")
Back to Journals » Journal of Inflammation Research » Volume 17
Development and Validation of Diagnostic Models for Transcriptomic Signature Genes for Multiple Tissues in Osteoarthritis
Authors Gao Q, Ma Y, Shao T, Tao X, Yang X, Li S, Gu J, Yu Z
Received 7 June 2024
Accepted for publication 20 July 2024
Published 31 July 2024 Volume 2024:17 Pages 5113—5127
DOI https://doi.org/10.2147/JIR.S472118
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Qichang Gao,1 Yiming Ma,1 Tuo Shao,1 Xiaoxuan Tao,2 Xiansheng Yang,1 Song Li,1 Jiaao Gu,1 Zhange Yu1
1Department of Spinal Surgery, The 1st Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang Province, People’s Republic of China; 2Department of Radiotherapy, The 3st Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang Province, People’s Republic of China
Correspondence: Zhange Yu, Email [email protected]
Background: Progress in research on expression profiles in osteoarthritis (OA) has been limited to individual tissues within the joint, such as the synovium, cartilage, or meniscus. This study aimed to comprehensively analyze the common gene expression characteristics of various structures in OA and construct a diagnostic model.
Methods: Three datasets were selected: synovium, meniscus, and knee joint cartilage. Modular clustering and differential analysis of genes were used for further functional analyses and the construction of protein networks. Signature genes with the highest diagnostic potential were identified and verified using external gene datasets. The expression of these genes was validated in clinical samples by Real-time (RT)-qPCR and immunohistochemistry (IHC) staining. This study investigated the status of immune cells in OA by examining their infiltration.
Results: The merged OA dataset included 438 DEGs clustered into seven modules using WGCNA. The intersection of these DEGs with WGCNA modules identified 190 genes. Using Least Absolute Shrinkage and Selection Operator (LASSO) and Random Forest algorithms, nine signature genes were identified (CDADC1, PPFIBP1, ENO2, NOM1, SLC25A14, METTL2A, LINC01089, L3HYPDH, NPHP3), each demonstrating substantial diagnostic potential (areas under the curve from 0.701 to 0.925). Furthermore, dysregulation of various immune cells has also been observed.
Conclusion: CDADC1, PPFIBP1, ENO2, NOM1, SLC25A14, METTL2A, LINC01089, L3HYPDH, NPHP3 demonstrated significant diagnostic efficacy in OA and are involved in immune cell infiltration.
Keywords: osteoarthritis, machine learning, immune cells infiltration, diagnostic model
Introduction
Osteoarthritis (OA) is a degenerative joint disease that is characterized by inflammation and pain.1 OA not only considerably diminishes life satisfaction for patients, but also increases the substantial financial burden on healthcare systems.2 Among the joints most frequently affected by OA, the knee joint encompasses various structural components including the meniscus, synovium, and articular cartilage.3
Meniscal damage marks an early pivotal event in knee OA development, exacerbating the load on the articular cartilage and potentially initiating inflammatory responses that hasten cartilage deterioration.4 Inflammation and hypertrophy of the synovium play vital roles in OA, and synovitis is recognized as a major contributor to OA-associated pain and its progression.5 Gradual wear and degradation of articular cartilage are essential pathological manifestations of OA, diminishing the cushioning ability of the joint, which results in pain and restricted mobility.6 The treatment of OA includes non-surgical approaches (health management, medication) and surgical interventions. With the advancement in the research of OA’s molecular mechanisms and the maturation of pluripotent stem cell research, new minimally invasive treatment methods have gradually increased. Although the long-term benefits are still inconclusive, intra-articular therapies such as hyaluronic acid (HA), platelet-rich plasma (PRP), and mesenchymal stem cells have become effective methods for early to mid-stage OA. Moreover, with the development of big data machine learning analysis models based on disease genomics and molecular phenotypes, novel diagnostic methods, distinct from traditional demographic and clinical imaging approaches, have demonstrated promising predictive capabilities and are being utilized in clinical practice.7,8
Recent research has increasingly illuminated the critical role of the immune system in OA development, particularly the significant impact of inflammatory responses in promoting damage to joint cartilage and accelerating disease progression.9 However, research on noninvasive biomarkers for OA diagnosis is challenging.10
In this study, we collectively analyzed the meniscus, synovium, and articular cartilage for the first time using various bioinformatics approaches to identify the distinctive genes associated with knee osteoarthritis. These genes have shown exceptional diagnostic efficacy as confirmed using external datasets and clinical tissue samples. We also explored the signaling pathways in which these genes are involved and their role in immune cell infiltration, aiming to deepen our understanding of the interplay between meniscal damage, synovial inflammation, and cartilage degeneration, while providing references for clinical diagnosis and treatment.
Methods and Materials
Clinical Samples
We collected six osteoarthritis (OA) tissue samples from patients who underwent total knee arthroplasty at the 1st Affiliated Hospital of Harbin Medical University and six normal tissue samples from patients who underwent reconstructive surgery due to trauma.
Microarray Data
Figure 1 illustrates the study. In this study, we downloaded three datasets, GSE206848, GSE98918, and GSE178557(http://www.ncbi.nlm.nih.gov/geo/).11–13 GSE206848 and GSE98918 (Including 19 healthy control groups and 19 disease groups) were designated as the training sets, whereas GSE178557 (Including 4 healthy control groups and 4 disease groups) was used as the validation dataset. The detailed characteristics of these datasets, including microarray platforms, composition of sample groups, and sample sizes (Table 1).
