was evaluated by measuring the levels of inflammatory cytokines, including TNF-α, IL-6, IL-1β, and IL-33, in the BALF. As shown in Figure 4E–H, both the AH2QDS and the erdosteine treatments significantly decreased the levels of these cytokines compared with those in the LPS+CS group, with AH2QDS resulting in a more pronounced reduction in IL-33 expression. These findings suggest that AH2QDS treatment significantly ameliorates airway inflammatory responses in COPD model rats.
AH2QDS Attenuates LPS+CSE-Induced Oxidative Stress in HBE Cells
To complement the in vivo findings and further investigate the cellular mechanisms, we established a parallel in vitro model using HBE cells exposed to CSE and LPS, mirroring the key pathological stimuli used in the animal experiments. As shown in Figure 5A–D, compared with those in the control group, the levels of SOD, CAT, and GSH-PX in the LPS+CSE group markedly decreased, and the MDA content in the LPS+CSE group increased. After treatment with AH2QDS, the expression levels of SOD, CAT, and GSH-PX increased, whereas the expression of MDA decreased. These results indicated that AH2QDS also attenuated oxidative stress damage in LPS+CSE-induced cells in vitro. As shown in Figure 5E and F, the ROS levels in the LPS+CSE-treated HBE cells were significantly reduced following AH2QDS treatment, whereas the MMP was notably increased. These findings suggest that AH2QDS can mitigate ROS production in LPS+CSE-induced cells and safeguard mitochondrial function.
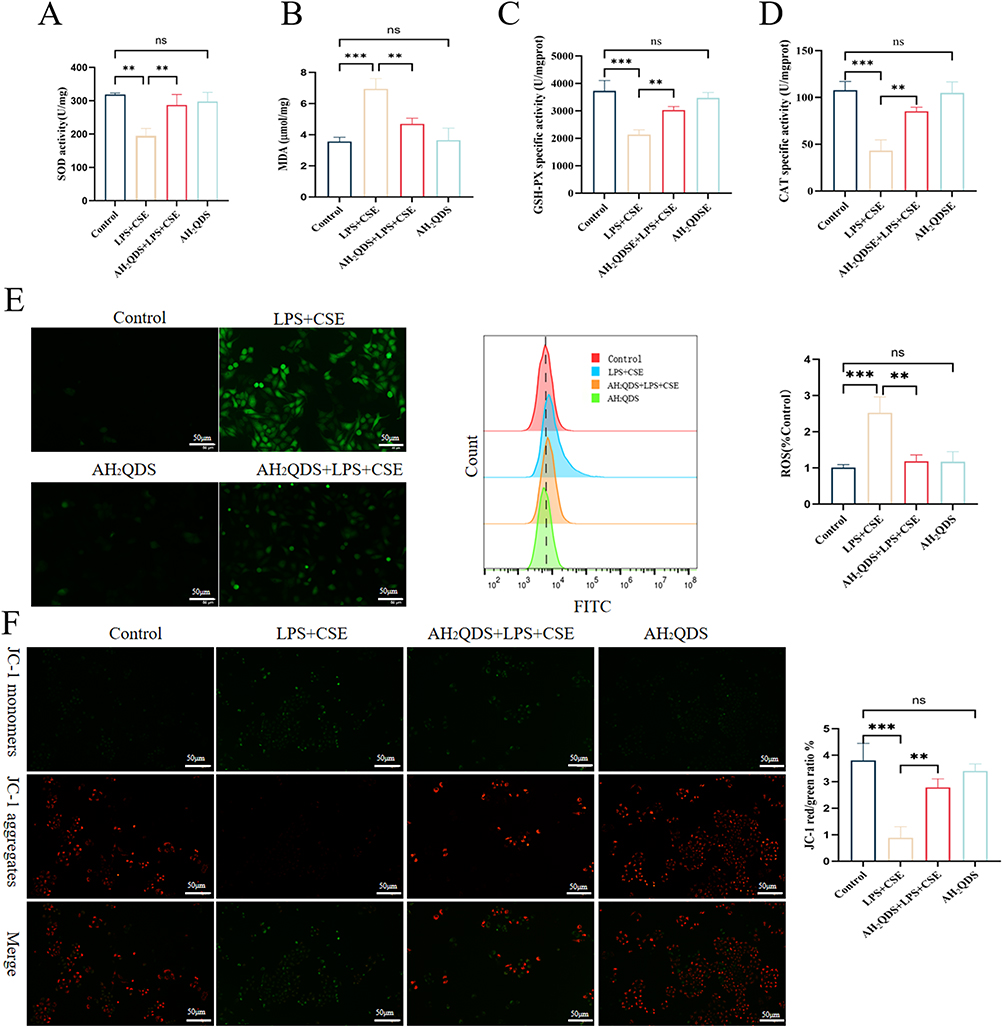 |
Figure 5 The impact of AH2QDS on oxidative stress-mediated injury in HBE cells. (A) SOD levels in the different experimental groups. (B) MDA levels. (C) GSH-PX levels. (D) CAT levels. (E) ROS levels were determined by fluorescence and flow cytometry, along with corresponding quantitative plots (scale bar = 50 μm). (F) MMP and the corresponding quantitative plots (scale bar = 100 μm). n ≥ 3, ns: not statistically significant, *P˂0.05, **P˂0.01, ***P˂0.001. |
Effects of AH2QDS on LPS+CSE-Induced HBE Cell Migration, Apoptosis and Inflammatory Factor Expression
The potential of AH2QDS to promote HBE cell migration under LPS+CSE induction was assessed through a Transwell assay. As shown in Figure 6A and B, AH2QDS treatment significantly increased the number of migrating cells in the LPS+CSE group, suggesting an increased capacity for cellular repair. Furthermore, as demonstrated in Figure 6C, the apoptosis rate in the LPS+CSE group was approximately 26%, which decreased to 16% following AH2QDS treatment, demonstrating its ability to mitigate LPS+CSE-induced apoptosis in HBE cells. Additionally, AH2QDS treatment notably decreased the levels of inflammatory cytokines, such as IL-6, IL-33, IL-1β, and TNF-α, as shown in Figure 6D–G.
