")
Back to Journals » Nature and Science of Sleep » Volume 17
Associations Between Obstructive Sleep Apnea and Metabolic Dysfunction-Associated Fatty Liver Disease: Insights from Comprehensive Mendelian Randomization and Gene Expression Analysis
Authors Ma T, Liao C, Chen W , Feng J
Received 17 December 2024
Accepted for publication 28 May 2025
Published 8 July 2025 Volume 2025:17 Pages 1571—1585
DOI https://doi.org/10.2147/NSS.S511115
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Ahmed BaHammam
Tianyu Ma,1 Chunyan Liao,2 Wenhui Chen,3 Jia Feng4,5
1School of Medicine, Jinan University, Guangzhou, Guangdong, 510630, People’s Republic of China; 2Department of Anesthesia and Surgery Center, The First Affiliated Hospital of Jinan University, Guangzhou, People’s Republic of China; 3Department of Metabolic and Bariatric Surgery, The First Affiliated Hospital of Jinan University, Guangzhou, People’s Republic of China; 4Department of Cellular Biology, Institute of Biomedicine, Jinan University, Guangzhou, Guangdong, 510632, People’s Republic of China; 5The First Affiliated Hospital, Jinan University, Guangzhou, Guangdong, 510630, People’s Republic of China
Correspondence: Jia Feng, Department of Cellular Biology, Institute of Biomedicine, Jinan University, No. 601 Huangpu Ave West, Guangzhou, Guangdong, 510632, People’s Republic of China, Tel/Fax +86 13535224413, Email [email protected] Chunyan Liao, Department of Anesthesia and Surgery Center, the First Affiliated Hospital of Jinan University, Guangzhou, People’s Republic of China, Tel +86 13725251007, Fax +86 020 38688580, Email [email protected]
Background: Obstructive sleep apnea (OSA) is linked to metabolic dysfunction-associated fatty liver disease (MAFLD), yet their exact causality and underlying mechanisms remain inconclusive. We aimed to investigate their causal associations and shared biomarkers using Mendelian randomization (MR) and bioinformatics approaches.
Methods: We used OSA-related and MAFLD-related GWAS data to explore their causal relationship and the role of body mass index (BMI) through two-sample and network MR analysis. Gene expression profiles were analyzed to identify intersection genes through differential expression analysis and weighted gene co-expression network analysis (WGCNA). Functional enrichment (GO and KEGG), protein–protein interaction (PPI) networks, and immune cell infiltration analyses (ssGSEA) were performed on the intersecting genes. We then conducted MR analysis to assess the relationship between immune cells and both diseases. Inverse variance weighting (IVW) served as the primary MR method, supplemented by MR-Egger regression, weighted median, and weighted mode.
Results: MR analysis revealed that OSA increased the risk of MAFLD [odds ratio (OR)=1.40, 95% CI 1.14– 1.73, p= 0.002], with OSA potentially mediating the effect of BMI on MAFLD, accounting for 62.3% of the mediation. Bioinformatics identified 42 intersection genes. Four hub genes (FOS, EGR1, NR4A1, JUN) were ultimately obtained by PPI network, which were strongly linked to immune cell infiltration. Additionally, three immune cell phenotypes (CD4RA on TD CD4+, HLA DR on CD14+ CD16-monocytes, and HLA DR on CD14+ monocytes) were found to be associated with both OSA and MAFLD.
Conclusion: OSA may causally influence MAFLD and mediate the effect of BMI on MAFLD. Four key genes and three immune cell phenotypes play crucial roles in the shared pathogenesis of both diseases.
Keywords: obstructive sleep apnea, metabolic dysfunction-associated fatty liver disease, OSA, Mendelian randomization, bioinformatics
Introduction
Obstructive sleep apnea (OSA) is an increasingly common sleep-related respiratory disorder, characterized by chronic intermittent hypoxia (CIH) and sleep fragmentation.1 It is estimated that nearly one billion people worldwide are affected by OSA.2 OSA is associated with a range of chronic metabolic comorbidities, including cardiovascular disorders, hypertension, type 2 diabetes, and metabolic dysfunction-associated fatty liver disease (MAFLD), contributing to a significant increase in morbidity and mortality.3–5 MAFLD, previously known as nonalcoholic fatty liver disease (NAFLD), is a most common chronic liver disease, with an incidence of 25–30% globally.6 The MAFLD encompasses a spectrum of conditions, ranging from isolated steatosis to the serious state of metabolic dysfunction-associated steatohepatitis (MASH), MASH-fibrosis, and cirrhosis.7 MASH is distinguished by hepatocellular ballooning and lobular inflammation, which may ultimately progress to end-stage complications, including cirrhosis and hepatocellular carcinoma.8 Unfortunately, the effective treatments for MAFLD/NASH have been challenging.9 Thus, a deeper understanding of the mechanisms driving MAFLD, especially its development into MASH, is crucial for improving treatment strategies in clinical practice.
Numerous emerging evidence consistently highlights OSA’s pivotal role in the pathogenesis and progression of MAFLD. Clinical studies suggest that CIH associated with OSA may exacerbate liver injury and accelerate the progression from NAFLD to MASH.10,11 For instance, a study by Fu et al based on biopsy-confirmed NAFLD demonstrated that OSA significantly increases the risk of NASH in patients with obesity.12 A meta-analysis, including nine studies with 2272 individuals, indicated that OSA was strongly associated with steatosis, lobular inflammation, ballooning degeneration, and fibrosis.13 Additionally, CIH induced rapid juvenile murine NASH model further suggested CIH can trigger and accelerate MASH.14 The underlying mechanisms linking OSA to NAFLD/MASH involve oxidative stress, inflammation, insulin resistance, lipid metabolism disorders, and gut microbiome dysbiosis.15 In a rat model of CIH, Chen et al identified differentially expressed genes (DEGs) primarily related to hepatic metabolism, apoptotic process, and oxidative stress.16 However, to date, the exact molecular mechanisms underlying the comorbidity of OSA and MAFLD have been poorly characterized.
