")
Back to Journals » Clinical Ophthalmology » Volume 18
Combination Low-Dose Pilocarpine/Diclofenac Sodium and Pilocarpine Alone for Presbyopia: Results of a Randomized Phase 2b Clinical Trial
Authors Farid M, Rowen SL, Moshirfar M, Cunningham D, Gaddie IB , Smits G, Ignacio T, Gupta PK
Received 3 May 2024
Accepted for publication 31 October 2024
Published 23 November 2024 Volume 2024:18 Pages 3425—3439
DOI https://doi.org/10.2147/OPTH.S476658
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Marjan Farid,1 Sheri L Rowen,2 Majid Moshirfar,3 Derek Cunningham,4 Ian B Gaddie,5 Gerard Smits,6 Teresa Ignacio,6 Preeya K Gupta7
1UCI Health, Irvine, CA, USA; 2NVISION Eye Centers, Newport Beach, CA, USA; 3Hoopes Vision Research Center, Hoopes Vision, Draper, UT, USA; 4Dell Laser Consultants, Austin, TX, USA; 5Gaddie Eye Centers, Louisville, KY, USA; 6Orasis Pharmaceuticals, Ponte Vedra, FL, USA; 7Triangle Eye Consultants, Raleigh, NC, USA
Correspondence: Marjan Farid, UCI Health, 850 Health Sciences Road, Irvine, CA, 92617, USA, Tel +1-949-824-2020, Fax +1-855-376-5057, Email [email protected]
Purpose: To evaluate the efficacy of 0.2% and 0.4% pilocarpine HCl (CSF-1) for the treatment of presbyopia and to determine the contributions of pilocarpine HCl and diclofenac sodium on the efficacy of fixed-dose combination (FDC) formulations.
Patients and Methods: This was a Phase 2b, multicenter, randomized, double-masked, parallel-group clinical trial. Adults (45– 64 years) with presbyopia were randomized 1:1:1 to 3 arms (Pilo arm: pilocarpine HCl; Pilo-Diclo FDC arm: pilocarpine HCl with 0.006% diclofenac sodium; Control arm: 0.006% diclofenac sodium). Participants in Pilo and Pilo-Diclo FDC arms received 0.2% pilocarpine HCl (0.2% Pilo or 0.2% Pilo FDC, respectively) from days 1– 8, and 0.4% pilocarpine HCl (CSF-1 or CSF-1-FDC, respectively) from days 8– 15. Primary efficacy endpoint was achievement of ≥ 3-line (15-letter) gain in mesopic, monocular distance-corrected near visual acuity (DCNVA) at 40 cm, 1 hour post-treatment of the study eye on days 8 and 15 in the per protocol (PP) population. Safety endpoints were assessed.
Results: One hundred and sixty-six participants were randomized (intent-to-treat, N = 166; PP, n = 160). There were no statistical differences between 0.2% Pilo or 0.2% Pilo FDC versus Control at 1 hour post-treatment on day 8. On day 15, 43.1% and 46.9% of participants receiving CSF-1-FDC (0.4% Pilo FDC) or CSF-1 (0.4% Pilo), respectively, achieved ≥ 3-line gain at 1 hour post-treatment in mesopic DCNVA compared with 16.1% of Control group in the PP population, meeting the primary endpoint (P = 0.0015 and P = 0.0002, respectively). All formulations were well tolerated.
Conclusion: CSF-1 demonstrated significant improvements in mesopic DCNVA and favorable safety. Pilocarpine HCl as a single active ingredient, at the concentration of 0.4% (CSF-1), provided a transient, therapeutic effect for presbyopia.
Keywords: pilocarpine hydrochloride, presbyopia, minimum effective concentration
Introduction
Presbyopia is an age-related vision disorder characterized by a progressive inability to focus on near objects due to the loss of lens elasticity, weakening of ciliary muscles, and an increase in lens rigidity.1 The prevalence and severity of presbyopia increases with age, so that nearly all people aged 40 years or older develop this condition.1 Whether presbyopia disrupts activities of leisure or professional life, patients consistently report that presbyopia has a considerable impact on quality of life and work productivity.2,3
There are multiple treatment approaches for presbyopia, and the options may vary depending on the progression of the condition, cost, and visual needs.1,3 The most common nonsurgical options for presbyopia correction are spectacles (reading, bifocal, progressive) and monovision or multifocal contact lenses, which can differ significantly in cost and technical complexities.3 Refractive surgeries (eg, laser-assisted in-situ keratomileusis [LASIK], photorefractive keratectomy [PRK], corneal inlays, and accommodative or multifocal intraocular lenses) have gained popularity but procedures can be cost prohibitive and post-surgical results can be inconsistent, and diminish over time.1,4 The limitations associated with the current treatments for presbyopia highlight a need for alternative strategies, such as pharmacologic treatments, which are effective in improving near visual acuity (VA) without compromising distance vision.5
Pilocarpine, a muscarinic agonist that primarily acts through cholinergic muscarinic M3 receptors on the iris sphincter, has been shown to constrict the pupil (miosis), thereby improving depth of focus.6,7 Pilocarpine has been investigated at different concentrations and in combination with other drugs for the treatment of presbyopia.6–8 The US Food and Drug Administration (FDA) approved the use of 1.25% pilocarpine hydrochloride (HCl) ophthalmic solution (VuityTM; Allergan) for once daily use in 2021 and twice daily use in 2023 as monotherapy for this condition.9,10 Muscarinic receptor agonists are known to induce some inflammation consistent with study results on the use of 1.25% pilocarpine HCl for presbyopia.6,11 The addition of a non-steroidal anti-inflammatory drug (NSAID) to pilocarpine could hypothetically reduce the inflammatory response potentially induced by pilocarpine and improve tolerability.7 Diclofenac sodium is a safe and effective NSAID prescribed for allergic conjunctivitis and the management of pain and inflammation in the eye following refractive and cataract surgeries.12–14 The Benozzi method of combination treatment with pilocarpine HCl (0.5% to 4%) and diclofenac sodium (0.1% to 0.5%) eyedrops for use in presbyopia has demonstrated up to 75% improvement from baseline in uncorrected near visual acuity (UNVA) with no changes in uncorrected distance visual acuity (UDVA) after eight years of use.15,16 A fixed-dose combination (FDC) treatment of pilocarpine nitrate 0.2% and diclofenac sodium 0.006% for the treatment of presbyopia was initially evaluated in two Phase 2a, randomized, placebo-controlled studies, which provided favorable safety and preliminary efficacy data.17,18 In this Phase 2b study, the salt of the active ingredient pilocarpine nitrate was changed to pilocarpine HCl because it is more readily available and consistent with the FDA-approved pilocarpine moieties. The purpose of this Phase 2b study was to determine the contribution of each active ingredient, pilocarpine HCl and diclofenac sodium, and the impact of different pilocarpine HCl concentrations (0.2% and 0.4%) on the efficacy and safety of the study drug for the temporary treatment of presbyopia.