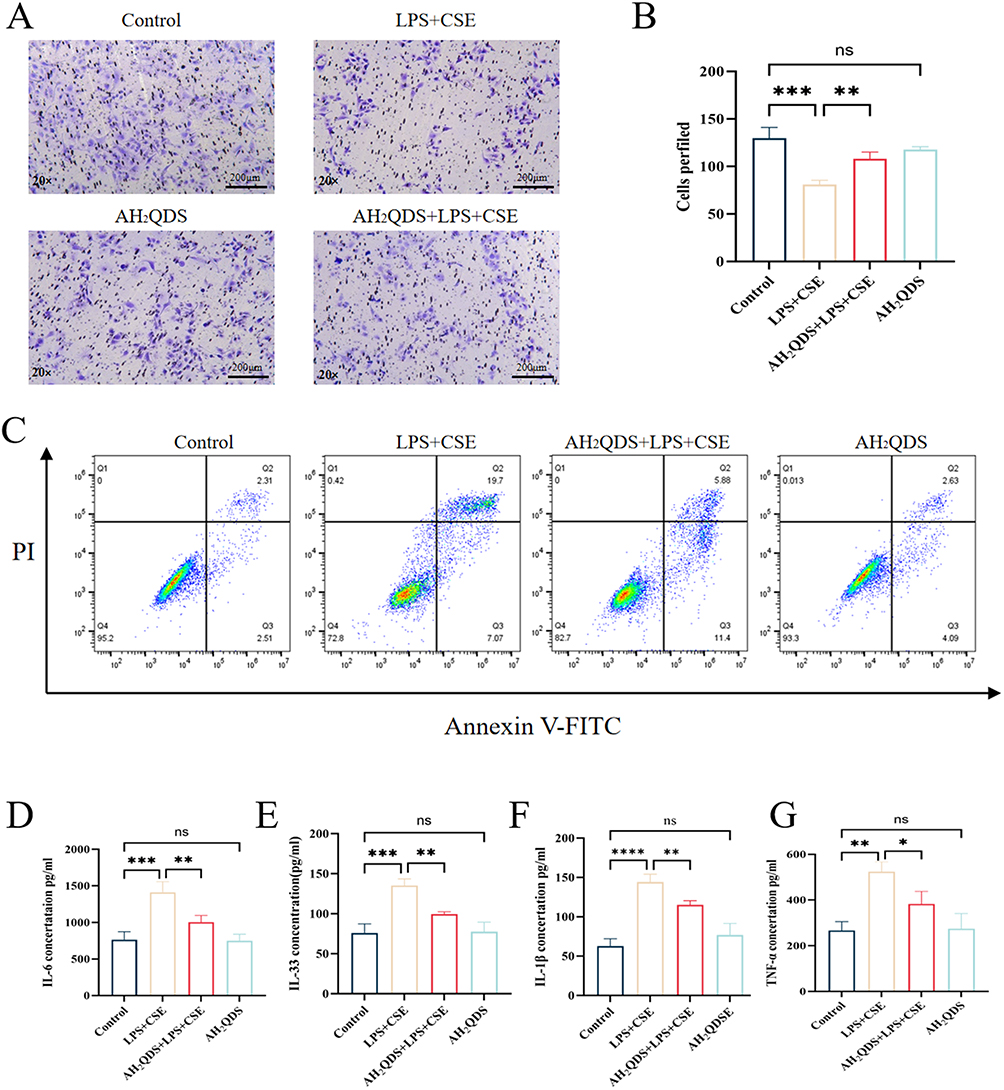 |
Figure 6 Effects of AH2QDS on the LPS+CSE-induced HBE cell migration, apoptosis and inflammatory factor expression. (A and B) Transwell and quantitative plots of each experimental group. (20×, scale bar=200μm). (C) Levels of apoptosis in the experimental groups. (D–G) Levels of IL-6, IL-33, IL-1β, and TNF-α. n≥3, ns: not statistically significant, *P˂0.05, **P˂0.01, ***P˂0.001, ****P˂0.0001. |
AH2QDS Promotes Antioxidant Properties by Activating the Keap1/Nrf2 Pathway
To explore the mechanism responsible for the AH2QDS-induced normalization of lung function, we hypothesized that AH2QDS alleviates oxidative stress-induced damage through the Keap1‒Nrf2 pathway. As shown in Figure 7A, B and D, RT‒qPCR, immunofluorescence, and Western blot analyses demonstrated that AH2QDS treatment attenuated the LPS+CSE-induced reduction in Nrf2 and promoted its nuclear translocation in HBE cells. As shown in Figure 7C and E, LPS+CSE treatment increased Keap1 mRNA and protein expression, an effect that was reversed by AH2QDS treatment. Furthermore, AH2QDS significantly increased the levels of HO-1 and NQO1, as shown in Figure 7F–G. Similarly, in vitro lung tissue experiments revealed reduced levels of Nrf2, HO-1, and NQO1, along with a significant increase in Keap1, which was reversed by AH2QDS treatment, as shown in Figure 8A–G. These findings indicate that AH2QDS regulates the Keap1‒Nrf2 pathway by binding to Keap1, thereby releasing Nrf2 into the nucleus and activating the intracellular antioxidant system. To further confirm the involvement of the Nrf2 pathway in regulating the antioxidant properties of AH2QDS, we pretreated HBE cells with ML385 (Details can be found in the supplementary files Figure S3), a specific Nrf2 inhibitor. After AH2QDS treatment, ML385 abolished the AH2QDS-mediated effects, including the LPS+CSE-induced upregulation of Nrf2 and HO-1 expression, increased SOD levels, and decreased MDA levels, as shown in Figure 9A-D. These findings indicate that the antioxidant properties of AH2QDS are regulated through the Keap1‒Nrf2 pathway.