 |
Table 1 Basic Information of Datasets |
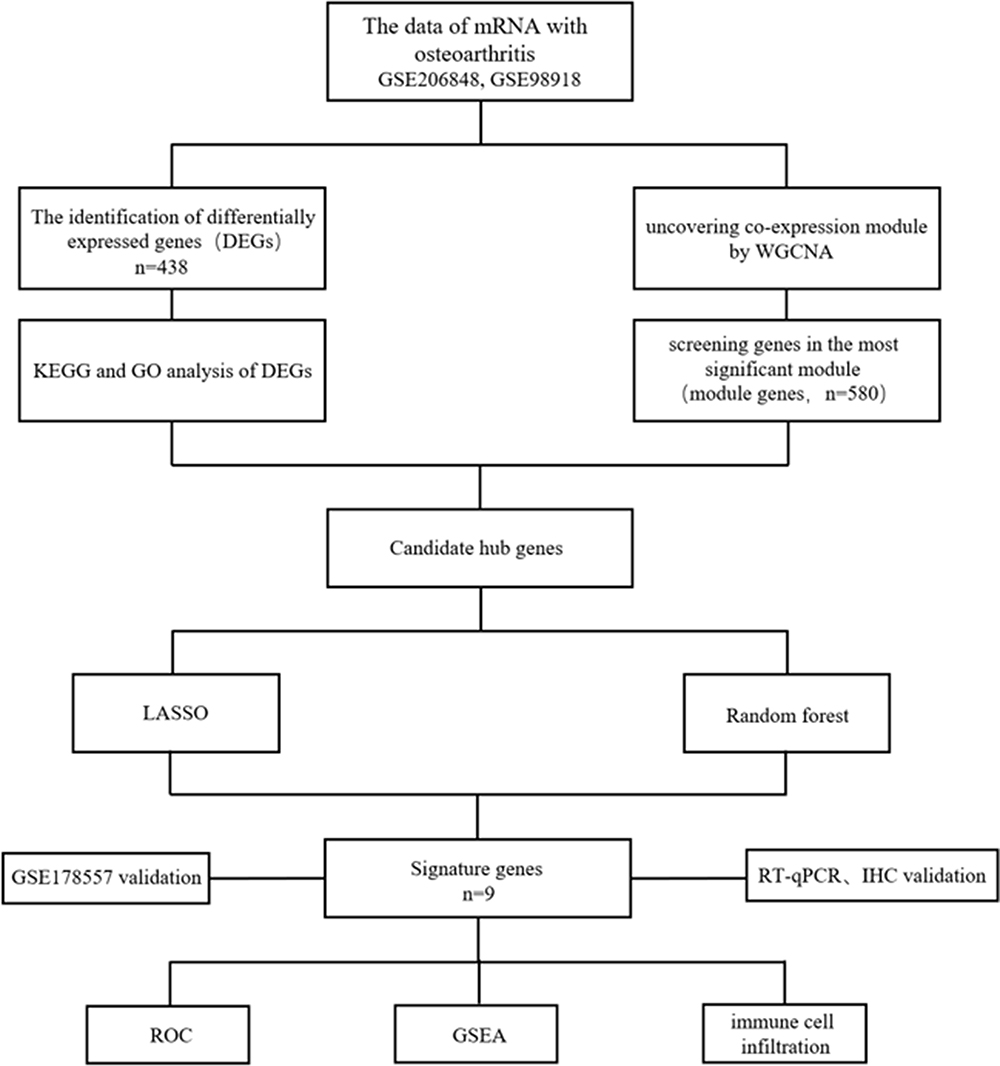 |
Figure 1 Designing of this research. |
Screening of DEGs
Initially, the training datasets were merged using R software’s insilomerging package.14 Subsequently, the method proposed by Johnson et al was used to eliminate batch effects.15 The limma package of R software (version 4.1.2) was used for differential analysis.16
Co-Expression Network Analysis
GSE206848 and GSE98918 datasets were processed using WGCNA.17 Briefly, the genes that exhibited the lowest 50% of the median absolute deviation values were discarded. The goodSamplesGenes method was used to exclude outlier genes and samples, thereby facilitating the construction of a scale-free network. The modules were subsequently delineated using dynamic tree cutting. WGCNA analysis was used to identify significant modules in OA.
Exploration of Functions
Gene profiling and visualization were conducted using the clusterProfiler package.18 Gene Ontology (GO) was used to explore the biological pathways involved in the DEGs.19 Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was used to analyze potential biological functions.20 Pathway association analysis of the characteristic genes was conducted using GSEA software. Significance was assessed based on the criterion of an adjusted p < 0.05.21
Protein Network
To further investigate the interactions between DEGs from OA patients, a protein network was analyzed using STRING (https://string-db.org/). Subsequently, the network was visualized using the CentiScape plugin in Cytoscape3.10.0.
Screening OA Diagnostic Genes
The LASSO and Random Forest algorithms were used to analyze the characteristic genes associated with osteoarthritis (OA). Data integration was performed using R software’s glmnet package.22 Subsequently, the randomForest package of R software was used to screen hub genes.23 The overlapping genes identified by both the machine learning models were used as characteristic genes for further analysis.
Immune Infiltration Analysis
The CIBERSORT algorithm was utilized to analyze correlations among immune cells, with the goal of investigating differences in immune infiltration in osteoarthritis (OA) patients and healthy patients. Additionally, the correlation between characteristic genes and immunity was assessed using Spearman’s method.24
RT-qPCR
Total RNA was extracted by the centrifuge column method (DP451; Invitrogen). An automatic medical PCR analysis system (SLAN-96S, China) and SYBR Green qPCR Mix (G3326-01; Servicebio) were used for RT-qPCR analysis. The mRNA expression levels were statistically analyzed using the housekeeping genes GAPDH. All experiments were repeated three times and significant differences were statistically analyzed using the normalized 2 −ΔΔ ct. Primers were designed as follows.