Materials and Methods
This was a randomized, double-masked, multicenter, parallel-group study conducted at 7 sites in the United States (ClinicalTrials.gov ID: NCT03885011). The study was performed in accordance with the ethical principles stated in the Declaration of Helsinki, the International Council for Harmonization for Good Clinical Practice, and all local applicable laws and regulations. The study protocol and data collection were carried out with approval from a central Institutional Review Board (IRB), Alpha IRB, and, prior to participation, all individuals signed a written informed consent form with details of the trial treatment, procedures, and potential risks. This study report follows Consolidated Standards of Reporting Trials (CONSORT) reporting guidelines.
Study Population
The inclusion criteria for this trial were individuals with presbyopia between 45 and 64 years of age, in general good health. Key inclusion criteria included distance-corrected near visual acuity (DCNVA) at 40 centimeters (cm) ≥0.40 and ≤0.90 logarithm of the minimum angle of resolution (logMAR, ~20/50-20/160 Snellen) in at least 1 eye at screening (visit 1) prior to placebo dosing and in the same eye on day 1 (visit 2 pre-treatment). At screening, individuals had between −4.50 and +2.00 diopter (D) of sphere in each eye determined by manifest refraction (MR) and ≤2.00 D difference between eyes, <2.00 D of cylinder in each eye determined by MR, and ≤0.04 logMAR (~20/20-2 Snellen or better) corrected distance visual acuity (CDVA) in each eye prior to placebo dosing. A complete list of the eligibility criteria is listed in the Supplementary Table.
Study Design
The study comprised of four office visits over approximately 2–4 weeks. Participants were screened during visit 1 (occurring on days −1 to −14), and potential participants received 1 drop of placebo (Refresh Classic®; Allergan, Irvine, CA). Eligible participants were randomized 1:1:1 via an interactive response technology system to 1 of the 3 treatment arms: Pilo, Pilo-Diclo-FDC, and Control (Figure 1). The Pilocarpine HCl alone arm (Pilo arm) received 0.2% pilocarpine HCl (Pilo 0.2%) on days 1–8, followed by 0.4% pilocarpine HCl (CSF-1; Orasis Pharmaceuticals, Ponte Vedra, FL) on days 8–15; the Pilo-Diclo-FDC arm received 0.2% pilocarpine HCl/0.006% diclofenac sodium (Pilo 0.2%-FDC) on days 1–8, followed by 0.4% pilocarpine HCl/0.006% diclofenac sodium (CSF-1-FDC) on days 8–15; and the Control arm received 0.006% diclofenac sodium alone on days 1–15. An independent, unmasked biostatistician who was not involved in the trial generated the complete randomized study drug kit list. The participant, study sponsor, investigators, and study staff were masked during the entire randomization process and throughout the study. Randomization was stratified by iris color (brown versus light [ie, blue, green, gray, and hazel]) and by baseline manifest refraction spherical equivalent (MRSE [−4.5 D to <-0.5 D; −0.5 D to +0.75 D; and >+0.75 D to +2.0 D]). Participants were instructed to dose twice daily (one drop in the morning and in the afternoon with at least 6 hours between doses) in both eyes until the afternoon before day 8 and record their doses in their paper dosing diary.
 |
Figure 1 Study design. |
On the morning of day 8, the study drug was administered once by study site personnel followed by efficacy and safety assessments for 8 hours. After day 8 assessments were completed, participants received a new study drug per their assigned treatment arm and a new dosing diary. The new study drug was instilled in-office by the study site personnel on day 8, after which the participants were instructed to continue the same dosing regimen (bilaterally twice-daily in the morning and afternoon) and record doses in their diary up to day 15 (visit 4).
After day 8, participants initially randomized to the Pilo-Diclo- FDC arm then received CSF-1-FDC; participants randomized to the Pilo arm then received CSF-1; and participants randomized to the Control arm continued dosing with a new kit of diclofenac sodium alone. On day 15, the morning dose of the study drug was administered by the study site personnel followed by efficacy and safety assessments for 8 hours. The participants exited the study after day 15 assessments were completed.
Study eye was defined as the eye that met all enrollment criteria. If both eyes met all enrollment criteria, the eye with the worse Day 1 (Visit 2) DCNVA was selected. If both eyes had the same DCNVA, the right eye was assigned as the study eye.
Study Endpoints, Safety, and Tolerability Assessments
The primary efficacy endpoints were the achievement of a ≥3-line (15-letter) gain from baseline in DCNVA at 40 cm, 1-hour post-treatment on day 8 (visit 3) and day 15 (visit 4) in the study eye using the Per Protocol (PP) population. The key secondary endpoints were the same as the primary efficacy endpoints using the Intent to Treat (ITT) population.
Secondary efficacy endpoints used the PP population and included: (1) ≥3-line (15-letter) gain from baseline in DCNVA at 40 cm at 20 minutes and 2, 4, 6, and 8 hours after treatment on day 8 (visit 3) and day 15 (visit 4), (2) ≥2-line (10-letter) gain from baseline in DCNVA at 20 minutes and 1, 2, 4, 6, and 8 hours post-treatment on day 8 (visit 3) and day 15 (visit 4), and (3) Mean change from baseline in pupil diameter under DCNVA testing conditions at 20 minutes and 1, 2, 4, 6, and 8 hours post-treatment on days 8 and 15.
DCNVA was measured at 40cm using customized scrambled Early Treatment Diabetic Retinopathy Study (ETDRS) charts (Precision Vision, Woodstock, Illinois). Multiple unique charts were used in a specified order to prevent memorization of letters. The near charts were calibrated for a testing distance of 40cm and used with retro-illuminated cabinets as specified by the manufacturer. Room lighting condition was measured using a light meter (Sper Scientific Light Meter; Sper Scientific Direct, Scottsdale, AZ) and recorded at a mesopic range of 0–135 lux. Each exam lane was certified before initiation of the study and confirmed to maintain the same distance and lighting conditions at every monitoring visit.
Pupil diameter was measured with a pupillometer (VIP®-300 Pupillometer; NeurOptics, Irvine, CA). Dark adapted pupillometry was taken at visit 1. Pre-treatment pupillometry measurements were done under DCNVA testing conditions prior to any near vision testing at visits 1, 2, 3, and 4 while post-treatment measurements under DCNVA testing conditions were taken at visits 3 and 4.
Safety and tolerability assessments included adverse events (AEs) reported, conjunctival redness grading, drop comfort assessment, slit lamp biomicroscopy, intraocular pressure (IOP), dilated indirect funduscopy, and CDVA at 4 meters (ETDRS chart; Precision Vision, Woodstock, IL). Conjunctival redness assessment was conducted at pre-treatment for all visits and at 20 minutes, 1, 2, 4, and 8 hours post-treatment on days 8 and 15 using an Ora Calibra 0–4 picture grading scale Ora Calibra® Redness Scale (#6.0). Ocular comfort was assessed using a 0–10 Ora Calibra® Drop Comfort Scale where 0 was very comfortable and 10 was very uncomfortable. Drop comfort assessments for each eye were conducted immediately upon drop instillation, at 30 seconds, and 1, 5, 10, 15, 30, and 60 minutes post-treatment on days 8 and 15. Slit lamp biomicroscopy and IOP measurement using Goldmann applanation tonometer were assessed at screening, day 8, and day 15. Dilated indirect funduscopy was performed on screening visit and on day 15.