Observational studies have attempted to determine the association between OSA and MAFLD, but the findings are susceptible to confounding factors. Mendelian randomization (MR) is commonly employed to infer the causal potential relationship between exposure and outcome, as it mitigates the effects of residual confounding and reverse causality.17 Previous MR analysis have investigated the causal link between OSA and MAFLD, suggesting that no direct causal relationship exists.18,19 Nevertheless, these studies did not rule out the possibility of an indirect effect of OSA on MAFLD via body mass index (BMI). As is well known, obesity and OSA often coexist. Moreover, these MR studies did not further investigate the interconnected relationships among BMI, OSA, and MAFLD. Thus, further investigation is needed to elucidate these relationships, which will facilitate a more comprehensive understanding of how OSA influences MAFLD. In this study, we aimed to conduct two-sample MR and network MR to explore the causal relationship between OSA and MAFLD, considering the role of BMI. Furthermore, we will employ bioinformatics approaches to investigate the mechanisms underlying this relationship between OSA and MAFLD, thereby providing new perspectives and strategies for understanding their shared pathogenesis.
Materials and Methods
Data Sources
The genome-wide association studies (GWAS) for OSA were acquired from a large-scale, recently conducted GWAS summary data from FinnGen R11 (https://r11.finngen.fi/), which includes 50200 OSA cases and 401484 controls normal controls of European individuals. The diagnosis of OSA in the database is based on the International Classification of Diseases (ICD) code (ICD-10: G47.3; ICD-9:3472), defined by subjective symptoms, clinical assessment, and sleep records, using AHI ≥ 5/h or respiratory event index ≥5/h. Summary-level statistics for MAFLD were extracted from a GWAS meta-analysis based on four European cohorts, comprising a total of 8434 NAFLD cases and 770,180 controls.20 The GWAS data for immune cells were available from the GWAS Catalog (https://www.ebi.ac.uk/gwas/) (IDs from GCST0001391 to GCST0002121).21 These datasets include 731 immunophenotypes, consisting absolute cell (AC) counts (n = 118), median fluorescence intensities (MFI) reflecting surface antigen levels (n = 389), morphological parameters (MP) (n = 32), and relative cell (RC) counts (n = 192).
Gene expression microarray data from the GSE135917 and GSE89632 datasets were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). The GSE135917 datasets (platform: GPL14951) include gene expression data from subcutaneous adipose tissue samples, comprising 34 OSA patients and 8 healthy controls. The GSE89632 dataset (platform: GPL14951) contained 63 liver tissue samples, consisting of 24 controls and 39 MAFLD patients.
MR Analysis
Before performing MR analysis, instrumental variables were selected. We identified genetic instruments with a p-value threshold of p < 5×10−8 for significant single nucleotide polymorphisms (SNPs) and removed SNPs with linkage disequilibrium (r2 > 0.001 and clumping window < 10Mb) according to the European 1000Genomes Project Phase 3 as reference panel for linkage disequilibrium. We also used the PhenoScanner (http://www.phenoscanner.medschl.cam.ac.uk/) to investigate and remove pleiotropic SNPs, including smoking behavior and alcohol consumption.
To increase the number of SNPs, the threshold was relaxed to p < 1×10−5 for immune cells. Furthermore, SNPs with F-statistics < 10, indicative of weak instrument associations, were excluded (F-statistic formula: F = β²/σ², where β represents the SNP-exposure effect estimate and σ is the standard deviation of the variant).22 After harmonizing data, we further filtered out SNPs strongly associated with the outcome (p < 5×10−8).
We conducted a two-sample MR analysis to explore the causal effects between OSA and MAFLD, following the Strengthening the Reporting of Mendelian Randomization Studies (STROBE-MR) statement.23 Multiplicative random-effects inverse variance weighting (IVW) was used as the primary analysis method We also applied three additional methods (MR-Egger, weighted median, and weighted mode) as supplements to ensure the robustness and validity of the results.22,24,25 Additionally, we further performed network MR analysis to explore the interconnected relationships among BMI, OSA, and MAFLD. The network MR analysis26 was based on three estimates: the total effect (c): MR results from exposure to outcome; the direct effect (a): MR results from exposure to mediator; the direct effect (b): MR results from mediator to outcome; Mediation effect (a*b): a*b/c.
For significant MR results, we used several sensitivity analyses to evaluate pleiotropy, heterogeneity, and stability. MR-Egger regression and MR-Pleiotropy Residual Sum and Outlier methods (MR-PRESSO) global tests were used to examine horizontal or directional pleiotropy.25,27 Cochrane’s Q statistic was applied to test for heterogeneity, with p > 0.05 indicating no heterogeneity.28 A leave-one-out analysis was conducted to assess the stability of the MR results by re-evaluating the data after omitting each SNP.
Differential Expression Analysis
We used the “limma” package in R software (R 4.4.1) to identify differentially expressed genes (DEGs) between control and disease groups, based on the standardized GSE135917 and GSE89632 datasets. The criteria for selecting DEGs were an adjusted p-value < 0.05 and |logFC| ≥ 0.585 across both datasets. Volcano plots and heatmaps were generated to visualize the differential expression results for each group.
Weighted Gene Co-Expression Network Analysis (WGCNA)
WGCNA was employed to screen the characteristic module genes that are highly correlated with OSA and MAFLD by constructing gene co-expression networks.29 For effective WGCNA, the following steps were taken:, (1) the genes with a median absolute deviation (MAD) was calculated, and the genes with MAD < 30% were removed; (2) sample hierarchical clustering was performed to identify outlier samples, and outliers were excluded; (3) the “pickSoftThreshold” function was utilized to determine an appropriate soft threshold, establishing a scale-free co-expression network; (4) a topological overlap matrix (TOM) was construct, and hierarchical clustering with the dynamic pruning-tree algorithm was employed to identify different gene modules; (5) Pearson correlation analysis was applied to examine the relationship between gene modules and clinical phenotypes.