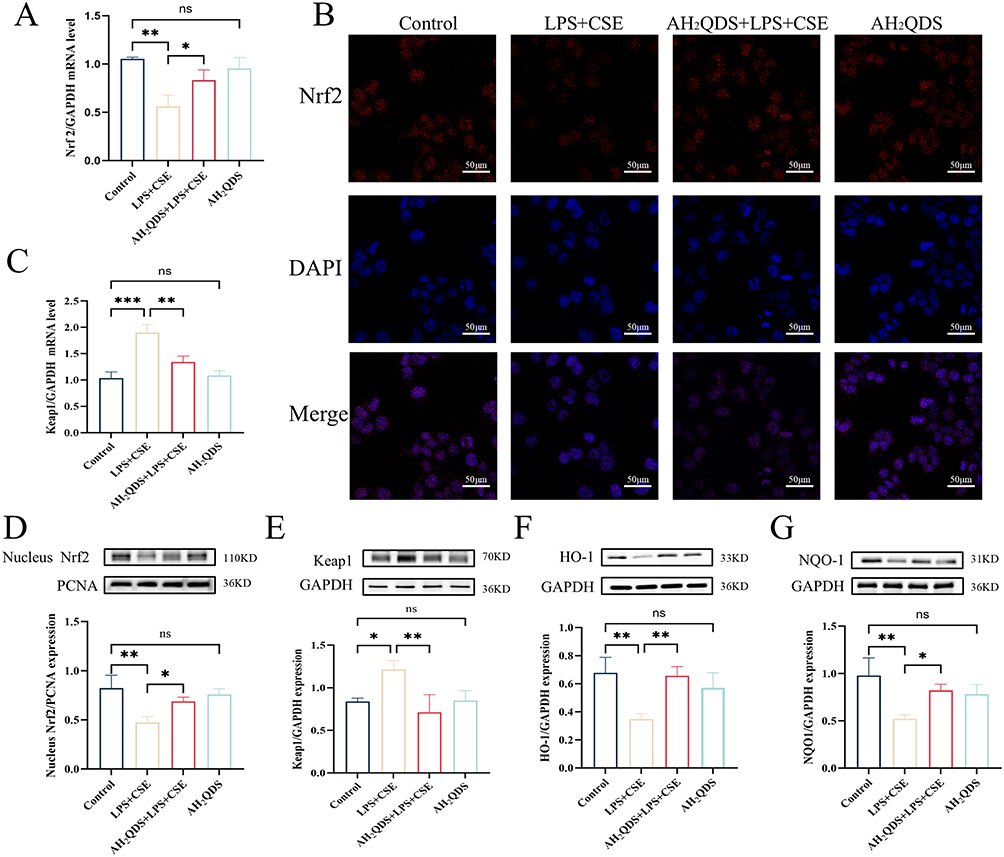 |
Figure 7 Effect of AH2QDS on the Nrf2 protein in vitro. In HBE cells, nuclear lysates were extracted for analysis of Nrf2 expression, and total lysates were extracted for analysis of Keap1, HO-1, and NQO1 protein expression. (A) mRNA expression of the Nrf2 protein in each experimental group. (B) Confocal microscopy was used to assess the immunofluorescence expression of the Nrf2 protein in each experimental group (scale bar=50 μm). (C) RT‒qPCR was used to determine the mRNA expression of Keap1 in the experimental groups. (D) Nrf2 protein expression in the nuclei of cells from each experimental group. (E) Keap1 protein expression in the cells of each experimental group. (F) HO-1 protein expression in each experimental group. (G) NQO1 protein expression in each experimental group. n≥3, ns: not statistically significant, *P˂0.05, **P˂0.01, ***P˂0.001. |
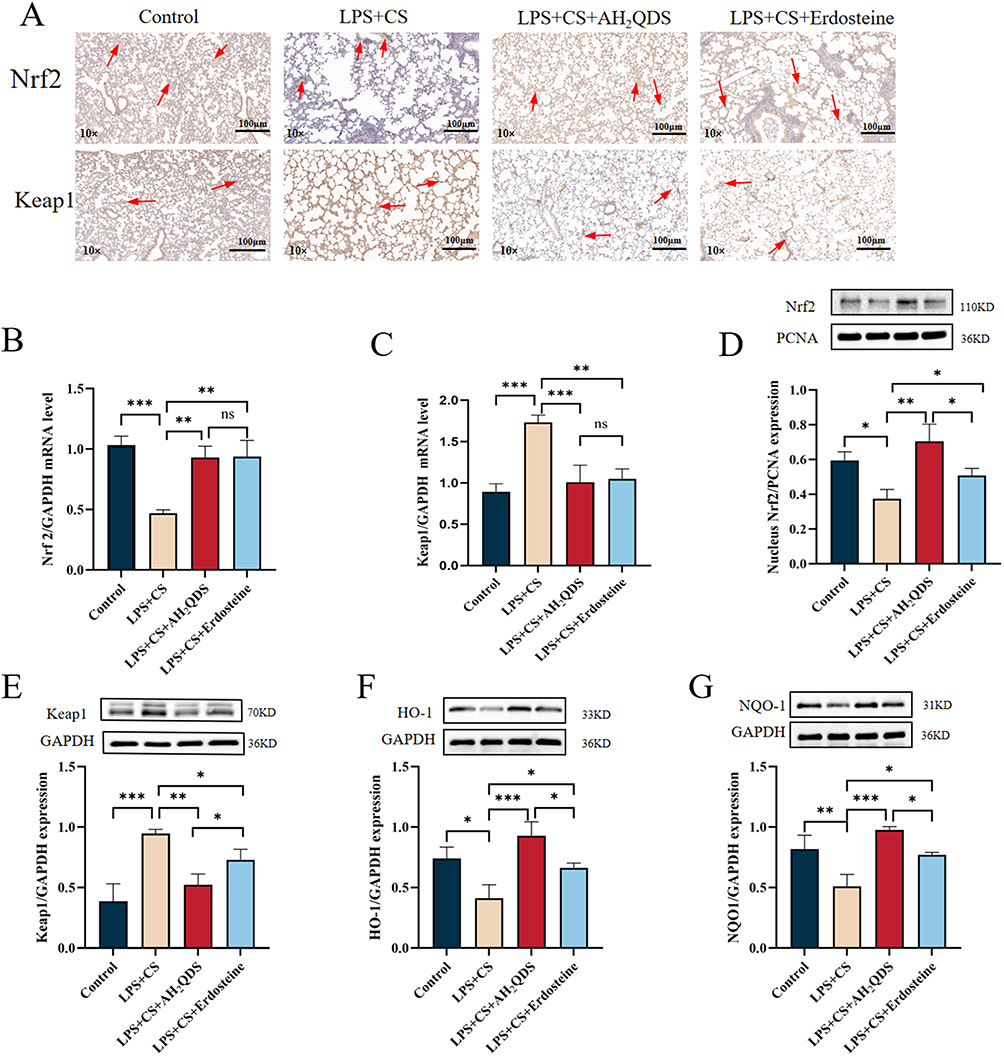 |
Figure 8 Effect of AH2QDS on the Nrf2 protein in rats with COPD. In lung tissue cells, nuclear lysates were extracted for analysis of Nrf2 expression, and total lysates were extracted for analysis of Keap1, HO-1, and NQO1 protein expression. (A) Immunohistochemistry was used to assess Nrf2 and Keap1 protein expression in lung tissues from each group of rats (10×, scale bar=100 μm). (B and C) RT‒qPCR analysis of Nrf2 and Keap1 mRNA expression in rat lung tissues. (D) Nrf2 protein expression in the nuclei of various groups of rat lung tissues. (E and G) Protein expression of Keap1, HO-1 and NQO1 in various groups of lung tissues from the rats. n≥3, ns: not statistically significant, *P˂0.05, **P˂0.01, ***P˂0.001. |
 |
Figure 9 ML385 eliminated the antioxidant effect of AH2QDS. ML385-treated HBE cells and extracted cell nucleus lysates were analysed for Nrf2 expression, and total lysates were extracted for HO-1 protein expression. (A) Nrf2 protein expression in the nucleus. (B) HO-1 protein expression. (C) SOD content in each experimental group. (D) MDA content. n≥3, *P ˂ 0.05, **P ˂ 0.01, ***P ˂ 0.001. |
AH2QDS Promotes the Dissociation of Nrf2 and Keap1 by Competing with Nrf2 for the Binding of Keap1 and Initiates the Cellular Antioxidant Damage Mechanism