SLC25A14: 5′-GCCCTGCCAGCCTTTCTACTC-3′, 5′-AACCACCCCACTGTTCTTCCATC-3′; NOM1: 5′-GGAGGCAGTGGTGAGGAAGTTC-3′; 5′-ATAAATGGGCAATGACGGTGAAC-3′; METTL2A1: 5′-ACGACCTGTGTGATGAAGAGAAGAG-3′, 5′-AGAAGCCTGCTCAGCCTGTTG-3′; NPHP3: 5′-CCTCTGCCTTGTCAATGTTAGTCAC-3′, 5′-AAGCAAGCCACATCCATAAGTCAAC-3′; ENO2: 5′-CGCACTTTCCACTTCTTCCTTTCTC-3′, 5′-TCTCTAACACCTCAGCACACCAAC-3′; PPFIBP1: 5′-TTGACCGTAGCACCTGGATGAAC-3′, 5′-CAATAGACAATCCGCTGCCTGTTG-3′; GAPDH: 5′-TGACATCAAGAAGGTGGTGAAGCAG-3′, 5′-GTGTCGCTGTTGAAGTCAGAGGAG-3′.
Immunohistochemistry Staining
The expression levels of PPFIBP1, ENO2, NPHP3, and METTL2A (13961-1-AP, 10149-1-AP, 22026-1-AP, 16983-1-AP; Proteintech) in both OA and normal tissues were confirmed using immunohistochemical staining. Briefly, tissues were initially fixed for 24 h, followed by stepwise dehydration using alcohol gradients, paraffin embedding, and sectioning at a thickness of 4 mm. Antigen retrieval was conducted and endogenous peroxidases were removed using 3% H2O2. Sections were subsequently blocked with goat serum (AR1009; Boster). Primary and secondary antibodies were then incubated sequentially. DAB (ZLI-9018, Origene) was used for chromogenic detection, and hematoxylin (G1004, Servicebio) was used for nuclear staining.
Statistical Analysis
All experiments were performed at least three times, with data presented as mean ± SD. Data were analyzed using GraphPad Prism software version 9.4.0. Comparisons between two groups were conducted using Student’s t-test, while multiple group comparisons were performed using ANOVA. *p < 0.05 was considered statistically significant.
Results
Identifying DEGs Between OA and Control
Following the removal of batch effects, the GSE206848 and GSE98918 datasets were merged (Supplementary Figure S1). Through differential analysis, we obtained all differentially expressed genes (Supplementary Table S1), including 123 upregulated and 315 downregulated genes (Figure 2a and b).
 |
Figure 2 Differential analysis of genes in OA. Notes: (a) Heatmaps and volcano plots were identified from the intersection dataset. (b) Upregulated and downregulated genes were shown in red and green triangles. |
Functional Enrichment Analysis
KEGG analysis revealed that the primary enriched pathways were “Adherens junction”, “Spliceosome and RNA degradation” (Figure 3a). GO analysis indicated that the main enriched terms in BP were “RNA splicing” and “positive regulation of DNA metabolic processes”. For CC, the primary enrichments were observed in “nuclear speckle”, “chromosomal region” and “ribonucleoprotein granule”. MF) analysis showed that “catalytic activity, acting on RNA” and “protein serine kinase activity” were the most significant features (Figure 3b).
 |
Figure 3 Enrichment analysis of DEGs in OA. Notes: (a) KEGG pathway analysis in OA. Significant pathways and genes were listed using different sizes and colors. (b) The top ten relevant functions were presented separately in the GO analysis. |
Gene Modularity Analysis
We utilized the R software’s WGCNA package for module classification, setting the soft-thresholding power at 7 for relatively better average connectivity (Figure 4a and b). Subsequently, eight clustered modules were generated and are represented in distinct colors (Figure 4c and d). The blue module, consisting of 580 genes, was significantly correlated with OA (correlation coefficient = 0.59, p = 4.2e-52) (Figure 4e and f).
 |
Figure 4 Module genes using WGCNA in OA. Notes: (a and b) C To pursue the smoothness of the curve, β = 7 was set as the standard. (c) Different gene modules were indicated in different colors. (d) Heatmap of eigengene adjacency. (e and f). The blue module was most associated with OA. |
Analysis of Intersection Between WCGNA and OA DEGs and PPI Network Construction
A total of 190 genes were screened for DEGs and key modules. (Figure 5a). KEGG analysis revealed that the primary enriched pathways were “Spliceosome”, “Ribosome biogenesis in eukaryotes” (Figure 5b). GO analysis indicated that the main enriched terms in BP were “organelle organization” and “the cell cycle”. For CC, the primary enrichments were observed in “nuclear part” and “nuclear lumen”. MF analysis showed that “nucleic acid binding” was more prominent (Figure 5c–e). Subsequently, we constructed a protein network using the intersecting genes, and visualized the most active modules using the MCODE plugin (Figure 5f and g).
 |
Figure 5 Analysis of the genes of DEGs and key modules. Notes: (a) The Venn diagram shows the 190 intersection genes. (b–e) Analysis of intersection genes. (f) The most related genes were displayed using cytoHubba plug-in. (g) Presentation of gene nodes. |
Identification of OA Signature Genes
The diagnostic model was constructed using two machine-learning algorithms. The LASSO regression algorithm identified 13 potential biomarkers (Figure 6a and b), whereas the random forest analysis identified 50 relatively important signature genes (Figure 6c and d). The identified signature genes are listed in Table 2. The convergence of these two algorithms identified nine signature genes: CDADC1, PPFIBP1, ENO2, NOM1, SLC25A14, METTL2A, LINC01089, L3HYPDH, and NPHP3 (Figure 6e).
