")
Back to Journals » Journal of Inflammation Research » Volume 18
Epigenetic Modulation by Lactylation in Sepsis: Linking Metabolism to Immune Dysfunction
Authors Chen Y , Hu H, Wang C, Wu J, Zan J, Liu Y
Received 11 February 2025
Accepted for publication 20 May 2025
Published 6 June 2025 Volume 2025:18 Pages 7357—7367
DOI https://doi.org/10.2147/JIR.S522081
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Yinghong Chen,1 Hanrong Hu,1 Congwei Wang,1 Junxuan Wu,1 Jie Zan,2 Yuntao Liu3
1Second Clinical Medical College, Guangzhou University of Chinese Medicine, Guangzhou, People’s Republic of China; 2School of Biomedical and Pharmaceutical Sciences, Guangdong University of Technology, Guangzhou, People’s Republic of China; 3State Key Laboratory of Traditional Chinese Medicine Syndrome, The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, People’s Republic of China
Correspondence: Yuntao Liu, Email [email protected] Jie Zan, Email [email protected]
Abstract: Lactate, traditionally viewed as a metabolic byproduct, is now recognized as a key regulator of immune and epigenetic processes in sepsis. A recently discovered post-translational modification, lactylation, utilizes lactate as a substrate and plays a crucial role in cellular regulation. Accumulating evidence suggests that elevated lactate levels contribute to immune dysfunction in sepsis by modulating the activity of various immune cells. This modification links metabolic changes to immune regulation, making it a crucial factor in sepsis progression. Understanding how lactylation is altered in sepsis unveils critical links between immunometabolism, epigenetic regulation, and disease pathophysiology. These insights also highlight the interplay between metabolic and epigenetic reprogramming during septic progression. As a result, lactylation has emerged as a promising biomarker and potential therapeutic target in sepsis. This review aims to summarize the latest findings on lactate metabolism, lactylation modifications, and their immunometabolic implications in sepsis.
Keywords: sepsis, lactate, histone, lactylation
Introduction
A dysregulated response to an infection can result in sepsis, which can induce organ dysfunction that is potentially fatal.1 With high rates of morbidity and mortality, it remains a major worldwide health concern.2 Up to now, no specific therapy for sepsis has yet been found. At present, sepsis is generally treated by anti-infection drugs, fluid resuscitation, mechanical ventilation, and other programs. One of the key metabolic alterations in sepsis is the accumulation of lactate, which serves not only as a metabolic byproduct but also as an important signaling molecule influencing immune responses. Clinically, numerous well-established routine laboratory tests can assess organ dysfunction in sepsis patients. However, the blood lactate level is the most widely utilized biomarker in indicating organ dysfunction. A study involving 86 patients diagnosed with sepsis and admitted to the ICU revealed that hyperlactatemia is prevalent among sepsis patients. However, there was no significant statistical difference in lactate levels between the deceased and survivor groups.3 Another study focusing on predicting mortality in pediatric patients with hospital-acquired infections, primarily within the context of sepsis, indicated that the lactate/albumin ratio can serve as an effective predictive indicator, with its predictive value being optimal in samples taken within 24 hours.4 Medical practitioners can guide resuscitation efforts in sepsis cases and assess the prognosis for patients with this potentially fatal illness by regularly monitoring blood lactate concentrations.5
Although lactate has historically been viewed as a metabolic waste product, recent research indicates that it also plays a regulatory role in immune function and inflammation. In sepsis, lactate accumulation can contribute to both metabolic adaptation and immune dysfunction, making it a critical factor in disease progression. Lactylation is a vital component of lactate function. Lactylation, a recently discovered post-translational modification, directly links metabolic changes to gene regulation. However, its role in sepsis remains underexplored. Understanding how lactylation affects immune response and metabolic reprogramming in sepsis could provide novel insights into disease pathophysiology and potential therapeutic targets.
Lactate Metabolism
Since lactate’s discovery in 1780, there has been a common misconception that it is a metabolic waste product that has several negative effects and is connected to low oxygen levels.6 Lactate Shuttle Theory, which describes the entire process of lactate transmembrane migration within cells,7,8 clarifies the function of lactate in the transport of oxidative and gluconeogenic substrates as well as cellular signaling. In 2019, Zhang et al made a groundbreaking discovery by demonstrating a novel epigenetic modification known as histone lysine lactylation. Furthermore, they showed that lactate might control protein expression and directly stimulate the synthesis of histone lactylation.9–12 Thus, the study of lactylation provides a new perspective for understanding the role of lactate and its role in the pathophysiology of sepsis, cancer, and so on. Increasing evidence suggests that, rather than just a waste product of anaerobic metabolism, lactate also plays a crucial role as a carbon source, signaling molecule, and immune modulator.