Functional Enrichment Analysis
The Venn diagram was constructed to obtain intersection genes based on DEGs and gene modules from both datasets. Then, functional enrichment analysis of these intersection genes was performed, including the Gene Ontology (GO)30 and Kyoto Encyclopedia of Genes and Genomes (KEGG).31 The GO analysis focused on Biological Process (BP), Molecular Function (MF), and Cellular Component (CC), with the top 10 GO terms visualized. A p < 0.05 was considered statistically significant.
Protein–Protein Interaction (PPI) Network Construction
For the intersection genes, a PPI network was constructed using the STRING database (http://string-db.org/), with a minimum interaction score of 0.4.32 The data from String database was further analyzed using Cytoscape software (version 3.9.1). Subsequently, four algorithms, including Maximal Clique Centrality (MCC), Maximum Neighborhood Component (MNC), Degree, and Edge Percolated Component (EPC), were selected to jointly detect hub expression genes. The top five intersection genes identified by each algorithm were considered as hub genes. A venn diagram was used to visualize the interaction of each algorithm with the hub genes. Violin plots were created to display the expression profiles of hub genes across the two databases. Receiver operating characteristic (ROC) curves and the area under the curve (AUC) were used to assess the diagnostic performance of the hub genes.
Immune Infiltration Analysis
To explore the role of immune cells in the pathogenesis of OSA and MASH, we performed immune infiltration analysis.33,34 The ssGSEA algorithm was utilized to quantify the infiltration levels of 24 immune cell types in both OSA and MAFLD compared to controls, based on immune cell-related expression matrices. Furthermore, Spearman correlations were employed to investigate the correlation between hub genes and immune-infiltrating cells. Box plots and correlation heatmaps were generated to present the results of immune infiltration.
Statistical Analysis
R software (version 4.3.1, R Foundation for Statistical Computing, Austria) was employed for statistical analyses and graph generation. A p-value of <0.05 considered to be nominally significant. However, the Bonferroni correction method was used to adjust for multiple testing. For immunophenotypes analyses, a significant p-value was set as at 6.8×10-5 (0.05/731).
Results
MR Analysis
The study framework is illustrated in Figure 1. After a rigorous screening process, a total of 19 SNPs were identified in the forward MR analysis, and 4 SNPs in the reverse MR analysis (Supplementary Table S1). In the main MR analysis, we found a suggestive association between OSA and an increased risk of MAFLD (OR: 1.40, 95% CI: 1.14–1.73, p = 0.002). Further reverse MR analysis exploring the effects of MAFLD on OSA (p>0.05). In sensitivity analyses, no significant heterogeneity was detected using the Cochran Q-test (all p>0.05). Both the MR Egger intercept test (all p >0.05) and the MR-PRESSO test (all global p>0.05) did not detect any evidence of pleiotropy. Finally, leave-one-out analyses indicated that the effects were not influenced by any single SNP (Figure 2A, Supplementary Table S2, Supplementary Figures S1 and S2).
 |
Figure 1 Overview and analysis process of our research. Abbreviations: OSA, Obstructive sleep apnea; MAFLD, metabolic dysfunction-associated fatty liver disease; MR, Mendelian randomization; GWAS, genome-wide association study; GEO, Gene Expression Omnibus; DEGs, differentially expressed genes; WGCNA, weighted gene co-expression network analysis; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, Protein–protein interaction; ROC, Receiver operating characteristic. |
 |
Figure 2 Results of Mendelian randomization. (A) Forward and reverse Mendelian randomization explored the associations between OSA and MAFLD; (B) Network Mendelian randomization study explored the interconnected relationships among BMI, OSA, and MAFLD. Abbreviations: OSA, Obstructive sleep apnea; MAFLD, metabolic dysfunction-associated fatty liver disease; BMI, Body mass index. CI, confidence interval; OR, odds ratio. SNP, single-nucleotide polymorphism. |
In network MR analyses, genetic variant of OSA was not associated with BMI (IVW: β= 0.04, p =406), but BMI was found a significant association with both MAFLD (IVW: β = 0.36, p <0.001) and OSA (IVW: β= 0.66, p < 0.001). These results indicated that BMI does not mediate the association between OSA and MAFLD (mediation effect: 4.2%). Conversely, OSA may act as a mediator in the pathway from BMI and MAFLD, with the mediated proportion of 62.3% (Figure 2B).
DEG Identification in OSA and MAFLD
A total of 380 DEGs were identified in OSA compared to controls, including 237 up-regulated genes and 143 down-regulated genes (Supplementary Table S3). Similarly, comparison of MAFLD and controls revealed 2289 DEGs, comprising 1021 up-regulated genes and 1268 down-regulated genes (Supplementary Table S4). Volcano plot was used to illustrate all DEGs (Figure 3A and B), and heatmaps were created to display the top 50 upregulated or downregulated DEGs (Figure 3C and D).