The primary mechanism for regulating Nrf2 activity involves its interaction with the Keap1 protein, which inhibits Nrf2 activity when bound to it. Upon dissociation, Nrf2 enters the nucleus to initiate the antioxidant response program.19 Multiple amino acid sites on Keap1 that bind to Nrf2 have been revealed.20–22 Through molecular docking analysis, the affinity between AH2QDS and the Keap1 protein was predicted by computer modelling, as shown in Figure 10A and B, where five hydrogen bond interactions were formed between the chemical three-dimensional structure of AH2QDS and the amino acid residues Tyr-334, Arg-380, Asn-382, and Asn-414 in the Keap1 protein (green dashed lines). As shown in Figure 10C, the results of AH2QDS were consistent with those of previous studies in that three amino acid binding sites, Tyr-334, Arg-380, and Asn-382, are involved in the amino acid binding site of Keap1/Nrf2. The stability of the AH2QDS-Keap1 protein complex was examined by 100 ns molecular dynamics simulation, as shown in Figure 10D and E. The root mean square deviation curve (RSMD) and the root mean square fluctuation curve (RMSF) both fluctuated little with good stability.
 |
Figure 10 Effect of AH2QDS on Keap1. (A) Molecular formula of AH2QDS. (B) Interaction of AH2QDS with the amino acid residues Tyr-334, Arg-380, Asn-382, and Asn-414 in the Keap1 protein. (C) AH2QDS cobinding amino acid Wayne plots with Nrf2 and Keap1. (D) Plot of the RSMD results. (E) Plot of the RMSF results. |
Discussion
This study aimed to demonstrate that AH2QDS activates the Keap1‒Nrf2 pathway, initiating intracellular antioxidant mechanisms, attenuating oxidative stress damage in the small airways of rats with LPS- and CS-induced COPD, reducing inflammatory factor expression in lung tissues, and restoring lung function (PEF and MMEF). As a potent antioxidant, AH2QDS competitively binds to Keap1, releasing Nrf2 and promoting its translocation into the nucleus, thereby activating Nrf2. These findings were confirmed via in vitro experiments in LPS- and CSE-stimulated HBE cells. Nrf2 expression was significantly increased in the AH2QDS-treated group. Furthermore, AH2QDS treatment increased the expression of antioxidant enzymes (SOD, CAT, and GSH-PX), reduced MDA levels, and significantly decreased ROS levels. Concurrently, the concentrations of proinflammatory cytokines (IL-6, IL-33, IL-1β, and TNF-α) also decreased. These results indicate that AH2QDS increases antioxidant factor levels and reduces inflammatory cytokine levels by activating the antioxidant system. Molecular docking and immunoblotting analyses revealed that AH2QDS has a high affinity for Keap1, promotes Nrf2 dissociation from Keap1, and promotes Nrf2 nuclear translocation through competitive binding.