Lactate, as a classical byproduct of glucose metabolism, is mainly produced by glycolysis. Once activated, the glycolysis pathway compensates for adenosine triphosphate (ATP) production during hypoxia, which inhibits the tricarboxylic acid (TCA) cycle. In particular, a series of well-known enzymatic processes convert glucose in the cytoplasm to pyruvate; lactate dehydrogenase (LDH) then reduces pyruvate to lactate without it entering mitochondria for oxidation.13 It should be highlighted that cancer cells use the Warburg effect, also known as aerobic glycolysis, to create lactate and ATP even when they are operating in fully aerobic conditions.14,15 This phenomenon is observed in both tumor cells and highly proliferative lymphocytes. Moreover, research has demonstrated that sepsis and other critical conditions cause a switch in glucose metabolism from oxidative phosphorylation to aerobic glycolysis.16 Interestingly, an excess of lactate in the serum can lead to potentially fatal lactic acidosis, causing a more dangerous result than other molecular fuels do.17 In sepsis patients, lactate levels are normally expected to increase, as reduced tissue perfusion results in hypoxic organs, which then resort to anaerobic glycolysis. Additionally, lactate levels may rise for other reasons in sepsis. Even without compromised tissue perfusion, red blood cells (which lack mitochondria) and some tissues with high glycolysis rates constantly produce lactate. The liver converts lots of lactate back into glucose and oxidizes the rest. Thus, reduced lactate clearance may result from hepatic dysfunction associated with sepsis.18 Systemic inflammation also accelerates anaerobic glycolysis, owing to that the increased rate of glucose metabolism often exceeds the oxidative capacity of mitochondria. Finally, it appears that sepsis patients’ tissues have mitochondrial malfunction, which raises lactic acid levels, through an unidentified mechanism.19 As shown in Figure 1, lactate metabolism involves both mitochondrial and cytoplasmic pathways, which are altered during sepsis-induced metabolic reprogramming. Beyond its metabolic role, lactate also acts as an immune modulator, affecting both innate and adaptive immunity. Understanding these mechanisms is crucial for exploring its role in sepsis.
 |
Figure 1 Lactate metabolism and lactylation in cells. Lactate is produced from the decomposition of glycolysis in the cytoplasm. In one pathway, pyruvate, produced during glycolysis, enters the mitochondria and is metabolized through the tricarboxylic acid cycle. In the other pathway, pyruvate produced by glycolysis is converted to lactate by the action of lactate dehydrogenase. Lactate is also the precursor of gluconeogenesis. Histones and nonhistone proteins are lactylated by lactyl-CoA, which is derived from lactate. In septic conditions, an increase in lactate is associated with mitochondrial dysfunction, systemic inflammation, decreased tissue perfusion, and liver dysfunction. (by figdraw.com). Abbreviations: HK2, hexokinase; PFK1, phosphofructokinase-1; TPI, triose-phosphate isomerase; PYK, pyruvate kinase; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase; TCA Cycle, tricarboxylic acid cycle. |
Lactate Modulates the Immune Response
Sepsis is a life-threatening syndrome caused by an abnormal infection-induced immune response. The host’s immune response progresses from an initial excessive inflammatory response against pathogenic factors to immunosuppression in the course of sepsis. In some sepsis patients who survive the initial hyperinflammatory phase, they become vulnerable to ‘secondary infections’ due to the impairment of immune cells’ ability to respond adequately to inflammatory stimuli. This predisposition can lead to fatal outcomes resulting from uncontrolled infections caused by immunosuppression. Immunosuppression can cause reinfection and organ overload in sepsis patients, and the extent of immunosuppression is in direct proportion to the mortality of sepsis patients. It is clear that immunosuppression is strongly linked to poor sepsis outcomes and that immunological dysfunction is a major factor in the development of sepsis.