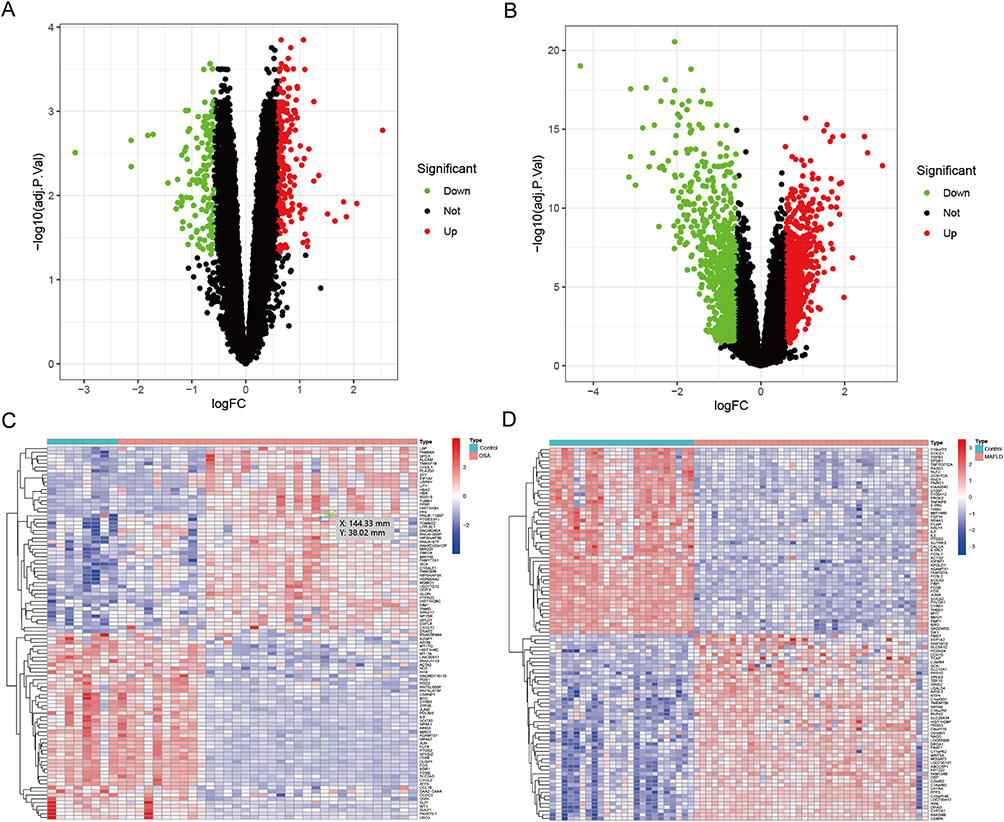 |
Figure 3 Differential gene expression analysis. ((A) OSA; (B) MAFLD) Volcano plots depict the differential expression genes (DEGs) in OSA and MAFLD; ((C) OSA; (D) MAFLD) Heatmaps illustrate the top 50 DEGs in OSA and MAFLD. |
Identification of Key Module Genes in OSA and MAFLD
We performed WGCNA analysis to identify the key module genes associated with OSA and MAFLD (Supplementary Table S5). Based on the WGCNA method, the optimal soft-thresholding power was set at 11 for GSE135917 dataset and 5 for GSE89632 dataset (Figure 4A and B). Cluster dendrograms were generated for OSA and MAFLD using a minimum module size of 50 and a merge cut height of 0.25 (Figure 4C and D). Finally, a total of 13 co-expression modules were identified for OSA dataset and 9 co-expression modules for MAFLD (Figure 4E and F). Consequently, we selected the modules MElightgreen (r = 0.63, p = 9e-6) and MEblue (r = 0.86, p = 1e-18), which indicated the highest positive correlation with OSA and MAFLD, respectively. Additionally, the modules MEturquoise (r = −0.55, p = 1e-4) and MEred (r = −0.67, p = 4e-09) were selected for their strong negative correlations.
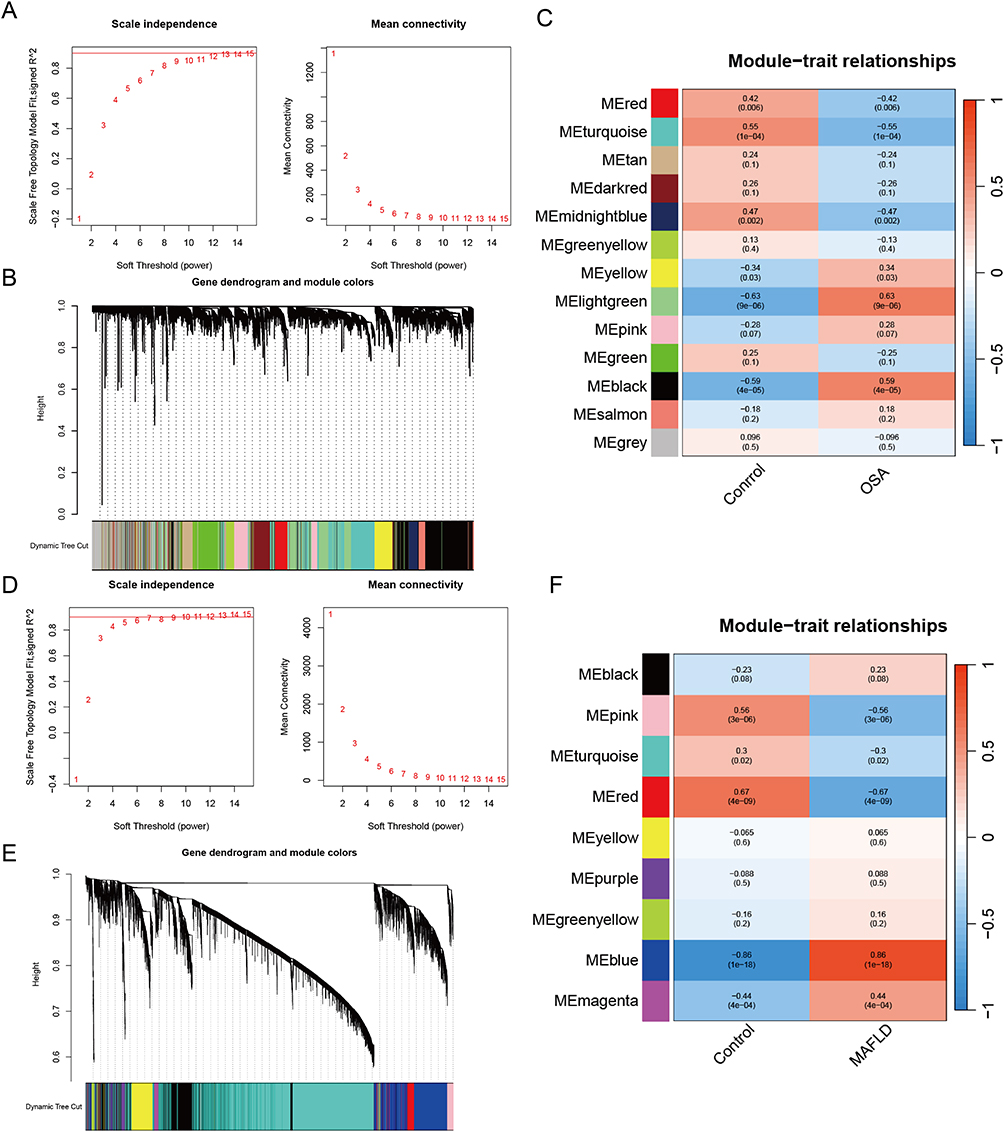 |
Figure 4 Weighted gene co-expression network analysis. ((A) OSA; (D) MAFLD) Determination of soft threshold powers in OSA and MAFLD. ((B) OSA; (E) MAFLD) Gene cluster trees in OSA and MAFLD. ((C) OSA; (F) MAFLD) Modular-feature relationship in OSA and MAFLD, with numbers in the modules representing correlation coefficients and p-values. |
Functional Enrichment Analysis of Intersection Genes
After intersecting the DEGs and key module genes, we obtained 42 intersection genes between the two diseases (Figure 5A). GO enrichment analysis of these intersection genes revealed significant enrichment in BP terms such as “response to lipopolysaccharide” and “T−helper 17 cell differentiation”, CC terms like “RNA polymerase II transcription regulator complex”, and “cytoplasmic side of plasma membrane”, and MF terms such as “DNA−binding transcription activator activity, RNA polymerase II−specific”, and “DNA−binding transcription activator activity”. KEGG pathway analysis showed significant enrichment in the “TNF signaling pathway” and “IL−17 signaling pathway” (Figure 5B and C).