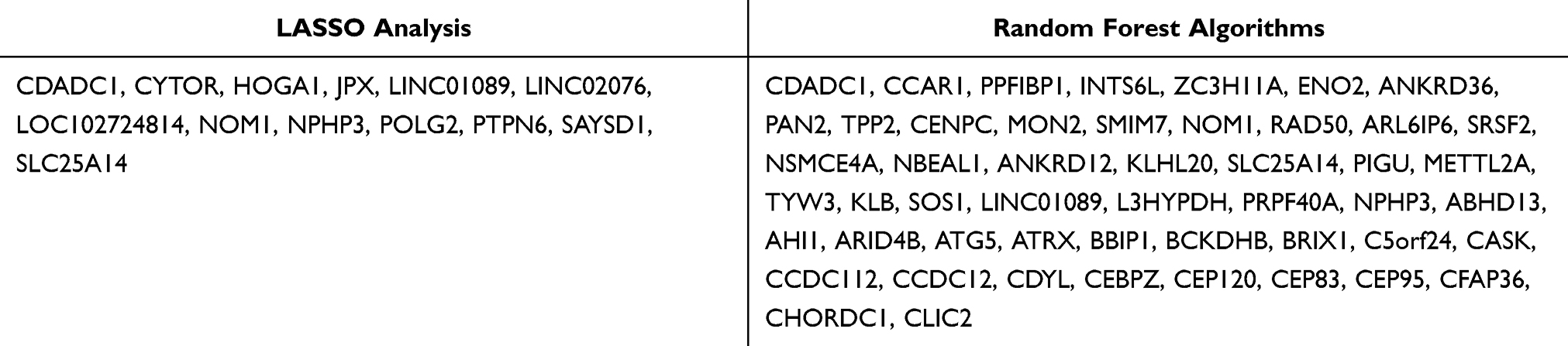 |
Table 2 Analysis Results of Machine Learning |
 |
Figure 6 Machine learning was used to screen the diagnostic genes of OA. Notes: (a and b) Thirteen diagnostic genes were identified through the LASSO model curve and optimization. (c and d) The top 20 candidate genes identified based on scoring statistics. (e) The Venn diagram shows the 9 intersection genes. |
Diagnostic Value Assessment
The identified genes were expressed at low levels in OA patients. (Figure 7a). The AUC values of the ROC analysis were 0.911 for CDADC1, 0.873 for ENO2, 0.925 for L3HYPDH, 0.853 for LINC01089, 0.701 for METTL2A, 0.781 for NPHP3, 0.783 for NOM1, 0.852 for SLC25A14, and 0.917 for PPFIBP1 (Figure 7b). Furthermore, the accuracy of each signature gene in predicting OA was evaluated using the external dataset GSE178557. The outcomes were in alignment with the trends observed in the GSE206848 and GSE98918 datasets (Figure 7c), demonstrating that these nine genes exhibited significant diagnostic efficiency in predicting OA. We extracted total RNA from three groups of clinical OA patients and healthy tissues. The RT-qPCR results were consistent with the expression predicted by the model. (Figure 7d).
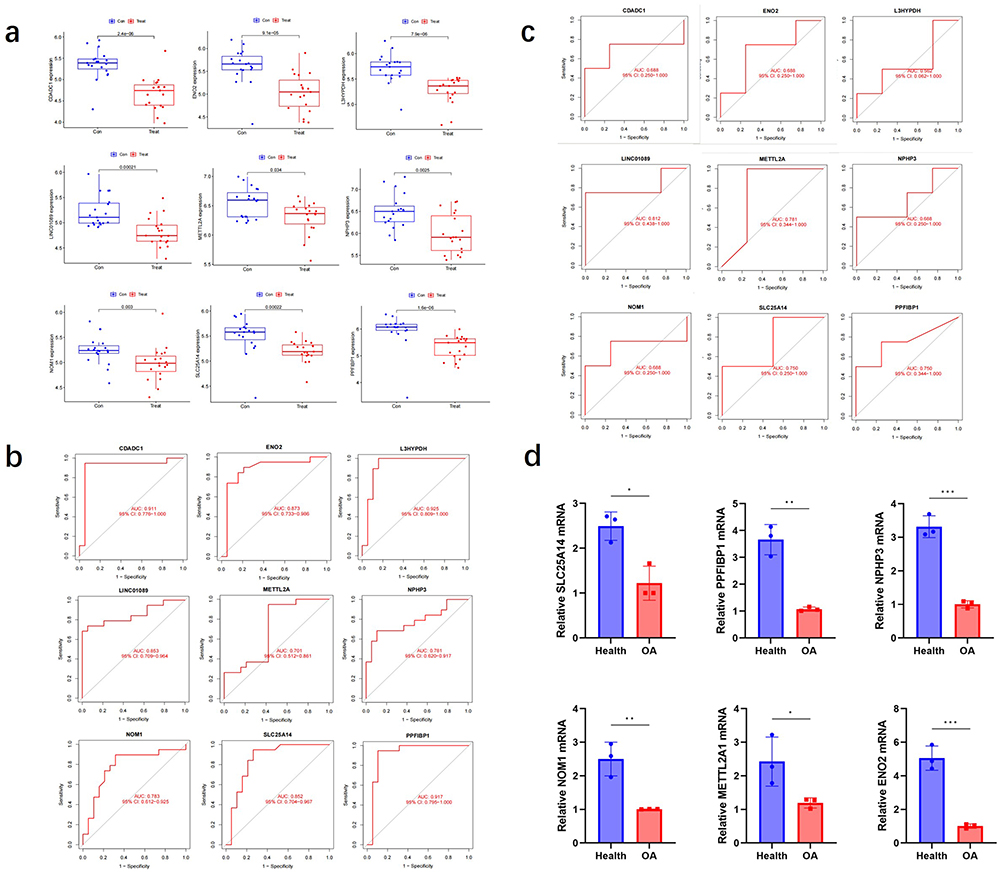 |
Figure 7 The model genes expression. Notes: (a) Gene expression in the training set. (b) The diagnostic efficiency of genes in the training set was evaluated. (c) The diagnostic efficacy of GSE178557 gene was evaluated. (d) Relevant mRNA expression levels were obtained by comparing Ct values. n=3, *p < 0.05, **p < 0.01, ***p < 0.001. |
The top ten pathways for each gene are shown in Figure 8. For instance, the expression of ENO2 is significantly correlated with RNA degradation, the cell cycle, and the Wnt signaling pathway. The expression of NOM1 correlated with the B-cell receptor pathway. The expression of PPFIBP1 significantly correlated with the cell cycle and olfactory transduction.
 |
Figure 8 Analysis of the model genes in OA. |
Immune Infiltration Analysis in OA
The immunological characteristics of the patients were illustrated in Figure 9a. Notably, OA patients exhibited higher levels of naive B cells and lower levels of activated NK cells (Figure 9b). We also found a positive correlation between resting NK cells and eosinophils, whereas resting dendritic cells were negatively correlated with plasma cells and memory B cells were negatively correlated with M2 macrophages (Figure 9c).