An imbalance between oxidative stress and antioxidant defences plays a pivotal role in the development of COPD.23,24 Promoting antioxidant defences can help reduce the development and progression of COPD. During the pathophysiological process of COPD, ROS exacerbate lipid peroxidation, impair antioxidant defences, and trigger the excessive release of inflammatory cytokines, leading to small airway epithelial cell damage and airway obstruction. In contrast, antioxidant enzymes such as SOD, GSH-PX, and CAT protect against small airway damage by neutralizing ROS.25–27 Conversely, inflammatory cytokines such as TNF-α, IL-33, IL-6, and IL-1β promote small airway wall thickening and structural remodelling by inducing fibroblast proliferation,28–30 exacerbating airflow limitation and ultimately reducing PEF and MMEF,31,32 which is consistent with the findings of this study. AH2QDS, a potent antioxidant, has been demonstrated to restores pulmonary vascular permeability and attenuate lung injury. Erdosteine is an approved pharmacological agent for the treatment of both acute and chronic pulmonary diseases.33 Accumulated evidence has demonstrated its potent antioxidant and anti-inflammatory properties.34,35 Clinical studies have consistently confirmed its efficacy in ameliorating airflow limitation in respiratory disorders.36 Therefore, erdosteine was selected as the positive control group in the present study. This study demonstrated that AH2QDS effectively reduces oxidative stress injury in lung tissue; restores PEF and MMEF by increasing SOD, GSH-PX, and CAT expression in a COPD rat model; promotes mitochondrial function; reduces apoptosis;37,38 and protects small airway epithelial cells by lowering ROS and cytokine levels.39 The comparison of the effects of AH2QDS and Erdosteine in the treatment of COPD revealed that AH2QDS demonstrated a distinct advantage in certain key indicators. In terms of antioxidant effects, although there were no significant differences in conventional oxidative stress markers such as SOD, MDA, and CAT between the two groups, the GSH-PX activity in the AH2QDS group was significantly higher than that in the Erdosteine group. This suggests that AH2QDS may exert antioxidant effects by specifically activating the glutathione system. Given the pivotal role of GSH-PX in hydrogen peroxide clearance, this advantage may be crucial in blocking the oxidative stress cascade in COPD. In terms of anti-inflammatory effects, although the levels of classical inflammatory factors, such as IL-1β, TNF-α, and IL-6, showed no significant differences between the two groups, AH2QDS exhibited a notably superior inhibitory effect on IL-33 compared to Erdosteine. IL-33, as an epithelial-specific alarmin, plays a crucial role in airway remodeling in COPD.40 These results suggest that AH2QDS may more effectively inhibit the epithelial-mesenchymal transition process through modulation of the IL-33/ST2 signaling pathway. Notably, in terms of pulmonary function improvement, the AH2QDS group showed a significantly greater improvement in MMEF and PEF compared to the Erdosteine group. This finding correlates with the molecular-level changes mentioned above. MMEF and PEF primarily reflect airway function, indicating that AH2QDS may have a specific protective effect on airway lesions.41,42 The differential therapeutic profile of AH2QDS may be attributed to its unique molecular structure, which appears to confer: selective targeting of the glutathione system, specific modulation of IL-33 signaling, enhanced protection of airway function. In addition, in vitro studies revealed that AH2QDS promotes cell migration and proliferation, thereby increasing cellular repair mechanisms. These findings highlight AH2QDS as a potential therapeutic agent for the prevention and management of COPD.
AH2QDS exerts its mechanism of action by activating the Keap1‒Nrf2 pathway, a well-established antioxidant pathway that regulates the transcription of antioxidant genes, modulates immune factors, and plays a crucial role in mitigating oxidative stress and impeding disease progression.9,43,44 In this study, treatment with ML385, a specific Nrf2 inhibitor, abolished the AH2QDS-induced upregulation of HO-1 and SOD expression and the reduction in MDA levels, further confirming that AH2QDS acts through the Keap1‒Nrf2 pathway. Additionally, molecular docking revealed that AH2QDS interacts with specific amino acid residues (Tyr-334, Arg-380, Asn-382, and Asn-414) in the Keap1 protein, further supporting the role of the Keap1‒Nrf2 pathway in mediating AH2QDS activity. LPS and CS exposure increased Keap1 levels while decreasing nuclear Nrf2 expression and downregulating its downstream targets HO-1 and NQO1. AH2QDS promoted the nuclear translocation of Nrf2, leading to the upregulation of downstream antioxidant proteins HO-1 and NQO-1. This molecular mechanism may explain the observed reduction in airway oxidative stress and lung damage in the COPD model. Multiple lines of evidence from our study support this explanation: Immunohistochemical analysis showed that, compared to the LPS and CS group, the accumulation of Nrf2 in the nucleus was significantly increased in the AH2QDS treatment group. After treatment, oxidative stress markers, such as MDA, were significantly reduced. Lung function parameters (including MMEF and PEF) in the AH2QDS treatment group improved, which may be associated with reduced alveolar destruction and airway remodeling in lung tissue. These findings reflect the potential antioxidant protective effect of AH2QDS.