 |
Figure 5 Functional and pathway enrichment analysis. (A) Venn diagram of intersecting analysis of DEGs and key module genes for OSA and MAFLD; (B) GO analysis of intersection genes; (C) KEGG enrichment of intersection genes. |
Selection of Hub Genes
We constructed a PPI network for the 42 intersection genes using the STRING database. After excluding 10 non-interaction genes, 32 genes were retained (Figure 6A). The CytoHubba plugin in Cytoscape, along with four algorithms (MCC, Degree, MNC, and EPC), was used to identify hub genes from the intersection genes (Figure 6B). Finally, four genes (FOS, ATF3, JUN, EGR1) were identified as hub genes. Expression analysis showed that all four hub genes exhibited significantly low expression in both OSA and MAFLD (Figure 6C and D). ROC curve analyses demonstrated satisfactory diagnostic performance for all hub genes, with AUC values >0.8 (Figure 6E and F).
 |
Figure 6 Construction of PPI network and identification of hub genes. (A) PPI network of intersection genes. (B) Hub genes determined by MCC, MNC, Degree, and EPC algorithms. (C and D) Expression profiles of hub genes in OSA and MAFLD; (E and F) ROC curves demonstrate the predictive performance of hub genes in OSA and MAFLD. |
Immune Cell Infiltration Analysis
Utilizing the ssGSEA algorithm, we quantified immune cell infiltration in OSA and MAFLD. Compared to the control group, the MAFLD group showed significantly higher levels of certain immune cells, such as “Activated.CD8.T.cell”, “CD56bright.natural.killer.cell”, “Gamma.delta.T.cell”. Conversely, the MAFLD group had lower levels of immune cells like “Activated.CD4.T.cell”, “Activated.dendritic.cell”, “CD56dim.natural.killer.cell”. In OSA, the results indicated higher infiltration of “Activated.CD8.T.cell”, “CD56bright.natural.killer.cell”, “Central.memory.CD8.T.cell”, but ower infiltration of “Eosinophil” and “Monocyte” (Figure 7A and B). Correlation analysis further showed a high consistency between the hub genes and some immune cells (Figure 7C and 7D).
 |
Figure 7 Immune cell infiltration analysis. ((A) OSA; (C) MAFLD) Composition of immune cells in OSA and MAFLD; ((B) OSA; (D) MAFLD) Analysis of the correlation between hub genes and various infiltrating immune cells, as well as among infiltrating immune cells. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. |
MR Analysis of the Effect Immune Cells on OSA and MAFLD
Given the close association of immune cells with both diseases, we further explored the causal relationships between 731 immune cell phenotypes and OSA as well as MAFLD using MR analysis. Detailed SNP information is provided in Supplementary Table S6. The results revealed that 103 immune phenotypes were related to OSA, and 27 immune phenotypes to MAFLD (Supplementary Table S7). After intersecting the immune cells, we obtained 4 immune phenotypes (CD4RA on TD CD4+, HLA DR on CD14+ CD16- monocyte, HLA DR on CD14+ monocyte and Myeloid DC %DC) that causally influenced both OSA and MAFLD. Among them,
Myeloid DC %DC was excluded due to its protective effect in MAFLD but risk factor in OSA. The remaining three phenotypes (CD4RA on TD CD4+, HLA DR on CD14+ CD16- monocyte and HLA DR on CD14+ monocyte) were associated with an increase in both OSA and MAFLD (all IVW OR >1.00, p<0.05) (Figure 8). Sensitivity analysis further confirmed the robustness of the results (Supplementary Table S2, Supplementary Figures S1 and S2).
 |
Figure 8 Results of Mendelian randomization of immune cell phenotypes and OSA and MAFLD. (A) Venn diagram of immunophenotypes that OSA and MAFLD; (B) Forest plots of immunophenotypes influencing both OSA and MAFLD. |
Discussion
There is a growing body of evidence linking obstructive sleep apnea (OSA) and metabolic-associated fatty liver disease (MAFLD), yet the exact causality and underlying molecular mechanisms remain unclear. To elucidate the complex associations between these two conditions, we conducted two-sample MR and network MR to explore the causal effect between OSA and MAFLD as well as the role of BMI. Our study found OSA were associated with the increased the risk of MAFLD and may serve as a mediator in the effect of BMI on MAFLD. Moreover, we further identified four hub genes (FOS, ATF3, JUN, EGR1) and 3 immune cell phenotypes (CD4RA on TD CD4+, HLA DR on CD14+ CD16- monocyte, HLA DR on CD14+ monocyte), which may play key roles in pathogenic mechanism linking OSA to MAFLD. These findings provide new insights into the physiological interactions between these two diseases.