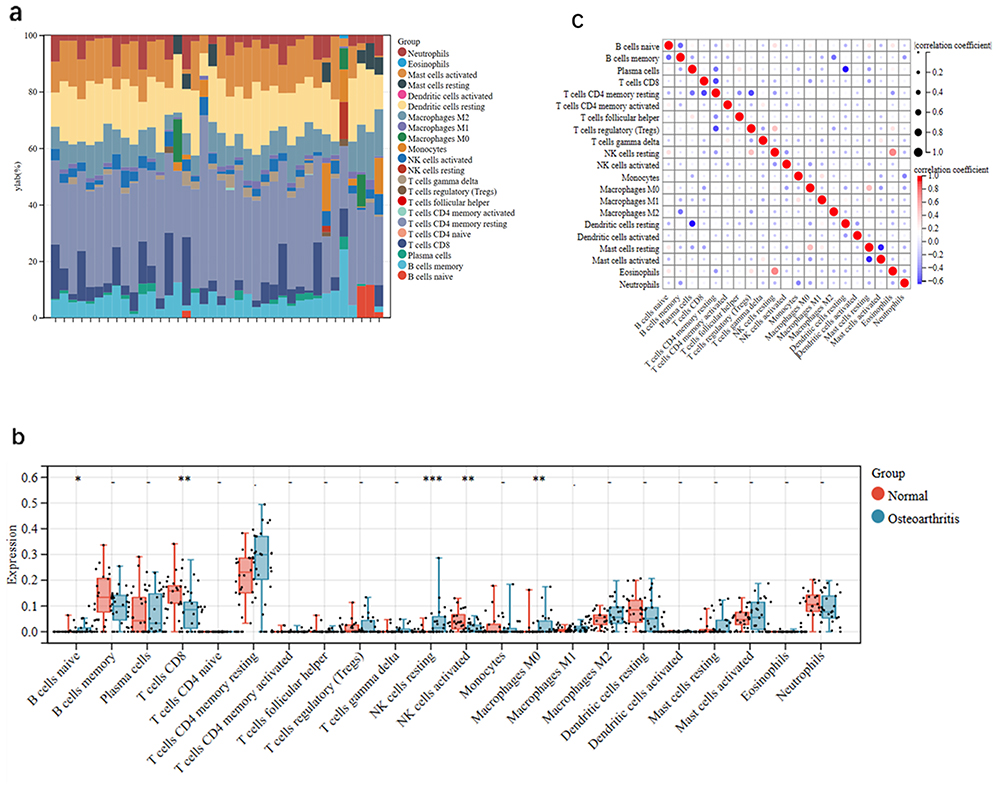 |
Figure 9 Immune cell infiltration analysis. Notes: (a) Trends in the distribution of immune cells between OA and healthy group samples. (b) The vioplot plot was used for immune correlation analysis. *p < 0.05, **p < 0.01, ***p < 0.001. (c) Correlations between different immune cell types. |
Expression of PPFIBP1, ENO2, NPHP3, and METTL2A in OA Tissues
Figure 10 illustrates the pathological features of synovium in the OA group. Marked proliferation and enlargement of synovial cells, neovascularization, pannus formation, and infiltration of various inflammatory cells including lymphocytes, macrophages, and neutrophils were observed. The compilation of target genes was also reduced along with the loss of normal synovial tissue. Additionally, immunohistochemical (IHC) analysis revealed that gene expression in the synovium of the OA group was consistent with the trends observed in the database.
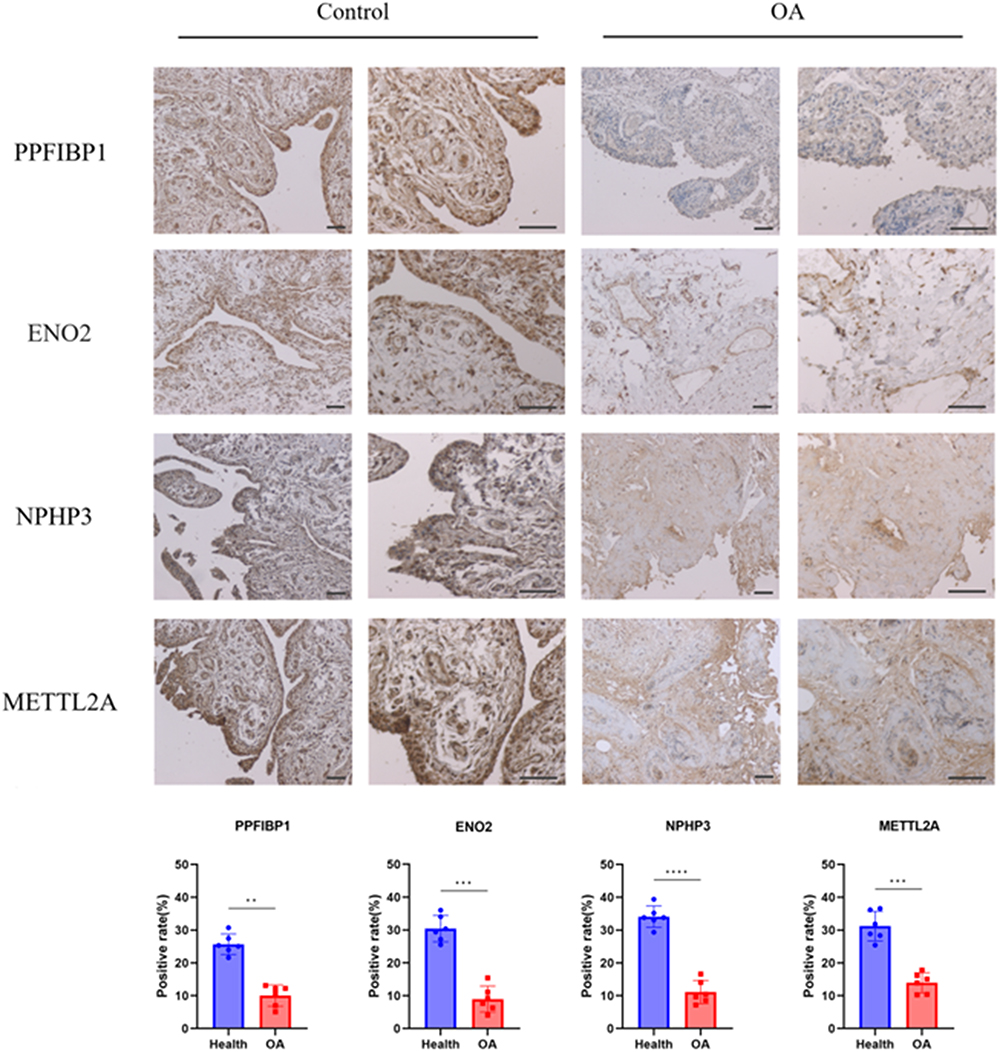 |
Figure 10 Typical IHC graphics of signature genes staining in synovium. Notes: The positive area ratio shows the expression of PPFIBP1, ENO2, NPHP3, and METTL2A in two groups (Microscope objectives: 4×, 10×. n=6, **p < 0.01, ***p < 0.001, ****p < 0.0001). |
Discussion
OA is a degenerative orthopedic disease that significantly affects the quality of life.25 The diagnosis of OA is predominantly based on clinical manifestations and radiological studies; however, specific diagnostic biomarkers are lacking. In recent years, several studies have conducted high-throughput sequencing on OA’s synovium, cartilage, and meniscus in an attempt to identify diagnostic biomarkers.26,27 However, no study has integrated these three components to explore the mechanisms underlying OA progression from a holistic perspective. The diagnostic value of this model for OA was assessed using several methods.
CDADC1 is expressed in multiple tissues of the human body, particularly in the heart, muscles and brain.28 GO analysis revealed that CDADC1 primarily participates in key biological processes, including the regulation of hydrolase activity, protein dimerization, cytidine deaminase activity, and DNA cytosine deamination. These biological functions are involved in OA development and development of OA. For example, hydrolases play a crucial role in joint cartilage self-repair by limiting excessive chondrocyte apoptosis and modulating inflammatory mediators to alleviate inflammatory reactions within the joints.29,30 These studies suggest that CDADC1 may be a potential target for alleviating OA.
ENO2 plays a pivotal role in the development of neural tissues.31 GO analysis indicated that ENO2 is involved in glucose metabolism and glycolysis. Recent studies have revealed the pivotal role of glycolysis in regulating the energy metabolism and inflammatory responses in OA chondrocytes.32 Glycolysis is correlated with the production of inflammatory mediators and aberrant chondrocyte differentiation under hypoxic conditions.33 Therefore, interventions targeting ENO2 may offer new therapeutic strategies for alleviating OA symptoms and delaying disease progression.