Although AH2QDS was initially shown to affect the development of COPD through the Keap1‒Nrf2 pathway, further investigation is needed to determine whether AH2QDS can also modulate the activity of other pathways, thereby affecting the pathophysiological processes of COPD. This issue is particularly important given that COPD is a heterogeneous disease involving the regulation of multiple signalling networks,45 such as RAGE/EGFR and other pathways. Although molecular docking analysis suggested a potential interaction between AH2QDS and Keap1, this study did not experimentally validate their binding (eg, via co-immunoprecipitation). In addition, no transcriptomic sequencing or promoter sequencing was performed on the AH2QDS treatment group during this study, which failed to provide a comprehensive understanding of the changes in the relevant signalling pathways, resulting in some limitations in terms of the mechanism of action of AH2QDS on COPD. AH2QDS demonstrates significant protective effects in experimental models of COPD through its ability to promote Nrf2 nuclear translocation and subsequently enhance cellular antioxidant defense mechanisms. While these preclinical findings are promising, their translation to clinical applications necessitates careful evaluation of several critical factors. First, potential interspecies differences in drug metabolism, target protein expression patterns, and disease pathogenesis must be thoroughly characterized. Current experimental evidence indicates favorable short-term safety profiles in rodent models. However, the potential risks associated with chronic administration remain to be elucidated. Although preliminary observations have not revealed significant adverse effects in animal studies, comprehensive safety assessments should include systematic proteomic and transcriptomic analyses to identify potential off-target interactions, longitudinal monitoring of oxidative stress biomarkers, regular evaluation of hepatic and renal function parameters to detect possible subclinical toxicity. As a novel therapeutic candidate targeting oxidative stress-related pulmonary pathologies, the clinical potential of AH2QDS ultimately depends on resolving several key challenges, optimization of dosing regimens through pharmacokinetic/pharmacodynamic studies, development of effective drug delivery systems tailored for pulmonary administration, comprehensive evaluation of long-term safety profiles in relevant animal models.
In summary, AH2QDS ameliorated oxidative stress injury in the lung tissues of rats with LPS+CS-induced COPD by binding to Keap1, activating Nrf2 and transferring it to the nucleus, regulating the HO-1 and NQO1 proteins, inhibiting oxidative stress, increasing the antioxidant capacity of tissue cells, reducing the level of inflammation, and restoring pulmonary function (PEF, MMEF) (Figure 11).
 |
Figure 11 Mechanism by which AH2QDS attenuates oxidative stress damage and inflammation induced by LPS+CS. |
Conclusion
AH2QDS promoted lung function, including PEF and MMEF, in rats with COPD by activating the Keap1/Nrf2 pathway. These findings indicate that AH2QDS may be a promising candidate compound for the treatment of COPD.
Abbreviations
AH2QDS, Anthrahydroquinone-2-6-Disulfonate; COPD, Chronic Obstructive Pulmonary Disease; SOD, Superoxide Dismutase; CSE, Cigarette Smoke Extract; CAT, Catalase; OD, Optical Density; TNF-α, Tumour Necrosis Factor Alpha; GSH-PX, Glutathione Peroxidase; MLI, Mean Linear Intercept; ANOVA, Analysis of Variance; LPS, Lipopolysaccharide; MDA, Malondialdehyde; IL-6, Interleukin-6; IL-1β, Interleukin-1β; IL-33, Interleukin-33; CS, Cigarette Smoke; ROS, Reactive Oxygen Species; SEM, Standard Error of the Mean; MAA, Mean Alveolar Area; FBS, Foetal Bovine Serum; PEF, Peak Expiratory Flow; CCK-8, Cell Counting Kit-8; RT‒qPCR, Real-Time Quantitative Polymerase Chain Reaction; MMEF, Maximum Mid-Expiratory Flow; FEV0.3/FVC, Ratio of 0.3-Second Forced Expiratory Volume to Forced Vital Capacity; ARE, Antioxidant Response Element.
Data Sharing Statement
The data associated with this research are available in the article and supplementary files.
Declarations
The experimental protocol was approved by the Ethics Committee of Hainan Medical University. All animal care and procedures were conducted in accordance with the guidelines specified in the Guide for the Care and Use of Laboratory Animals (No. HYLL-2024-063).
Author Contributions
All authors have made significant contributions to the reported work, including conception, study design, execution, data acquisition, analysis, and interpretation. They participated in drafting, revising, or critically reviewing the manuscript, approved the final version for publication, and agreed on the journal to which the article was submitted. Each author reviewed and consented to all versions of the manuscript, including pre-submission drafts, revisions, the final accepted version, and any significant changes introduced during the proofing stage. All authors take full responsibility for the content of the article.
Funding
This study was funded by the National Natural Science Foundation of China (grant no. 81960351), the National Key Research and Development Program Project (grant no. 2023YFC3011800), the High‑Level Talent Fund of Hainan (grant no. 822RC835) and Hainan Provincial General Program (Grant No. 825MS199).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. De Luca SN, Vlahos R. Targeting accelerated pulmonary ageing to treat chronic obstructive pulmonary disease-induced neuropathological comorbidities. Brit J Pharmacol. 2024;181(1):3–20. doi:10.1111/bph.16263
2. Heijink IH, Brandenburg SM, Postma DS, van Oosterhout AJ. Cigarette smoke impairs airway epithelial barrier function and cell-cell contact recovery. Eur Respir J. 2012;39(2):419–428. doi:10.1183/09031936.00193810
3. Cha SR, Jang J, Park SM, Ryu SM, Cho SJ, Yang SR. Cigarette smoke-induced respiratory response: insights into cellular processes and biomarkers. Antioxidants-Basel. 2023;12(6):1210.