The association between OSA and MAFLD has long been an ongoing debate topic. Observational studies suggest that OSA is associated with MAFLD, independently of obesity.10,12,35 Nevertheless, obesity is a well-known risk factor for OSA. Epidemiological studies showed up to 93.5% of patients with obesity have OSA, and approximately 70% of individuals with OSA were obese.36,37 So, we cannot ignore the role of BMI in the relationship between the two diseases. Zhang et al conducted univariable MR analyses and reported the causal link between OSA and MAFLD was observed,19 which was consistent with our study. Notably, while Zhang et al proposed that BMI might mediate the OSA-MAFLD link, our network MR analysis provides stronger evidence that OSA itself may mediate the effect of BMI on MAFLD. Thus, it would reasonably be speculated that both OSA and BMI may jointly promote the occurrence and development of MAFLD.
OSA is known to induce a range of physiological disturbances, such as oxidative stress, systemic inflammation, intermittent hypoxia, hypercapnia, and sympathetic hyperactivity.1 However, the exact mechanisms by which these factors contribute to MAFLD remain unclear. In this study, we identified four molecular signatures (FOS, ATF3, JUN, EGR1) that might participate toward the development of MAFLD in the context of OSA. FOS, an activator protein 1 (AP-1) transcription factor subunit plays an important role in regulating various physiological processes, including control of stress responses, organogenesis, and immune responses.38 JUN, an essential component of AP-1, binds to Fos and forms the AP-1 transcription factor complex, involving in inflammation regulation, immune regulatory function.39 ATF3 belongs to a member of the ATF/cAMP response element–binding family of transcription factors, and plays a crucial role in inflammation, apoptosis, oxidative stress, and endoplasmic reticulum stress, which has significant implications in the progression of MAFLD.40–42 EGR1 is a zinc-finger transcription factor involved in cell growth, metastasis, apoptosis, DNA repair, immune response, and fibrosis.43,44 Recently, studies have also shown EGR1 contributes to MAFLD pathogenesis.45 As mentioned above, the function of hub genes widely involved in immune response. Consistent with these results, we also found four hub genes was a close association with special immune cell infiltration. Thus, these findings contribute to our understanding of the complex interactions between four genes and immune cell populations in the pathogenesis of MAFLD.
OSA-related CIH activates pathways such as NF-κB and HIF-1α, leading to immune cell activation and infiltration, such as lymphocytes, macrophages and monocytes, which results in the development of OSA-related disease.1,46 In MAFLD, various pathogenic factors trigger inflammation, leading to lipid overload, lipotoxicity, oxidative stress and ER stress in hepatic, which directly or indirectly result in immune cell activation and infiltration, and damage hepatocytes.47 Thus, the immune cells play an integral role in pathogenesis and progression of MAFLD. The previous review described that OSA may mediate the occurrence and development of MAFLD through immune cells.48 In the study, our study found that 103 immune phenotypes were causally related to OSA, while 27 were linked to MAFLD. We obtained three common immune cell phenotypes (CD4RA on TD CD4+, HLA DR on CD14+ CD16- monocyte, HLA DR on CD14+ monocyte) between OSA and MAFLD. Three common immune cell phenotypes may play a crucial role in immune-mediated mechanisms in MAFLD caused by OSA, but further research is required to validate the findings. Notably, our findings also underscore the importance of clinical interventions targeting OSA itself to prevent or ameliorate MAFLD. For instance, orthopedic treatments such as rapid maxillary expansion and mandibular advancement have shown promise in pediatric OSA management by improving airway patency and reducing hypoxia severity, which may indirectly attenuate metabolic liver injury.49 Similarly, oral appliances designed to advance the mandible during sleep have demonstrated efficacy in select adult OSA populations, particularly for responders identified through tailored protocols.50 Future studies should evaluate whether these interventions—by alleviating OSA severity—reduce MAFLD risk or improve hepatic outcomes, particularly in high-risk cohorts.
Strengths and Limitations
Our study firstly employed comprehensive MR to explore causal impact of OSA on MAFLD and the role of BMI, combining bioinformatics methods to further investigate underlying mechanisms between them. Our findings provide new guidance for future etiology against these two diseases. However, our study had several limitations. Firstly, our MR analysis only used GWAS data from the European population, resulting in potential biases when generalizing to other ancestry. We could not determine the degree of sample overlap between exposure and outcome populations among GWAS datasets, which may lead to the bias of results. Secondly, the lack of individual information, we could not further evaluate the effect of OSA severity and BMI stratification on MAFLD. Thirdly, we recognize that the relatively small number of instruments, particularly for MAFLD (n=4), may still impose non-negligible restrictions on pleiotropy-robust methods, and this limitation should be considered when interpreting causal estimates. Fourthly, although we have employed various methods to align with the core hypothesis of MR as much as possible, we are still unable to eliminate potential horizontal pleiotropy or biological pathways, such as inflammatory pathways and insulin resistance. Finally, lack of external experimental validation in vivo and in vitro, our findings from bioinformatics should be cautiously interpreted. Given the limitations, further experimental studies are required to elucidate their molecular mechanisms in these diseases.
Conclusion
In this study, our MR analyses provide a causal relationship between OSA and MAFLD, and suggest that OSA may mediate the effect of BMI on MAFLD. Four common hub genes and three common immune cell phenotypes may play crucial roles in common pathogenesis for both diseases, shedding new light on the mechanistic interactions of OSA-MAFLD, thereby offering novel insights for future basic research and clinical therapeutic markers.
Data Sharing Statement
The datasets for MR analysis are publicly available GWAS summary data (https://gwas.mrcieu.ac.uk/). The datasets for bioinformatics analysis can be found in the GEO database: GSE135917 and GSE38792.