Lactate and Innate Immunity
Critical illness such as sepsis usually leads to the shift from mitochondrial oxidative phosphorylation to aerobic glycolysis, indicating lactate production, multiple organ dysfunction, and poor outcomes. As a result of sepsis, the immune system undergoes metabolic reprogramming, triggering both hyperinflammation and immunosuppression and therefore disrupting innate and adaptive immune response. Generally, the activation of immune cells relies on aerobic glycolytic metabolism, which leads to an increased production of lactate. Lactate produced during aerobic glycolysis, however, may have an immunosuppressive effect on sepsis patients, thereby modifying their immune response.20
The immune dysregulation during sepsis affects the function of the innate immune system. Early in sepsis, activated innate immune cells stimulate innate immune and inflammatory responses against invading pathogens. Inadequate management of the early response can result in heightened innate immune responses and inflammatory reactions, which can seriously jeopardize organ function21,22 and ultimately raise the risk of sepsis.23
Lactate has the potential immunomodulatory effects on innate immune cells, mainly on macrophages and dendritic cells (DCs). In the adaptive response, these cells serve as antigen-presenting cells and gatekeepers for lymphocyte B and T cell activation. One of the main characteristics of protracted sepsis is the improper development of immune tolerance to infections.24,25 Notably, the immunomodulatory function of lactate is closely associated with its modulation of dendritic cell phenotypic polarization. As illustrated in Figure 2A, immature dendritic cells differentiate into tolerogenic dendritic cells under the influence of an immunosuppressive microenvironment characterized by high lactate levels. This process re-establishes immune tolerance homeostasis through the secretion of inhibitory cytokines such as IL-10 and TGF-β, which promote the differentiation of initial T cells into regulatory T cells.26 Considerable metabolic dysfunction in these cells may be the reason for this change.27,28 Toll-like receptors (TLRs), which are critical in the induction of innate immune and inflammatory responses, can interact with their ligands, comprising bacterial components and endogenous ligands. This binding starts a chain reaction of cellular signaling cascades that eventually alter the transcriptional control of the cell.29,30 As illustrated in Figure 2B, lipopolysaccharide (LPS) activates the MyD88-dependent signaling pathway by specifically binding to Toll-like receptor 4 (TLR4) on the surfaces of DCs and macrophages. This interaction triggers a phosphorylation cascade involving TAK1, ultimately resulting in the nuclear translocation of NF-κB (p50/p65) mediated by the IKK complex (IκB kinase), which drives the secretion of pro-inflammatory cytokines. This inflammatory response is further accompanied by significant metabolic reprogramming, characterized by a transition from oxidative phosphorylation to aerobic glycolysis.31–33 Macrophage polarization is an important factor in the development and course of inflammation-related illnesses, which include sepsis and acute injuries, among other things.34,35 The progression of different diseases in pathological situations might be greatly aided by the shift in macrophage phenotype towards either M1 or M2 polarization.36,37 Research is beginning to show that lactate is mainly responsible for polarizing macrophages.38–40 It has been determined that GPR81, a member of the G protein-coupled receptor (GPCR) family, is a lactate receptor.41 Interestingly, lactate causes macrophages to change from the M1 to the M2 phenotype via GPR81 in a sepsis animal model39,42–44 (Figure 2C). Studies have shown that lactate also may polarize macrophages into M2-like phenotypes through epigenetic mechanisms, such as histone lysine lactic acid and acetylation9 (Figure 2D). Macrophages mostly take on an immunosuppressive M2 phenotype during the terminal phase of sepsis, which may be crucial in the emergence of immunological dysfunction.45,46 Excessive activation of the M2 phenotype can lead to immune suppression and aggravation of sepsis.