Monocular CDVA was measured under mesopic room light and low luminance conditions at pre-treatment for each visit, and at 20 minutes, 1, 2, 4, 6, and 8 hours post-treatment on days 8 and 15. Low luminance CDVA was assessed using a 2.0 neutral density filter as specified and standardized by the manufacturer (Large mesopic filter for 2425 ETDRS Illuminator Cabinet; Precision Vision, Woodstock, IL) over the ETDRS chart.
Statistical Analysis
Fifty participants in each of the treatment arms (N = 150) yielded 85% power to establish the superiority of CSF-1 and CSF-1-FDC versus control in the two primary endpoints (≥3-line gain from baseline in DCNVA at 40 cm at 1-hour post-treatment on day 8 and day 15 in the study eye). The PP population was used for the analysis of the primary and secondary efficacy endpoints. The primary endpoints were analyzed using the ITT population as key supportive secondary analyses. The Safety population included all participants who received at least one dose of the study drug.
Statistical analyses were performed using SAS® version 9.4 (SAS Institute, Cary, NC). The study was considered a success if either of the 2 primary endpoint null hypotheses was rejected in favor of their corresponding alternative hypothesis on either day 8 or day 15. All 3 secondary endpoints and safety endpoints were tested without adjustment for multiplicity.
Testing of the primary efficacy variables was completed using a logistic (binomial distribution and logit link) model estimated by Generalized Estimating Equation (GEE) methods. The analysis of the first two secondary endpoints [percentage of study eyes of participants with a ≥3-line (15-letter) gain and a ≥2-line (10-letter) gain from baseline for each timepoint (20 minutes, 2, 4, 6, and 8 hours post-treatment) was performed using Pearson’s chi-squared test. McNemar’s test was conducted to investigate differences in effects of the 0.4% and 0.2% dose levels within a treatment arm. These analyses were performed using the PP population. For the third secondary endpoint, the mean change from baseline in pupil diameter under DCNVA at 40 cm testing conditions was analyzed in the PP population using an analysis of co-variance model. Paired Student’s t-tests were computed at each timepoint to investigate differences in effect between the 0.4% and 0.2% dose levels within a treatment arm.
All safety analyses were conducted using the safety population. All AEs were coded using Medical Dictionary for Regulatory Activities (MedDRA) version 21.1 and summarized. Safety assessments were summarized descriptively.
Post-Hoc Analyses
Since pilocarpine in higher concentrations can potentially affect distance VA, a post hoc analysis was conducted on the primary endpoint with the additional criterion of no loss of ≥1 line (5 letters) in CDVA at 4 m (ETDRS chart) from baseline. Pearson chi-square tests were used for analysis within each treatment comparison and timepoint.
A second post hoc analysis, whereby sustained duration of improvement of ≥3-lines from baseline across consecutive timepoints, starting at the 1-hour timepoint, was conducted in the ITT population. This differs from the standard analysis, which only requires ≥3-line gain at a specific timepoint.
Differences in treatment effect, measured by change in DCNVA letters read at 40 cm, was assessed for age, sex, iris color, and baseline DCNVA at 1-hour post-treatment on Day 8. Results were analyzed using analysis of variance, which included a treatment by covariate interaction term.
Results
Demographic and Baseline Characteristics
A total of 166 participants were randomized and included in the ITT and safety populations (Supplementary Figure); the PP population comprised of 160 participants. Six participants were discontinued from the study. Two participants (Pilo arm, n = 2) discontinued after an AE of coronary artery disease (n = 1) that led to death and was deemed not related to the study drug, and migraine (n = 1) that was mild and resolved on the same day. Three participants (n = 1, each arm) discontinued for a protocol violation, and 1 participant (Pilo arm) withdrew. This study was conducted between February 14, 2019, and July 26, 2019. Demographics and baseline characteristics were overall similar across treatment arms (Table 1). The median age of participants was 54.5 years (range 45–64 years). Majority of participants were female (61.4%), White (80.7%), and did not identify as Hispanic or Latino (97.6%). The most common eye color was brown (42%).
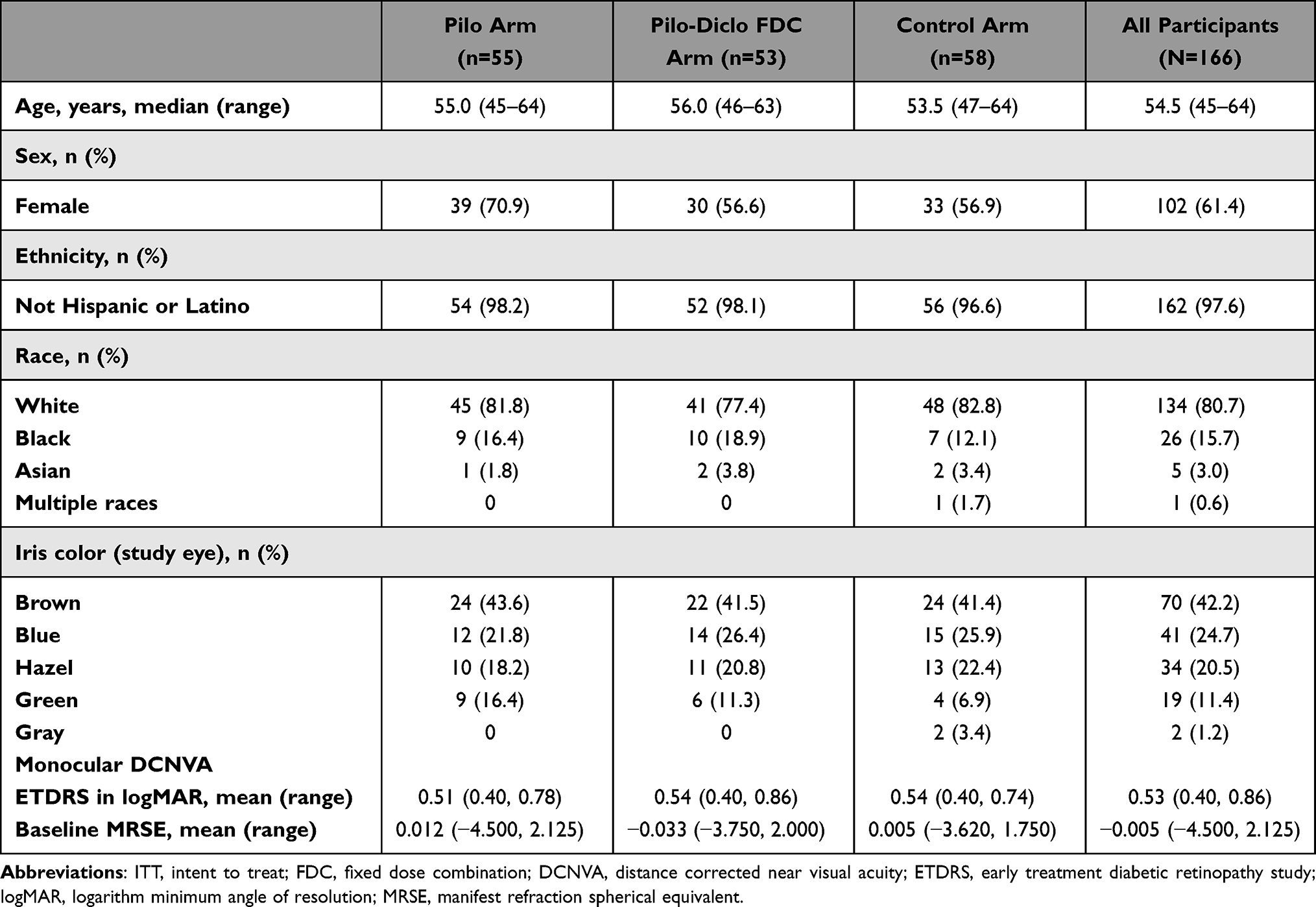 |
Table 1 Demographics and Baseline Characteristics of the Study Participants (ITT Population) |