L3HYPDH regulates dietary proteins and other proteins such as collagen IV.34 GO analysis revealed that L3HYPDH primarily participates in the hydrolase activity. GO annotations related to METTL2A (Methyltransferase 2A) include methyltransferase and tRNA (cytidine) methyltransferase activities.35 NOM1 (Nucleolar protein with MIF4G domain1) is involved in the biogenesis of ribosomal small subunits, while another diagnostic gene, SLC25A14, participates in ion flux across the mitochondrial membrane and the regulation of membrane potential.36,37 The functions of these genes are closely and intricately associated with the induction of inflammation in articular cartilage and degradation of the extracellular matrix. Identifying the specific connections between these regulatory factors and the OA phenotype is of paramount importance for future work.38
NPHP3 is primarily involved in biological pathways, including organelle biogenesis, maintenance, and cargo trafficking to the periciliary membrane, and can also act as a molecular switch within the Wnt signaling pathway.39 Abnormal activation of the Wnt pathway affects changes in the subchondral tissue and synovial inflammation.40
PPFIBP is crucial for regulating focal adhesion disassembly and mediating protein-protein interactions at synapses.41 The coordination of internal and external signal transduction is a key factor in the perception of stress transmission.42 The pervasive presence of PPFIBP1 in the synovium, meniscus, and articular cartilage of OA patients highlights its potential as a diagnostic biomarker.43 This indicates that PPFIBP1 may be instrumental in understanding the pathological processes of OA and may serve as a novel target for therapeutic interventions.
Naïve B cells, resting NK cells, and M0 macrophages play distinct roles in OA pathophysiological progression. Naive B cells differentiate into plasma cells that produce antibodies that target self-antigens within the joint.44 This activity promotes inflammation and accelerates the tissue degradation. Resting NK cells play a dual role in OA. They can sometimes exacerbate local inflammation and, in special cases, promote knee joint repair through the secretion of growth factors.45 M1 macrophages drive inflammation and tissue damage, whereas M2 macrophages facilitate resolution of inflammation and tissue repair.46 The progression of OA may lead to a shift in balance, thereby influencing disease outcomes.
Our study has certain limitations. First, the data were sourced from public databases with a relatively small sample size, which might introduce selection bias and impact the diagnostic value. However, the reliability of our analysis was confirmed by using external datasets and immunohistochemistry. Secondly, larger clinical sample sizes and follow-up studies are required to validate our findings. Third, further investigation is required to elucidate the role of hub genes in immune response.
Conclusion
This study screened out Nine signature genes (CDADC1, PPFIBP1, ENO2, NOM1, SLC25A14, METTL2A, LINC01089, L3HYPDH, NPHP3) had a significant diagnostic value for OA. Additionally, we explored the characteristics of immune cell infiltration in patients with OA to provide new insights into OA treatment.
Ethics Approval
This study was conducted in accordance with the principles of the Declaration of Helsinki. The human-related data, surgical samples involved in this study have been approved by the Ethics Committee of the First Affiliated Hospital of Harbin Medical University.