4. Kume H, Yamada R, Sato Y, Togawa R. Airway smooth muscle regulated by oxidative stress in COPD. Antioxidants-Basel. 2023;12(1):142. doi:10.3390/antiox12010142
5. Rahman I, Biswas SK, Kode A. Oxidant and antioxidant balance in the airways and airway diseases. Eur J Pharmacol. 2006;533(1–3):222–239. doi:10.1016/j.ejphar.2005.12.087
6. Lee J, Jang J, Park SM, Yang SR. An update on the role of Nrf2 in respiratory disease: Molecular mechanisms and therapeutic approaches. Int J Mol Sci. 2021;22(16):8406.
7. Piotrowska M, Swierczynski M, Fichna J, Piechota-Polanczyk A. The Nrf2 in the pathophysiology of the intestine: Molecular mechanisms and therapeutic implications for inflammatory bowel diseases. Pharmacol Res. 2021;163:105243. doi:10.1016/j.phrs.2020.105243
8. Zhang Z, Fu C, Liu J, et al. Hypermethylation of the Nrf2 promoter induces ferroptosis by inhibiting the Nrf2-GPX4 axis in COPD. Int J Chronic Obstr. 2021;16:3347–3362.
9. Barnes PJ. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020;33:101544. doi:10.1016/j.redox.2020.101544
10. Li N, Yi Y, Chen J, et al. Anthrahydroquinone‑2,6‑disulfonate attenuates PQ‑induced acute lung injury through decreasing pulmonary microvascular permeability via inhibition of the PI3K/AKT/eNOS pathway. Int J Mol Med. 2024;54(1):63. doi:10.3892/ijmm.2024.5387
11. Zhang Q, Liu J, Duan H, Li R, Peng W, Wu C. Activation of Nrf2/HO-1 signaling: An important molecular mechanism of herbal medicine in the treatment of atherosclerosis via the protection of vascular endothelial cells from oxidative stress. J Adv Res. 2021;34:43–63. doi:10.1016/j.jare.2021.06.023
12. Li Q, Qian J, Huang QF, et al. Research progress on the molecular mechanisms of lung injury induced by paraquat poisoning. J Hainan Med Univ. 2022;28(4):309–314.
13. Li C, Chen F, Lin L, Li J, Zheng Y, Chen Q. CSE triggers ferroptosis via SIRT4-mediated GNPAT deacetylation in the pathogenesis of COPD. Resp Res. 2023;24(1):301. doi:10.1186/s12931-023-02613-0
14. Ding H, Yan L, Wang Y, et al. Astaxanthin attenuates cigarette smoke-induced small airway remodeling via the AKT1 signaling pathway. Resp Res. 2024;25(1):148. doi:10.1186/s12931-024-02768-4
15. Upadhyay P, Wu CW, Pham A, et al. Animal models and mechanisms of tobacco smoke-induced chronic obstructive pulmonary disease (COPD). J Toxicol Env Heal B. 2023;26(5):275–305. doi:10.1080/10937404.2023.2208886
16. Liang GB, He ZH. Animal models of emphysema. Chinese Med J-Peking. 2019;132(20):2465–2475. doi:10.1097/CM9.0000000000000469
17. Labaki WW, Rosenberg SR. Chronic obstructive pulmonary disease. Ann Intern Med. 2020;173(3):ITC17–32. doi:10.7326/AITC202008040
18. Brightling C, Greening N. Airway inflammation in COPD: progress to precision medicine. Eur Respir J. 2019;54(2):1900651. doi:10.1183/13993003.00651-2019
19. Ulasov AV, Rosenkranz AA, Georgiev GP, Sobolev AS. Nrf2/Keap1/ARE signaling: towards specific regulation. Life Sci. 2022;291:120111. doi:10.1016/j.lfs.2021.120111
20. Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–223. doi:10.1038/ncb2021
21. Yamamoto T, Suzuki T, Kobayashi A, et al. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28(8):2758–2770. doi:10.1128/MCB.01704-07
22. Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol. 2006;26(8):2887–2900. doi:10.1128/MCB.26.8.2887-2900.2006
23. Yang IA, Jenkins CR, Salvi SS. Chronic obstructive pulmonary disease in never-smokers: risk factors, pathogenesis, and implications for prevention and treatment. Lancet Respir Med. 2022;10(5):497–511. doi:10.1016/S2213-2600(21)00506-3
24. Keir HR, Chalmers JD. Neutrophil extracellular traps in chronic lung disease: implications for pathogenesis and therapy. Eur Respir Rev. 2022;31(163):210241. doi:10.1183/16000617.0241-2021
25. Fujii J, Homma T, Osaki T. Superoxide radicals in the execution of cell death. Antioxidants-Basel. 2022;11(3). doi:10.3390/antiox11030501
26. Militello R, Luti S, Gamberi T, Pellegrino A, Modesti A, Modesti PA. Physical activity and oxidative stress in aging. Antioxidants-Basel. 2024;13(5). doi:10.3390/antiox13050557
27. Islam MN, Rauf A, Fahad FI, et al. Superoxide dismutase: an updated review on its health benefits and industrial applications. Crit Rev Food Sci. 2022;62(26):7282–7300. doi:10.1080/10408398.2021.1913400
28. Riera-Martinez L, Canaves-Gomez L, Iglesias A, Martin-Medina A, Cosio BG. The role of IL-33/ST2 in COPD and its future as an antibody therapy. Int J Mol Sci. 2023;24(10):8702. doi:10.3390/ijms24108702
29. Barnes PJ. Oxidative stress in chronic obstructive pulmonary disease. Antioxidants-Basel. 2022;11(5). doi:10.3390/antiox11050965
30. Chung KF. Cytokines in chronic obstructive pulmonary disease. Eur Respir J. 2001;18(34 suppl):50s–59s. doi:10.1183/09031936.01.00229701
31. Fan J, Fang L, Cong S, et al. Potential pre-COPD indicators in association with COPD development and COPD prediction models in Chinese: a prospective cohort study. Lancet Reg Health-W. 2024;44:100984.