Efficacy
Treatment comparisons for the primary and secondary endpoints used logistic models performed separately by day. Data are presented as a percentage of responders in the study eye. In the PP population, the primary efficacy endpoint was met on day 15 (visit 4), when a significantly higher percentage of participants receiving CSF-1 (P < 0.01; n = 52) and CSF-1-FDC (P < 0.01; n = 52) achieved a ≥3-line gain in DCNVA from baseline 1-hour post-treatment compared with the Control group (n = 56) (Figure 2). The Pilo 0.2% (n = 52) and Pilo 0.2%-FDC (n = 52) groups failed to demonstrate statistical significance (P = 0.27 and P = 0.84, respectively) compared with Control (n = 56) on day 8.
 |
Figure 2 Percentage of participants with a gain of ≥3 lines (15 letters) in mesopic DCNVA at 1 hour post-treatment on day 8 and day 15 in per protocol population (primary endpoint). |
This analysis was performed using the ITT population as a key secondary analysis. The results in the ITT population were similar to those of the PP population, where a significantly larger proportion of participants receiving CSF-1 (n = 55) or CSF-1-FDC (n = 53) achieved a ≥3-line gain in DCNVA from baseline on day 15 compared with the control group (n = 58) (P < 0.01 on all); neither the Pilo 0.2% (n = 55) nor Pilo 0.2%-FDC (n = 53) groups differed significantly from the Control group (n = 58) (P = 0.26 and P = 0.93, respectively) on day 8.
The percentage of participants in the PP population who achieved a ≥3-line improvement in DCNVA at prespecified timepoints between 20 minutes and 8 hours on day 8 and day 15 are shown in Table 2. There was no significant difference in the proportion of participants with a ≥3-line gain in DCNVA compared with those receiving Pilo 0.2% or Pilo 0.2%-FDC with the Control group in any of the tested timepoints on day 8. On day 15, CSF-1 treatment had a significantly higher percentage of participants with a ≥3-line improvement in DCNVA compared with the Control group at 20 minutes (P < 0.01), 1 hour (P < 0.01), and 2 hours (P < 0.01) post-dose, but the differences were not significant at later timepoints. Similar results were seen among participants in the CSF-1-FDC group on day 15 (P < 0.01).
 |
Table 2 Number and Percentage of Participants Who Achieved a ≥3-Line or ≥2-Line Improvement in Mesopic DCNVA by Timepoint on Day 8 (Visit 3) or Day 15 (Visit 4) in PP Population |
With in-arm analysis, a dose-level comparison was performed at each timepoint with a McNemar’s test to determine if there was a difference between the percentage of study eyes with a ≥3-line gain in DCNVA on day 8 (after Pilo 0.2% or Pilo 0.2%- FDC was administered) and day 15 (after CSF-1 or CSF-1-FDC was administered). The comparison demonstrated a significant improvement of ≥3-lines in DCNVA associated with CSF-1 compared with Pilo 0.2% at 20 min (P < 0.05), 1 hour (P < 0.01), 2 hours (P < 0.01), and at 8 hours (P < 0.05) but not at 4 and 6 hours (P > 0.05 at both timepoints). Similar results were observed with CSF-1-FDC compared with Pilo 0.2%-FDC with significant results only from 20 minutes to 2 hours (P < 0.01 on both timepoints), and at 8 hours post-treatment (P < 0.05).
In the PP population, the proportion of participants achieving a ≥2-line improvement in DCNVA from baseline on day 8 approached significance only at 1-hour post-treatment with Pilo 0.2% (P < 0.10) and at 20 minutes (P < 0.10) post-treatment with Pilo 0.2%-FDC. There was no significant difference between Pilo 0.2% and Pilo 0.2%-FDC versus the Control group at all the other timepoints. On day 15, there was a significantly higher proportion of participants receiving CSF-1 with a ≥2-line improvement in DCNVA compared with the Control arm at 20 minutes (P < 0.01), 1 hour (P < 0.01), and 2 hours (P < 0.01) post-treatment, but this difference was not detected at later timepoints. CSF-1-FDC was associated with a greater proportion of participants with a ≥2-line gain in DCNVA from baseline compared with the Control arm at 20 minutes (P < 0.01) and 1 hour (P < 0.01) post-treatment. There was no statistical difference from the Control group at later timepoints.
Dose-level comparisons within the Pilo arm demonstrated superiority of the CSF-1 formulation compared with Pilo 0.2% up to 8 hours, except at 20 minutes (P = 0.10) and 4 hours post-treatment (P = 0.39). Among participants in the Pilo-Diclo-FDC arm, there was a significantly higher proportion of participants achieving a ≥2-line improvement in DCNVA with CSF-1-FDC compared with Pilo 0.2%-FDC at 1 hour, 4 hours, and 6 hours (all P < 0.01), but not at other timepoints after study drug administration.