Informed Consent
The study was conducted with informed consent from patients. All patients signed informed consent forms approved by the institutional review Committee.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tang S, Yao L, Ruan J, et al. Single-cell atlas of human infrapatellar fat pad and synovium implicates APOE signaling in osteoarthritis pathology. Sci Transl Med. 2024;16(731):eadf4590. doi:10.1126/scitranslmed.adf4590
2. Li J, Gui T, Yao L, et al. Synovium and infrapatellar fat pad share common mesenchymal progenitors and undergo coordinated changes in osteoarthritis. J Bone Miner Res. 2024. doi:10.1093/jbmr/zjad009
3. Van kooten NJT, Blom AB, Teunissen van Manen IJ, et al. S100A8/A9 drives monocytes towards M2-like macrophage differentiation and associates with M2-like macrophages in osteoarthritic synovium. Rheumatology. 2024. doi:10.1093/rheumatology/keae020
4. Lee K, Niku S, Koo SJ, Belezzuoli E, Guma M. Molecular imaging for evaluation of synovitis associated with osteoarthritis: a narrative review. Arthritis Res Ther. 2024;26(1):25. doi:10.1186/s13075-023-03258-6
5. Ma J, Yu P, Ma S, et al. Bioinformatics and integrative experimental method to identifying and validating Co-expressed ferroptosis-related genes in OA articular cartilage and synovium. J Inflamm Res. 2024;17:957–980. doi:10.2147/JIR.S434226
6. Chen Y, Zeng D, Wei G, et al. Pyroptosis in osteoarthritis: molecular mechanisms and therapeutic implications. J Inflamm Res. 2024;17:791–803. doi:10.2147/JIR.S445573
7. Tian R, Su S, Yu Y, et al. Revolutionizing osteoarthritis treatment: how mesenchymal stem cells hold the key. Biomed Pharmacother. 2024;173:116458. doi:10.1016/j.biopha.2024.116458
8. Foster NE, Eriksson L, Deveza L, Hall M. Osteoarthritis year in review 2022: epidemiology & therapy. Osteoarthritis Cartilage. 2023;31(7):876–883. doi:10.1016/j.joca.2023.03.008
9. Li H, Yuan Y, Zhang L, Xu C, Xu H, Chen Z. Reprogramming macrophage polarization, depleting ROS by astaxanthin and thioketal-containing polymers delivering rapamycin for osteoarthritis treatment. Adv Sci. 2024;11(9):e2305363. doi:10.1002/advs.202305363
10. Ma K, Pham T, Wang J, et al. Nanoparticle-based inhibition of vascular endothelial growth factor receptors alleviates osteoarthritis pain and cartilage damage. Sci Adv. 2024;10(7):eadi5501. doi:10.1126/sciadv.adi5501
11. Yu J, Wang W, Jiang Z, Liu H. TPX2 upregulates MMP13 to promote the progression of lipopolysaccharide-induced osteoarthritis. PeerJ. 2024;12:e17032. doi:10.7717/peerj.17032
12. Brophy RH, Zhang B, Cai L, Wright RW, Sandell LJ, Rai MF. Transcriptome comparison of meniscus from patients with and without osteoarthritis. Osteoarthritis Cartilage. 2018;26(3):422–432. doi:10.1016/j.joca.2017.12.004
13. Liu Y, Yang Y, Lin Y, et al. N(6) -methyladenosine-modified circRNA RERE modulates osteoarthritis by regulating β-catenin ubiquitination and degradation. Cell Prolif. 2023;56(1):e13297. doi:10.1111/cpr.13297
14. Su Y, Yu G, Li D, et al. Identification of mitophagy-related biomarkers in human osteoporosis based on a machine learning model. Front Physiol. 2023;14:1289976. doi:10.3389/fphys.2023.1289976
15. Geng Y, Shao R, Xu T, Zhang L. Identification of a potential signature to predict the risk of postmenopausal osteoporosis. Gene. 2024;894:147942. doi:10.1016/j.gene.2023.147942
16. Feng L, Chen Y, Mei X, Wang L, Zhao W, Yao J. Prognostic signature in osteosarcoma based on amino acid metabolism-associated genes. Cancer Biother Radiopharm. 2024. doi:10.1089/cbr.2024.0002
17. Liu J, Lu Y, Liu Y, et al. A gene signature linked to fibroblast differentiation for prognostic prediction of mesothelioma. Cell Biosci. 2024;14(1):33. doi:10.1186/s13578-023-01180-7
18. Duan X, Xing F, Zhang J, et al. Bioinformatic analysis of related immune cell infiltration and key genes in the progression of osteonecrosis of the femoral head. Front Immunol. 2023;14:1340446. doi:10.3389/fimmu.2023.1340446
19. Tang H, Xie J, Du YX, Tan ZJ, Liang ZT. Osteosarcoma neutrophil extracellular trap network-associated gene recurrence and metastasis model. J Cancer Res Clin Oncol. 2024;150(2):48. doi:10.1007/s00432-023-05577-2
20. Wang X, Liang Z, Liu Q, et al. Identification of PIK3R5 as a hub in septic myocardial injury and the cardioprotective effects of Psoralidin. Phytomedicine. 2024;122:155146. doi:10.1016/j.phymed.2023.155146
21. Yuan H, Yang S, Han P, Sun M, Zhou C. Drug target genes and molecular mechanism investigation in isoflurane-induced anesthesia based on WGCNA and machine learning methods. Toxicol Mech Methods. 2024;34(3):319–333. doi:10.1080/15376516.2023.2286619
22. Lee KS, Park H. Machine learning on thyroid disease: a review. Front Biosci. 2022;27(3):101. doi:10.31083/j.fbl2703101
23. Tang J, Henderson A, Gardner P. Exploring AdaBoost and Random Forests machine learning approaches for infrared pathology on unbalanced data sets. Analyst. 2021;146(19):5880–5891. doi:10.1039/D0AN02155E
24. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–457. doi:10.1038/nmeth.3337
25. Swain S, Sarmanova A, Mallen C, et al. Trends in incidence and prevalence of osteoarthritis in the United Kingdom: findings from the Clinical Practice Research Datalink (CPRD). Osteoarthritis Cartilage. 2020;28(6):792–801. doi:10.1016/j.joca.2020.03.004
26. Mathiessen A, Cimmino MA, Hammer HB, Haugen IK, Iagnocco A, Conaghan PG. Imaging of osteoarthritis (OA): what is new? Best Pract Res Clin Rheumatol. 2016;30(4):653–669. doi:10.1016/j.berh.2016.09.007
27. Demehri S, Guermazi A, Kwoh CK. Diagnosis and longitudinal assessment of osteoarthritis: review of available imaging techniques. Rheum Dis Clin North Am. 2016;42(4):607–620. doi:10.1016/j.rdc.2016.07.004
28. Xu Y, Li L, Li J, Liu Q. Structural and biological function of NYD-SP15 as a new member of cytidine deaminases. Gene. 2016;583(1):36–47. doi:10.1016/j.gene.2016.02.048
29. Wu Y, Shen S, Chen J, et al. Metabolite asymmetric dimethylarginine (ADMA) functions as a destabilization enhancer of SOX9 mediated by DDAH1 in osteoarthritis. Sci Adv. 2023;9(6):eade5584. doi:10.1126/sciadv.ade5584
30. Zheng L, Zhang Z, Sheng P, Mobasheri A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res Rev. 2021;66:101249. doi:10.1016/j.arr.2020.101249
31. Chai G, Brewer JM, Lovelace LL, Aoki T, Minor W, Lebioda L. Expression, purification and the 1.8 angstroms resolution crystal structure of human neuron specific enolase. J Mol Biol. 2004;341(4):1015–1021. doi:10.1016/j.jmb.2004.05.068
32. Matsuoka K, Bakiri L, Bilban M, et al. Metabolic rewiring controlled by c-Fos governs cartilage integrity in osteoarthritis. Ann Rheum Dis. 2023;82(9):1227–1239. doi:10.1136/ard-2023-224002
33. Arra M, Swarnkar G, Ke K, et al. LDHA-mediated ROS generation in chondrocytes is a potential therapeutic target for osteoarthritis. Nat Commun. 2020;11(1):3427. doi:10.1038/s41467-020-17242-0
34. Visser WF, Verhoeven-Duif NM, de Koning TJ. Identification of a human trans-3-hydroxy-L-proline dehydratase, the first characterized member of a novel family of proline racemase-like enzymes. J Biol Chem. 2012;287(26):21654–21662. doi:10.1074/jbc.M112.363218
35. Mao XL, Li ZH, Huang MH, et al. Mutually exclusive substrate selection strategy by human m3C RNA transferases METTL2A and METTL6. Nucleic Acids Res. 2021;49(14):8309–8323. doi:10.1093/nar/gkab603
36. Heus HC, Hing A, van Baren MJ, et al. A physical and transcriptional map of the preaxial polydactyly locus on chromosome 7q36. Genomics. 1999;57(3):342–351. doi:10.1006/geno.1999.5796
37. Gorgoglione R, Porcelli V, Santoro A, et al. The human uncoupling proteins 5 and 6 (UCP5/SLC25A14 and UCP6/SLC25A30) transport sulfur oxyanions, phosphate and dicarboxylates. Biochim Biophys Acta Bioenergy. 2019;1860(9):724–733. doi:10.1016/j.bbabio.2019.07.010
38. Zhao Z, Wang Z, Pei L, Zhou X, Liu Y. Long non-coding ribonucleic acid AFAP1-AS1 promotes chondrocyte proliferation via the miR-512-3p/matrix metallopeptidase 13 (MMP-13) axis. Bioengineered. 2022;13(3):5386–5395. doi:10.1080/21655979.2022.2031390
39. Olbrich H, Fliegauf M, Hoefele J, et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet. 2003;34(4):455–459. doi:10.1038/ng1216
40. Shang X, Böker KO, Taheri S, Hawellek T, Lehmann W, Schilling AF. The Interaction between microRNAs and the Wnt/β-Catenin signaling pathway in osteoarthritis. Int J Mol Sci. 2021;22(18):9887. doi:10.3390/ijms22189887
41. Kriajevska M, Fischer-Larsen M, Moertz E, et al. Liprin beta 1, a member of the family of LAR transmembrane tyrosine phosphatase-interacting proteins, is a new target for the metastasis-associated protein S100A4 (Mts1). J Biol Chem. 2002;277(7):5229–5235. doi:10.1074/jbc.M110976200
42. Wu TJ, Chang SL, Lin CY, et al. IL-17 facilitates VCAM-1 production and monocyte adhesion in osteoarthritis synovial fibroblasts by suppressing miR-5701 synthesis. Int J Mol Sci. 2022;23(12). doi:10.3390/ijms23126804
43. Steinberg J, Southam L, Fontalis A, et al. Linking chondrocyte and synovial transcriptional profile to clinical phenotype in osteoarthritis. Ann Rheum Dis. 2021;80(8):1070–1074. doi:10.1136/annrheumdis-2020-219760
44. Domínguez Conde C, Xu C, Jarvis LB, et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science. 2022;376(6594):eabl5197. doi:10.1126/science.abl5197
45. Yang J, Fan Y, Liu S. ATF3 as a potential diagnostic marker of early-stage osteoarthritis and its correlation with immune infiltration through bioinformatics analysis. Bone Joint Res. 2022;11(9):679–689. doi:10.1302/2046-3758.119.BJR-2022-0075.R1
46. Kou L, Huang H, Tang Y, et al. Opsonized nanoparticles target and regulate macrophage polarization for osteoarthritis therapy: a trapping strategy. J Control Release. 2022;347:237–255. doi:10.1016/j.jconrel.2022.04.037
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.