Ethics Approval
The study adheres to the Declaration of Helsinki. All genome-wide association studies and GEO database included in this study had been approved by a relevant review board, and participants had provided written informed consent. The study had also been approved by the ethics committee of the First Affiliated Hospital of Jinan University. Full ethical review was not required by the ethics committee of the First Affiliated Hospital of Jinan University in adherence to your institutional guidelines and regulations for secondary analysis of open-access data.
Acknowledgments
The authors are grateful to the authors of the original studies for sharing the genome-wide association studies (GWASs) summary statistics and GEO database.
Author Contributions
Tianyu Ma: Data curation, Investigation, Methodology, Project administration, Software, Visualization, Writing – original draft; Chunyan Liao: Conceptualization, Formal analysis, Funding acquisition, Writing – original draft; Wenhui Chen: Data curation, Methodology, Project administration, Writing – review and editing; Jia Feng: Conceptualization, Project administration, Writing – review and editing.
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation; all authors took part in drafting, revising or critically reviewing the article; all authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding was received.
Disclosure
The authors declared no conflict of interest in this work.
References
1. Lv R, Liu X, Zhang Y, et al. Pathophysiological mechanisms and therapeutic approaches in obstructive sleep apnea syndrome. Signal Transduct Target Ther. 2023;8(1):218. doi:10.1038/s41392-023-01496-3
2. Benjafield AV, Ayas NT, Eastwood PR, et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: a literature-based analysis. Lancet Respir Med. 2019;7(8):687–698. doi:10.1016/S2213-2600(19)30198-5
3. Lee JJ, Sundar KM. Evaluation and management of adults with obstructive sleep apnea syndrome. Lung. 2021;199(2):87–101. doi:10.1007/s00408-021-00426-w
4. Li M, Li X, Lu Y. Obstructive sleep apnea syndrome and metabolic diseases. Endocrinology. 2018;159(7):2670–2675. doi:10.1210/en.2018-00248
5. Mysliwiec V, Martin JL, Ulmer CS, et al. The management of chronic insomnia disorder and obstructive sleep apnea: synopsis of the 2019 U.S. department of veterans affairs and U.S. department of defense clinical practice guidelines. Ann Intern Med. 2020;172(5):325–336. doi:10.7326/M19-3575
6. Henry L, Paik J, Younossi ZM. Review article: the epidemiologic burden of non-alcoholic fatty liver disease across the world. Aliment Pharmacol Ther. 2022;56(6):942–956. doi:10.1111/apt.17158
7. European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO). EASL-EASD-EASO clinical practice guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J Hepatol. 2024;81(3):492–542. doi:10.1016/j.jhep.2024.04.031
8. Tacke F, Puengel T, Loomba R, Friedman SL. An integrated view of anti-inflammatory and antifibrotic targets for the treatment of NASH. J Hepatol. 2023;79(2):552–566. doi:10.1016/j.jhep.2023.03.038
9. Harrison SA, Allen AM, Dubourg J, Noureddin M, Alkhouri N. Challenges and opportunities in NASH drug development. Nat Med. 2023;29(3):562–573. doi:10.1038/s41591-023-02242-6
10. Zhang L, Zhang X, Meng H, et al. Obstructive sleep apnea and liver injury in severely obese patients with nonalcoholic fatty liver disease. Sleep Breath. 2020;24(4):1515–1521. doi:10.1007/s11325-020-02018-z
11. Hany M, Abouelnasr AA, Abdelkhalek MH, et al. Effects of obstructive sleep apnea on non-alcoholic fatty liver disease in patients with obesity: a systematic review. Int J Obes. 2023;47(12):1200–1213. doi:10.1038/s41366-023-01378-2
12. Fu Y, Zhang N, Tang W, et al. Chronic intermittent hypoxia contributes to non-alcoholic steatohepatitis progression in patients with obesity. Hepatol Int. 2022;16(4):824–834. doi:10.1007/s12072-022-10347-2
13. Jin S, Jiang S, Hu A. Association between obstructive sleep apnea and non-alcoholic fatty liver disease: a systematic review and meta-analysis. Sleep Breath. 2018;22(3):841–851. doi:10.1007/s11325-018-1625-7
14. Zhou J, Zhao Y, Guo YJ, et al. A rapid juvenile murine model of nonalcoholic steatohepatitis (NASH): chronic intermittent hypoxia exacerbates Western diet-induced NASH. Life Sci. 2021;276(119403):119403. doi:10.1016/j.lfs.2021.119403
15. Wang L, Liu H, Zhou L, et al. Association of obstructive sleep apnea with nonalcoholic fatty liver disease: evidence, mechanism, and treatment. Nat Sci Sleep. 2024;16:917–933. doi:10.2147/NSS.S468420
16. Chen LD, Chen Q, Lin XJ, et al. Effect of chronic intermittent hypoxia on gene expression profiles of rat liver: a better understanding of OSA-related liver disease. Sleep Breath. 2020;24(2):761–770. doi:10.1007/s11325-019-01987-0
17. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:1.