Pupil Diameter
A significantly greater decrease from baseline in mean pupil diameter was noted in the Pilo 0.2% and Pilo 0.2%-FDC groups at 20 minutes post-treatment (P < 0.01 for both groups), 1, 2, 4, 6, and 8 hours post-treatment compared with the Control group (P < 0.05 at all timepoints on both groups) on day 8 (Figure 3). Similar results were demonstrated on day 15 in the CSF-1 and CSF-1-FDC groups. This result was indicative of the efficacy of both treatment arms toward producing an immediate and sustained reduction in pupil size.
 |
Figure 3 Mean pupil size (diameter) under mesopic DCNVA testing conditions at different timepoints for (A) Pilo 0.2% and Pilo 0.2% FDC on day 8 and (B) CSF-1 and CSF-1-FDC on day 15 and associated P values (PP population). |
When comparing Pilo 0.2% and CSF-1, the paired Student’s t-test yielded significant results in favor of CSF-1 from 20 minutes to 6 hours post-treatment (all P < 0.01). When comparing the doses in the Pilo-Diclo-FDC arm, the paired Student’s t-test yielded results in favor of CSF-1-FDC compared with Pilo 0.2%-FDC at 20 minutes (P < 0.01), 1 hour (P < 0.01), 2 hours (P < 0.01), 4 hours (P < 0.05), 6 hours (P < 0.01), and 8 hours post-treatment (P < 0.05).
Post Hoc Analyses
≥3-Line (15-Letter) Gain in Mesopic DCNVA at 40cm, 1-Hour Post-Treatment, with an Additional Criterion of No Loss of ≥5 Letters in CDVA at 4m from Baseline
A post hoc analysis with the additional criterion of no loss of ≥1 line (5 letters) in CDVA to the primary efficacy endpoint showed a significantly higher percentage of participants in the CSF-1 (42.9%, P < 0.01) and CSF-1-FDC groups (43.1%, P < 0.01) achieved a ≥3-line gain in DCNVA without loss of ≥1 line (5-letter) in CDVA 1-hour post-treatment versus the Control group (Table 3). On day 8, only 24.0% in the Pilo 0.2% and 19.6% of participants in the Pilo 0.2%-FDC treatment groups compared with 14.3% in the Control group (P = 0.20 and P = 0.46, respectively) achieved a ≥3-line gain in DCNVA with no loss of ≥1 line (5 letters) in CDVA, which was not statistically significant.
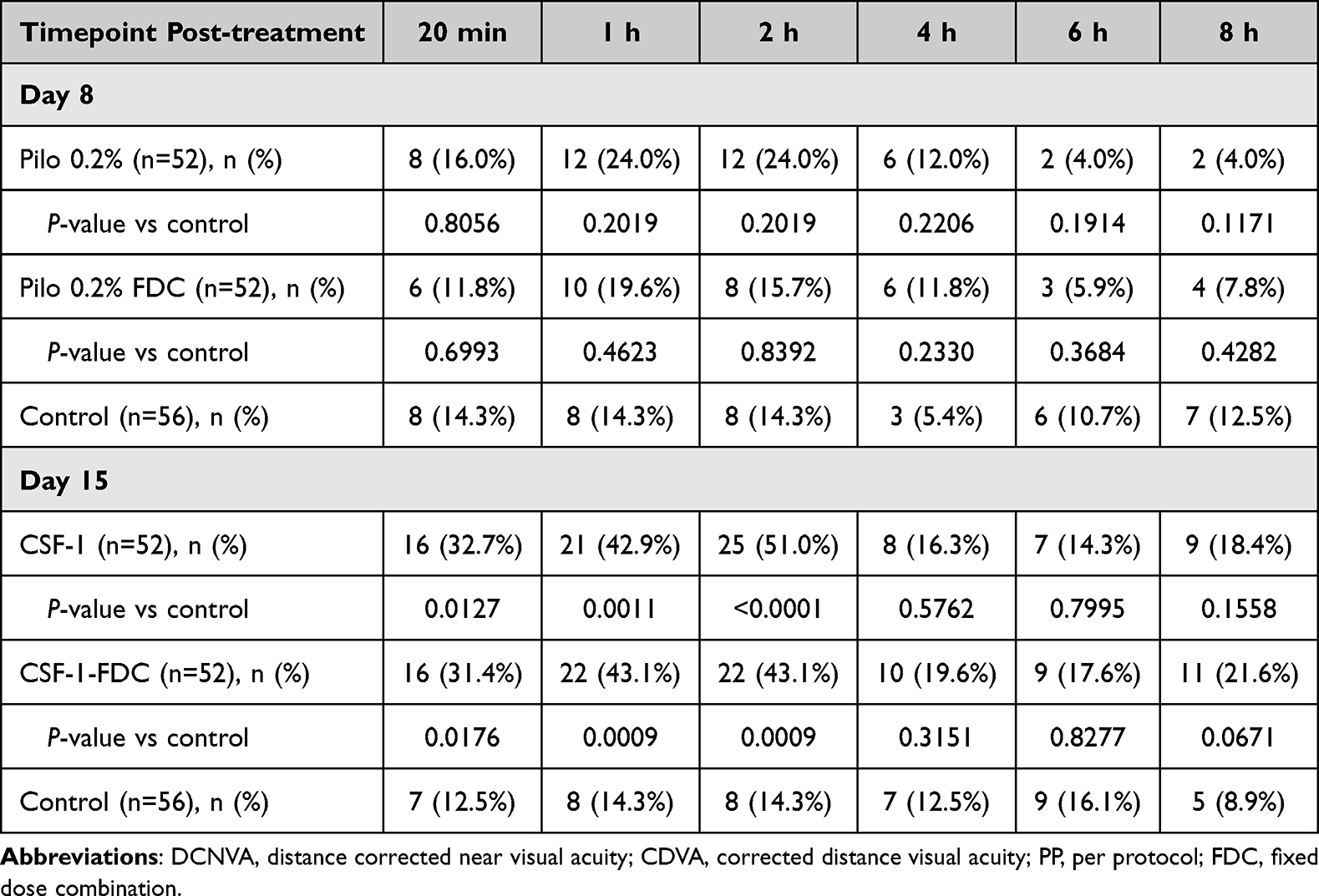 |
Table 3 Number and Percentage of Participants Who Achieved a ≥3-Line Improvement in Mesopic DCNVA Without Loss of ≥1-Line in CDVA by Timepoint on Day 8 and Day 15 in the PP Population |
Sustained Gain of ≥3-Line (15-Letter) Post-Treatment Across Consecutive Timepoints
On day 15, the Pearson’s chi-squared test yielded statistically significant improvement of ≥3-lines in monocular DCNVA at 40 cm at 1-hour post-treatment in the CSF-1 group (P < 0.01) and CSF-1-FDC group (P < 0.01) compared with the Control group in the ITT population (Figure 4). This statistically significant improvement was maintained at each tested timepoint up to 4 hours post-treatment in the CSF-1 group (P < 0.05) and CSF-1-FDC group (P < 0.01). Pilo 0.2% and Pilo 0.2%-FDC did not show a statistically significant ≥3-line improvement from baseline compared with the Control group at 1-hour post-treatment and had no sustained gains at later timepoints. The sustained post-hoc analysis (Figure 4) differs from standard analysis at specific timepoints with the difference in the requirement for achieving the endpoint and the number of participants analyzed (ie, sustained analysis N = 113; standard analysis N = 160).
 |
Figure 4 Percentage of participants with a sustained gain of ≥3-lines (15 letters) in mesopic DCNVA post-treatment by timepoint for (A) Pilo 0.2% and Pilo 0.2% FDC on day 8 and (B) CSF-1 and CSF-1-FDC on day 15 and associated P values (ITT population). |
Modification of Treatment Effect by Age, Sex, Iris Color, and Baseline VA
There was no statistically significant difference in treatment effect based on age, sex, iris color or baseline DCNVA at 1-hour post-treatment on day 8.