32. Cen J, Weng L. Comparison of peak expiratory Flow(PEF) and COPD assessment test (CAT) to assess COPD exacerbation requiring hospitalization: a prospective observational study. Chron Resp Dis. 2022;19:231461117. doi:10.1177/14799731221081859
33. Cazzola M, Page C, Rogliani P, Calzetta L, Matera MG. Multifaceted beneficial effects of erdosteine: more than a mucolytic agent. Drugs. 2020;80(17):1799–1809.
34. Kim SJ, Park C, Lee JN, et al. Erdosteine protects HEI-OC1 auditory cells from cisplatin toxicity through suppression of inflammatory cytokines and induction of Nrf2 target proteins. Toxicol Appl Pharm. 2015;288(2):192–202.
35. Park JS, Park MY, Cho YJ, et al. Anti-inflammatory effect of erdosteine in lipopolysaccharide-stimulated RAW 264.7 cells. Inflammation. 2016;39(4):1573–1581. doi:10.1007/s10753-016-0393-4
36. Calverley PM, Page C, Dal negro RW, et al. Effect of erdosteine on COPD exacerbations in COPD patients with moderate airflow limitation. Int J Chronic Obstr. 2019;14:2733–2744.
37. Jing Q, Zhou C, Zhang J, et al. Role of reactive oxygen species in myelodysplastic syndromes. Cell Mol Biol Lett. 2024;29(1):53.
38. Di Rienzo M, Romagnoli A, Refolo G, et al. Role of AMBRA1 in mitophagy regulation: emerging evidence in aging-related diseases. Autophagy. 2024;20(12):2602–2615. doi:10.1080/15548627.2024.2389474
39. Jain A, Kim BR, Yu W, et al. Mitochondrial uncoupling proteins protect human airway epithelial ciliated cells from oxidative damage. Proc Natl Acad Sci U S A. 2024;121(10):e1976196175.
40. Zhou Y, Xu Z, Liu Z. Role of IL-33-ST2 pathway in regulating inflammation: current evidence and future perspectives. J Transl Med. 2023;21(1):902. doi:10.1186/s12967-023-04782-4
41. Almeshari MA, Alobaidi NY, Sapey E, Stockley RA, Stockley JA. Small airways dysfunction: the importance of utilising Z-scores to define MMEF abnormalities in clinical practice. Heliyon. 2023;9(10):e20744. doi:10.1016/j.heliyon.2023.e20744
42. Csonka LL, Tikkakoski A, Vuotari L, Karjalainen J, Lehtimaki L. Relation of changes in peak expiratory flow (PEF) and forced expiratory volume in 1 s (FEV(1)) during bronchoconstriction. Clin Physiol Funct I. 2024;44(6):447–453. doi:10.1111/cpf.12898
43. Boutten A, Goven D, Artaud-Macari E, Boczkowski J, Bonay M. NRF2 targeting: a promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol Med. 2011;17(7):363–371. doi:10.1016/j.molmed.2011.02.006
44. van der Horst D, Carter-Timofte ME, van Grevenynghe J, Laguette N, Dinkova-Kostova AT, Olagnier D. Regulation of innate immunity by Nrf2. Curr Opin Immunol. 2022;78:102247. doi:10.1016/j.coi.2022.102247
45. Abdo M, Pedersen F, Kirsten AM, et al. Association of airway inflammation and smoking status with IL-33 level in sputum of patients with asthma or COPD. Eur Respir J. 2024;64(3):2400347. doi:10.1183/13993003.00347-2024
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Efficacy of Vitamin C Supplementation on Chronic Obstructive Pulmonary Disease (COPD): A Systematic Review and Meta-Analysis
Lei T, Lu T, Yu H, Su X, Zhang C, Zhu L, Yang K, Liu J
International Journal of Chronic Obstructive Pulmonary Disease 2022, 17:2201-2216
Published Date: 10 September 2022
The Relationship Between BMI and Lung Function in Populations with Different Characteristics: A Cross-Sectional Study Based on the Enjoying Breathing Program in China
Tang X, Lei J, Li W, Peng Y, Wang C, Huang K, Yang T
International Journal of Chronic Obstructive Pulmonary Disease 2022, 17:2677-2692
Published Date: 18 October 2022
CC16 as an Inflammatory Biomarker in Induced Sputum Reflects Chronic Obstructive Pulmonary Disease (COPD) Severity
Chen M, Xu K, He Y, Jin J, Mao R, Gao L, Zhang Y, Wang G, Gao P, Xie M, Liu C, Chen Z
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:705-717
Published Date: 27 April 2023
Clinical and Functional Effects of Inhaled Dual Therapy Umeclidinium/Vilanterol in Patients with Chronic Obstructive Pulmonary Disease: A Real-Life Study
Pelaia C, Ferrante Bannera A, Rotundo FL, Tropea FG, Armentaro G, Maglio A, Sciacqua A, Vatrella A, Pelaia G
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:995-1002
Published Date: 26 May 2023
Can Blood Biomarkers Be Used to Assess Oxidative Stress in COPD Patients After Pulmonary Rehabilitation
Beykumül A, Ersoy Y, Gülbaş G, Neselioglu S
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:2179-2186
Published Date: 5 October 2023