18. Ding X, Zhao L, Cui X, Qi L, Chen Y. Mendelian randomization reveals no associations of genetically-predicted obstructive sleep apnea with the risk of type 2 diabetes, nonalcoholic fatty liver disease, and coronary heart disease. Front Psychiatry. 2023;14(1068756). doi:10.3389/fpsyt.2023.1068756
19. Zhang Z, Li M, Ji G, Zhang L. Causal relationship between sleep apnea and non-alcoholic fatty liver disease: a Mendelian randomization study. Eur J Clin Invest. 2024;54(3):e14116. doi:10.1111/eci.14116
20. Ghodsian N, Abner E, Emdin CA, et al. Electronic health record-based genome-wide meta-analysis provides insights on the genetic architecture of non-alcoholic fatty liver disease. Cell Rep Med. 2021;2(11):100437. doi:10.1016/j.xcrm.2021.100437
21. Orrù V, Steri M, Sidore C, et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat Genet. 2020;52(10):1036–1045. doi:10.1038/s41588-020-0684-4
22. Bowden J, Del Greco MF, Minelli C, et al. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I 2 statistic. Int J Epidemiol. 2016;45(6):1961–1974. doi:10.1093/ije/dyw220
23. Skrivankova VW, Richmond RC, Woolf BAR, et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization: the STROBE-MR statement. JAMA. 2021;326(16):1614–1621. doi:10.1001/jama.2021.18236
24. Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology. 2017;28(1):30–42. doi:10.1097/EDE.0000000000000559
25. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through egger regression. Int J Epidemiol. 2015;44(2):512–525. doi:10.1093/ije/dyv080
26. Carter AR, Sanderson E, Hammerton G, et al. Mendelian randomisation for mediation analysis: current methods and challenges for implementation. Eur J Epidemiol. 2021;36(5):465–478. doi:10.1007/s10654-021-00757-1
27. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–698. doi:10.1038/s41588-018-0099-7
28. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–314. doi:10.1002/gepi.21965
29. Pei G, Chen L, Zhang W. WGCNA application to proteomic and metabolomic data analysis. Methods Enzymol. 2017;585:135–158.
30. Alexa A, Rahnenführer JJBI. Gene set enrichment analysis with topGO. Bioconductor Improv. 2009;27(1–26):776.
31. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–D61. doi:10.1093/nar/gkw1092
32. Szklarczyk D, Gable AL, Nastou KC, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–d12. doi:10.1093/nar/gkaa1074
33. Huby T, Gautier EL. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat Rev Immunol. 2022;22(7):429–443. doi:10.1038/s41577-021-00639-3
34. Jiang Y, Lin C, Xu M, et al. Differences and risk factors of peripheral blood immune cells in patients with obstructive sleep apnea. Nat Sci Sleep. 2024;16:737–749. doi:10.2147/NSS.S458098
35. Kim T, Choi H, Lee J, Kim J. Obstructive sleep apnea and nonalcoholic fatty liver disease in the general population: a cross-sectional study using nationally representative data. Int J Environ Res Public Health. 2022;19(14):8398.
36. Loo GH, Rajan R, Mohd Tamil A, Ritza Kosai N. Prevalence of obstructive sleep apnea in an Asian bariatric population: an underdiagnosed dilemma. Surg Obes Relat Dis. 2020;16(6):778–783. doi:10.1016/j.soard.2020.02.003
37. Chen W, Feng J, Wang Y, Wang C, Dong Z. Development and validation of a nomogram for predicting obstructive sleep apnea in bariatric surgery candidates. Nat Sci Sleep. 2021;13:1013–1023. doi:10.2147/NSS.S316674
38. Cordier F, Creytens D. New kids on the block: FOS and FOSB gene. J Clin Pathol. 2023;76(11):721–726. doi:10.1136/jcp-2023-208931
39. Ren FJ, Cai XY, Yao Y, Fang GY. JunB: a paradigm for Jun family in immune response and cancer. Front Cell Infect Microbiol. 2023;13(1222265). doi:10.3389/fcimb.2023.1222265
40. Willemsen N, Arigoni I, Studencka-Turski M, Krüger E, Bartelt A. Proteasome dysfunction disrupts adipogenesis and induces inflammation via ATF3. Mol Metab. 2022;62(101518):101518. doi:10.1016/j.molmet.2022.101518
41. Wen Y, Liu Y, Li Q, et al. Spatiotemporal ATF3 expression determines VSMC fate in abdominal aortic aneurysm. Circ Res. 2024;134(11):1495–1511. doi:10.1161/CIRCRESAHA.124.324323
42. Hu S, Li R, Gong D, et al. Atf3-mediated metabolic reprogramming in hepatic macrophage orchestrates metabolic dysfunction-associated steatohepatitis. Sci Adv. 2024;10(30):eado3141. doi:10.1126/sciadv.ado3141
43. Pan M, Luo M, Liu L, et al. EGR1 suppresses HCC growth and aerobic glycolysis by transcriptionally downregulating PFKL. J Exp Clin Cancer Res. 2024;43(1):35. doi:10.1186/s13046-024-02957-5
44. Ma ZG, Yuan YP, Fan D, et al. IRX2 regulates angiotensin II-induced cardiac fibrosis by transcriptionally activating EGR1 in male mice. Nat Commun. 2023;14(1):4967. doi:10.1038/s41467-023-40639-6
45. Guo Y, Miao X, Sun X, et al. Zinc finger transcription factor Egf1 promotes non-alcoholic fatty liver disease. JHEP Rep. 2023;5(6):100724. doi:10.1016/j.jhepr.2023.100724
46. Polasky C, Steffen A, Loyal K, et al. Redistribution of monocyte subsets in obstructive sleep apnea syndrome patients leads to an imbalanced PD-1/PD-L1 cross-talk with CD4/CD8 T cells. J Immunol. 2021;206(1):51–58. doi:10.4049/jimmunol.2001047
47. Peiseler M, Schwabe R, Hampe J, et al. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease - novel insights into cellular communication circuits. J Hepatol. 2022;77(4):1136–1160. doi:10.1016/j.jhep.2022.06.012
48. Tarantino G, Citro V, Finelli C. What non-alcoholic fatty liver disease has got to do with obstructive sleep apnoea syndrome and viceversa? J Gastrointestin Liver Dis. 2014;23(3):291–299. doi:10.15403/jgld.2014.1121.233.gvt
49. Lima Illescas MV, Aucapiña Aguilar DC, Vallejo Ledesma LP. A review on the influence of rapid maxillary expansion and mandibular advancement for treating obstructive sleep apnea in children. J Clin Pediatr Dent. 2023;47(1):9–16. doi:10.22514/jocpd.2022.035
50. Segù M, Cosi A, Santagostini A, Scribante A. Efficacy of a trial oral appliance in OSAS management: a new protocol to recognize responder/nonresponder patients. Int J Dent. 2021;2021(8811700):1–9. doi:10.1155/2021/8811700
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.