Safety and Tolerability
Treatment-Emergent Adverse Events (TEAEs)
All TEAEs in any group are summarized separately for ocular and non-ocular events in the safety population (n = 166) and shown in Table 4. There were 38 participants (22.9%) who had at least 1 TEAE. Of the total safety population, most TEAEs reported in the study were of mild intensity (ocular: 15.7%; non-ocular: 6.6%) with few participants (ocular: 0.6%; non-ocular: 1.2%) reporting a TEAE of moderate severity. There were no severe ocular TEAEs during the study. There was 1 non-ocular TEAE rated severe that was not related to the study drug.
 |
Table 4 Ocular and Non-Ocular Adverse Events Occurring in ≥1 Participant in the Safety Population |
Ocular TEAE
The most frequently reported ocular TEAE was instillation site pain, reported in 9.4% of participants in the Pilo 0.2%-FDC group. Most of the instillation site pain, described as burning or stinging, was reported immediately after instillation, classified as mild, and resolved within 30 seconds. Other ocular TEAEs ≥5% are conjunctival hyperemia (5.7%) in the Pilo 0.2%-FDC group and vision blurred (5.5%) in the Pilo 0.2% group described as transient and mild and reported upon instillation of the drug. No ocular TEAE resulted in study discontinuation.
Non-Ocular TEAE
The most frequently reported non-ocular TEAE was headache in all treatment arms (Pilo 0.2%, n = 4 [7.3%] and CSF-1, n = 1 [1.9%]; Pilo 0.2%-FDC, n = 1 [1.9%] and CSF-1-FDC, n = 0; Control, n = 2 [3.5%] by day 8, and n = 0 by day 15). All reported headaches were classified as mild in severity. Two non-ocular TEAEs, coronary artery disease and migraine, resulted in study discontinuation. The coronary artery disease event was graded as severe and resulted in death. It was considered not related to the study drug. The TEAE of migraine, which occurred in a participant with a medical history of migraine, was reported as mild and resolved on the same day.
Conjunctival Redness
The Ora Redness Scale #6.0 has a grading from 0 to 4, with zero indicating no redness and 4 extremely severe redness. The mean conjunctival redness scores were similar at baseline ranging from 0.5 to 0.7 for all treatment groups (Figure 5). The mean change from baseline in the conjunctival redness score was similar in all treatment groups by 1-hour post-instillation and at the end of each visit (8 hours post-instillation), with values ranging from −0.2 to 0.2.
 |
Figure 5 Mean conjunctival redness in the study eye (Safety population) on day 8 (A) and day 15 (B). |
Drop Comfort
Drop comfort scores were low overall, indicating that the study drugs were well tolerated. On day 8, comfort scores of 2 or lower were reported in >70% of participants immediately after instillation and in more than 80% of participants at 1-minute post-treatment in all treatment groups. More than 85% of participants reported scores of 1 or lower at all timepoints thereafter in all treatment groups. On day 15, comfort scores of 2 or lower were reported immediately after instillation by 70.6% of participants receiving CSF-1, 69.2% of participants receiving CSF-1-FDC, and 87.7% of participants receiving the control. A higher percentage of participants reported comfort score ≤2 by 1-minute post-treatment (CSF-1, 90.2%; CSF-1-FDC, 86.5%; Control, 87.7%).
Slit Lamp Biomicroscopy
At baseline, slit lamp biomicroscopy assessments for anterior chamber, cornea, conjunctiva, iris, eyelids, and lens were reported as normal for the majority of eyes (>75%). Lens findings at baseline were considered abnormal but not clinically significant (NCS) for 21.8% of the Pilo-only groups (Pilo 0.2% and CSF-1), 20.8% of the Pilo-Diclo-FDC groups (Pilo 0.2%-FDC and CSF-1-FDC) and 32.8% of the Control group. The majority of the abnormal NCS lens found at baseline was cataract. No clinically significant (CS) changes were noted in the study. A participant experienced a change from normal corneal findings to abnormal trace superficial punctate keratitis in the CSF-1 group throughout all visits in the study. No other changes were noted.
Intraocular Pressure
IOP levels were comparable in all groups at baseline and at study exit. Thus, changes from baseline results were also similar between the groups at the study end.
Dilated Indirect Funduscopy
At baseline, dilated indirect funduscopy assessments for the posterior pole, which includes the vitreous, retina, macula, choroid, and optic nerve, were reported as normal for the majority of eyes. One participant in the CSF-1 treatment group (a 64-year-old female) developed a CS finding of posterior vitreous detachment in the fellow eye on day 15. No other participants had a change from normal to abnormal findings reported. Funduscopic findings were generally comparable in all treatment groups throughout the study.
Corrected Distance Visual Acuity at 4m
The mean change from baseline results were similar across all treatment groups under both lighting conditions (Figure 6). There was no clinically meaningful change in monocular CDVA noted from baseline to exit in all treatment groups under both lighting conditions.
 |
Figure 6 Assessment of monocular CDVA under mesopic and low luminance lighting conditions on day 8 (A and C) and day 15 (B and D): Mean change in letters in study eye (Safety population). |
Discussion
The initial choice to use a 0.2% concentration in phase 2a studies was based on a gradual increase from the lower pilocarpine concentration (0.1%) that is known to induce miosis. Other concentrations of pilocarpine have been effective in treating dilated pupils after cataract surgery and Adie’s pupil, and other systemic conditions.19–23 This stepwise approach determined the selection to use 0.2% and 0.4% concentrations in this dose range finding Phase 2b study.
The results from this phase 2b study demonstrated that 0.4% pilocarpine HCl groups (CSF-1 and CSF-1-FDC) met the primary efficacy endpoint of ≥3-line gain in DCNVA 1 hour post-treatment on day 15, while the 0.2% pilocarpine HCl groups (Pilo 0.2% and Pilo 0.2%-FDC) on day 8 did not. The results also showed a higher percentage of participants achieving ≥3-line improvement from baseline in DCNVA in the CSF-1 and CSF-1-FDC groups compared to the Control group up to 2-hours post-treatment and at 8 hours post-treatment on day 15. Results of the secondary endpoint of ≥2-line gain from baseline showed CSF-1 and CSF-1-FDC demonstrated superiority over Pilo 0.2% and Pilo 0.2%-FDC up to 8 hours and 6 hours, respectively. The results of the primary efficacy endpoint and the secondary efficacy endpoints are an indication that 0.4% pilocarpine HCl is the minimum concentration needed to achieve ≥3-line and ≥2-line gain from baseline, which are improvements that are considered clinically significant and clinically meaningful for presbyopes.6 These results also informed the study design of the two Phase 3 studies evaluating the efficacy and safety of CSF-1.
All pilocarpine HCl−containing study drugs were associated with significantly smaller pupil diameter starting at 20 minutes and up to 8 hours compared with diclofenac alone. Although the Pilo 0.2% and Pilo 0.2%-FDC achieved this secondary endpoint, in-arm analyses yielded results in favor of CSF-1 and CSF-1-FDC. The miotic effect observed with pilocarpine HCl confirms that pupillary constriction enhances depth of focus, thus improving near vision. Although higher concentrations of pilocarpine can cause ciliary muscle contraction that increases accommodation and impacts distance vision,24,25 the assessment of the primary efficacy variable showed that CSF-1 and CSF-1-FDC maintained their benefits without any loss of ≥1-line in mean CDVA. This suggests that 0.4% pilocarpine HCl is the minimum concentration needed to achieve clinically significant near vision improvement without compromising distance VA.
There was no serious ocular AE reported in this study. When comparing 0.2% and 0.4% treatment concentrations, there were more TEAEs reported during treatment with Pilo 0.2% and in Pilo 0.2%-FDC than with CSF-1 and CSF-1-FDC treatment. Pilo 0.2%-FDC was associated with the two most frequently reported ocular TEAEs: instillation site pain and conjunctival hyperemia (9.4% and 5.7%, respectively). Overall, CSF-1 and CSF-1-FDC had a similar incidence of TEAEs from day 8 to day 15. This observation suggests that diclofenac sodium had no anti-inflammatory effect in our study, as noted by the higher conjunctival hyperemia rate reported in the Pilo 2%-FDC and CSF-1-FDC arms. The same concentration of diclofenac sodium, 0.006%, either as a fixed dose combined with pilocarpine HCl or as a control was used and no other concentrations of diclofenac sodium were evaluated in this study. The known anti-inflammatory effect of diclofenac sodium may require a higher concentration than 0.006%.
Headache is a common AE associated with pilocarpine HCl and is consistent with the results in this phase 2b study, with the highest frequency observed in participants receiving Pilo 0.2% (7.3%). The headaches reported in this study were all classified as mild, and the majority were intermittent. Retinal detachment is another AE commonly associated with higher concentrations of pilocarpine.26 Retinal pathology was not reported in any treatment groups of this study. An overall higher rate of ocular and non-ocular TEAEs reported by participants receiving Pilo 0.2% and Pilo 0.2%-FDC during day 1 through day 8 may be due to a more diligent reporting of AEs at the beginning of the study or the participants’ initial symptoms diminished and were no longer noticeable over time.
Even though instillation site pain was reported, participants still reported the drops as very comfortable based on the drop comfort assessment. The high tolerability may be attributed to the preservative-free formulation of the drug at a near-neutral pH. CDVA assessments of the 0.4% groups (CSF-1 and CSF-1-FDC) under different lighting conditions demonstrated a mean improvement of up to 3.5 letters in CDVA from baseline at all timepoints in the study eye. Even though this gain is not clinically meaningful, it demonstrates that distance vision was not compromised.
Some limitations of the study include assessments performed after only one dose even though the participants dosed twice daily at home. This does not reflect the real-world intended use of the drug. Assessments after twice daily bilateral dosing have been adapted in the phase 3 study design. Another limitation is that participants randomized in the pilocarpine containing groups started with a 0.2% pilocarpine HCl concentration. The results may have been different if some participants started with 0.4% pilocarpine HCl concentration and if a washout period between visit 3 and visit 4 had been included in the study design. The short duration of follow-up is another limitation of the study. Lastly, the participant sample size is too small to detect rare safety events and is probably the reason that no significant difference was seen in the post hoc subgroup analyses. Further subgroup analysis to determine differences in therapeutic response between demographic groups will be performed in the Phase 3 studies and reported in a separate paper. Even with the study limitations, the study informed us of the minimum effective concentration of pilocarpine HCl necessary to achieve clinically significant efficacy, and that diclofenac is not necessary in the formulation.
Conclusion
The overall results of this study demonstrated that CSF-1 (0.4% pilocarpine HCl) as a single active ingredient with its proprietary formulation was effective in improving near VA with a favorable safety and tolerability profile. The results also showed that diclofenac sodium had no significant contribution and, therefore, CSF-1 was the formulation used for the phase 3 (NEAR-1 [ClinicalTrial.gov ID: NCT04599933] and NEAR-2 [ClinicalTrial.gov ID: NCT04599972]) studies and eventually approved by the FDA for commercial use.
Acknowledgments
This study was supported by Orasis Pharmaceuticals, Ltd. Orasis participated in the study design, analysis, interpretation of data, and the review and approval of the publication. Neither honoraria nor payments were made for authorship.
The authors acknowledge the following investigators for their participation in the study: Ralph Chu, MD (Chu Vision Institute); William C. Christie, MD (Scott and Associates PC); David Evans, OD (The Clinical Trials Center at Total Eye Care); Lance Forstot, MD (Colorado Eye Consultants); Jack Greiner, DO, PhD (Andover Eye Associates); Majid Morshifar, MD (Hoopes, Durrie, Rivera Research); David L. Wirta, MD (Eye Research Foundation).
Disclosure
M.F. is a consultant/advisor for Alcon, Aldeyra, Allergan, Bausch + Lomb, Bio-Tissue, CorneaGen, Dompé, Glaukos, Johnson & Johnson Vision, Kala Pharmaceuticals, Novartis, Orasis Pharmaceuticals, Oyster Point, Sun Pharma, Tarsus Pharmaceuticals, ZEISS; is a member of the Medical Advisory Board for Orasis Pharmaceuticals. S.R. is a consultant for Abbvie/Allergan, Ace Vision Group, Alcon, Azura, Bausch & Lomb, Centricity, Dompé, Glia, Ivizia, Johnson & Johnson Vision, Kala Pharmaceuticals, Novartis, Ocuterra, Orasis Pharmaceuticals, Visus; Oyster Point, PRN Nutriceuticals, RxSight, Science Based Health, Sight Sciences, Stuart Pharmaceuticals, Tarsus Pharmaceuticals, Thea, Viatris, Visus, Zeiss; received honoraria for speakers bureau and/or travel reimbursement from Orasis Pharmaceuticals; Support for attending meetings and/or travel: Orasis Pharmaceuticals. M.M is an employee of Hoopes Vision, Durrie, and Rivera Research. D.C. is a consultant for Orasis Pharmaceuticals, Johnson & Johnson and holds Orasis Pharmaceuticals stock options. I.B.G. is a consultant for Orasis Pharmaceuticals and holds Orasis Pharmaceuticals stock options. G.S. is a consultant for Orasis Pharmaceuticals. T.I. is an employee of Orasis Pharmaceuticals. P.G. is a consultant for Alcon, Aldeyra Therapeutics, Allergan, Azura, Expert Opinion, HanAll Biopharma, Johnson & Johnson Vision, Kala Pharmaceuticals, New World Medical, Novartis, Ocular Science, Ocular Therapeutix, Orasis Pharmaceuticals, Oyster Point Pharma, Santen, Sight Sciences, SpyGlass Pharma, Sun Pharma, Surface Ophthalmics, Inc., Tarsus, TearLab, TearClear, TissueTech, Inc., Visionology, ZEISS; Holds stock options for Orasis Pharmaceuticals and Tarsus Pharmaceuticals. The authors report no other conflicts of interest in this work.
References
1. Katz JA, Karpecki PM, Dorca A, et al. Presbyopia - A review of current treatment options and emerging therapies. Clin Ophthalmol. 2021;15:2167–2178. doi:10.2147/OPTH.S259011
2. Stokes J, Shirneshan E, Graham CA, Paulich M, Johnson N. Exploring the experience of living with and managing presbyopia. Optom Vis Sci. 2022;99(8):635–644. doi:10.1097/OPX.0000000000001913
3. Kandel H, Khadka J, Goggin M, Pesudovs K. Impact of refractive error on quality of life: a qualitative study. Clin Exp Ophthalmol. 2017;45(7):677–688. doi:10.1111/ceo.12954
4. Moussa K, Jehangir N, Mannis T, Wong WL, Moshirfar M. Corneal refractive procedures for the treatment of presbyopia. Open Ophthalmol J. 2017;11(1):59–75. doi:10.2174/1874364101711010059
5. Grzybowski A, Markeviciute A, Zemaitiene R. A review of pharmacological presbyopia treatment. Asia Pac J Ophthalmol. 2020;9(3):226–233. doi:10.1097/APO.0000000000000297
6. Waring GO 4th, Price FWJ, Wirta D, et al. Safety and efficacy of AGN-190584 in individuals with presbyopia: the GEMINI 1 phase 3 randomized clinical trial. JAMA Ophthalmol. 2022;140(4):363–371. doi:10.1001/jamaophthalmol.2022.0059
7. Haghpanah N, Alany R. Pharmacological treatment of presbyopia: a systematic review. Eur J Transl Myol. 2022;32(3):10781. doi:10.4081/ejtm.2022.10781
8. Grzybowski A, Ruamviboonsuk V. Pharmacological treatment in presbyopia. J Clin Med. 2022;11(5):1385. doi:10.3390/jcm11051385
9. Meghpara BB, Lee JK, Rapuano CJ, Mian SI, Ho AC. Pilocarpine 1.25% and the changing landscape of presbyopia treatment. Curr Opin Ophthalmol. 2022;33(4):269–274. doi:10.1097/ICU.0000000000000864
10. Vuity [package insert]. North Chicago, IL; Allergan, an AbbVie Company; 2022.
11. Kanarr S, El-Harazi SM, Moshirfar M, et al. Safety and efficacy of twice-daily pilocarpine HCl in presbyopia: the virgo phase 3, randomized, double-masked, controlled study. Am J Opthalmol. 2023;253:189–200. doi:10.1016/j.ajo.2023.05.008
12. Laibovitz RA, Koester J, Schaich L, Reaves TA. Safety and efficacy of diclofenac sodium 0.1% ophthalmic solution in acute seasonal allergic conjunctivitis. J Ocul Pharmacol Ther. 1995;11(3):361–368. doi:10.1089/jop.1995.11.361
13. Flach AJ, Dolan BJ, Donahue ME, Faktorovich EG, Gonzalez GA. Comparative effects of ketorolac 0.5% or diclofenac 0.1% ophthalmic solutions on inflammation after cataract surgery. Ophthalmology. 1998;105(9):1775–1779. doi:10.1016/S0161-6420(98)99053-4
14. Parker J, Tandon A, Shtein RM, et al. Management of pain with diclofenac after femtosecond-assisted laser in situ keratomileusis. J Cataract Refract Surg. 2011;37(3):569–573. doi:10.1016/j.jcrs.2010.09.020
15. Benozzi G, Perez C, Leiro J, Facal S, Orman B. Presbyopia treatment with eye drops: an eight year retrospective study. Trans Vision Sci Technol. 2020;9(7):25.
16. Benozzi JL. Ophthalmic compositions of parasympathetic stimulants and anti-inflammatories for use in the treatment of presbyopia (US Patent 8,524,758 B2). U.S. Patent and Trademark Office. 2013.
17. ClinicalTrials.gov. Safety, tolerability and efficacy of presbidrops (CSF-1) in presbyopic subjects. 2016. Available from: https://ClinicalTrials.gov/show/NCT02965664;.
18. Socea S, Mimouni M, Andreja V, Blumenthal EZ. Drops for presbyopia: results of CSF-1, a multicenter randomized double-masked placebo-controlled crossover study [abstract]. Invest Ophthalmol Vis Sci. 2019;60(9):1385.
19. Patel JI, Jenkins L, Benjamin L, Webber S. Dilated pupils and loss of accommodation following diode panretinal photocoagulation with sub-tenon local anaesthetic in four cases. Eye. 2002;16(5):628–632. doi:10.1038/sj.eye.6700004
20. Lana-Peixoto MA, Campos WR, Reis PA, Campos CM, Rodrigues CA. Tonic pupil in leprosy. Arq Bras Oftalmol. 2014;77(6):395–396. PMID: 25627189. doi:10.5935/0004-2749.20140098
21. Bowie EM, Givre SJ. Tonic pupil and sarcoidosis. Am J Ophthalmol. 2003;135(3):417–419. PMID: 12614777. doi:10.1016/s0002-9394(02)01959-1
22. Chemmanam T, Pandian JD, Kadyan RS, Bhatti SM. Anhidrosis: a clue to an underlying autonomic disorder. J Clin Neurosci. 2007;14(1):94–96. PMID: 17070054. doi:10.1016/j.jocn.2005.11.041
23. Leavitt JA, Wayman LL, Hodge DO, Brubaker RF. Pupillary response to four concentrations of pilocarpine in normal subjects: application to testing for Adie tonic pupil. Am J Ophthalmol. 2002;133(3):333–336. PMID: 11860969. doi:10.1016/s0002-9394(01)01420-9
24. Price FW Jr, Hom M, Moshirfar M, et al. Combinations of pilocarpine and oxymetazoline for the pharmacological treatment of presbyopia: two randomized phase 2 studies. Ophthalmol Sci. 2021;1(4):100065. doi:10.1016/j.xops.2021.100065
25. Lutjen-Drecoll E, Tamm E, Kaufman PL. Age-related loss of morphologic responses to pilocarpine in rhesus monkey ciliary muscle. Arch Ophthalmol. 1988;106(11):1591–1598. doi:10.1001/archopht.1988.01060140759051
26. Pape LG, Forbes M. Retinal detachment and miotic therapy. Am J Ophthalmol. 1978;85(4):558–566. doi:10.1016/s0002-9394(14)75255-9
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.