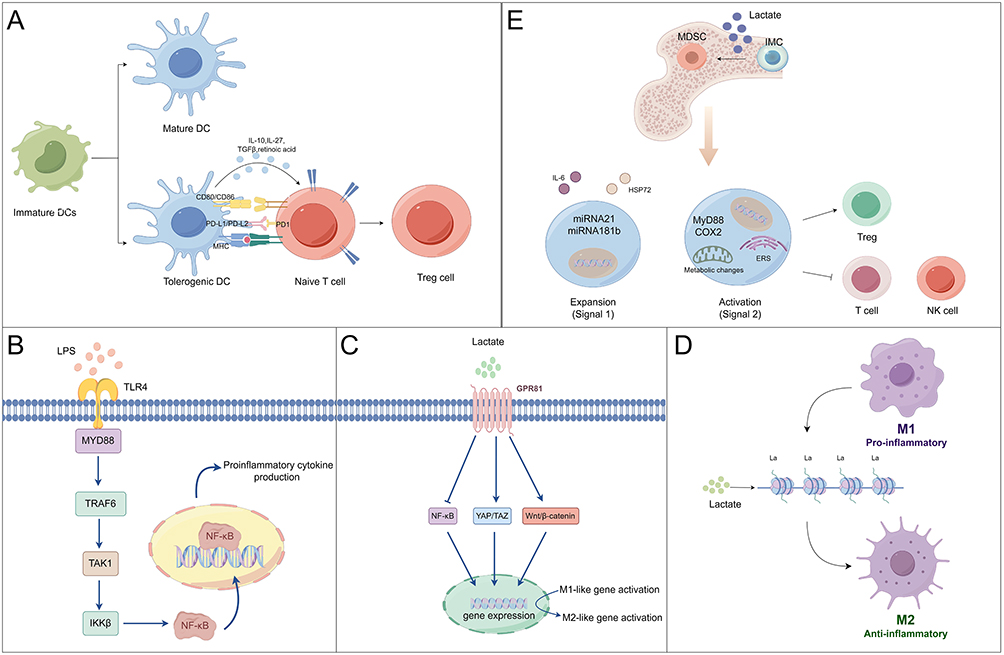 |
Figure 2 Potential mechanisms by which lactate modulates the innate immune responses. (A) Immature dendritic cells differentiate into a tolerogenic phenotype in high lactate environments, activating inhibitory signaling pathways such as TGF-β/Smad3 through the secretion of IL-10 and TGF-β. This process drives the differentiation of naive T cells into regulatory T cells. (B) LPS activates the MyD88-dependent signaling pathway by binding to TLR4 on the surface of dendritic cells and macrophages, triggering the TAK1 phosphorylation cascade that ultimately induces the nuclear translocation of NF-κB (p50/p65) mediated by the IKK complex. This translocation drives the secretion of pro-inflammatory cytokines, including TNF-α and IL-6. (C) Lactate binds to the GPR81 receptor on the surface of macrophages, activating downstream signaling pathways that inhibit the expression of M1-type pro-inflammatory genes while inducing the transcription of M2-type repair genes. (D) Lactate induces histone lysine lactylation and acetylation modifications, reshaping chromatin openness and promoting the transcriptional activation of M2-type repair genes. (E) A high-lactate environment induces the differentiation of bone marrow stem cells into MDSCs, significantly promoting their proliferation and activation. Activated MDSCs inhibit the immune functions of T cells and NK cells while inducing the expansion of regulatory T cells, thereby enhancing immune suppression. (by figdraw.com). |
Studies have reported that lactate affects the maturation process of bone marrow stem cells. Lactate added to the growing media significantly increases the production of myeloid-derived suppressor cells (MDSCs), a diverse group of cells that mostly perform immunosuppressive tasks, before bone marrow stem cell differentiation is induced.47 The presence of abnormal signaling pathways contributes to the extensive proliferation and activation of MDSCs, which then hinder the regular operation of immune cells through intricate molecular mechanisms (Figure 2E). The underlying processes of persistent immunological dysfunction have been associated with elevated levels of MDSCs in sepsis.48,49 In sepsis, elevated lactate levels may exacerbate the immunosuppressive state by promoting the proliferation and activation of MDSCs, which contribute to secondary organ damage.
Lactate and Adaptive Immunity
Apart from the influence on the innate immune system, lactate is also able to modulate adaptive immune responses. In the inflammatory response, all T cells dramatically increase energy production following activation due to rapid proliferation and the enhanced demand for the biosynthesis of nucleic acids, lipids, and proteins. Aerobic glycolysis is a major source of energy for pro-inflammatory CD4+ effector T cells, such as Th1 and Th17 lymphocytes, which results in increased lactate generation.50 Given that metabolic alterations have a major impact on T cell cytokine output, this indicates a strong relationship between metabolic activity and T cell effector function.51,52
Lactate accumulation at sites of inflammation is likely to have an extensive impact on T cell function, but the exact influences of lactate on T cells are still under debate. According to recent studies, effector molecules in T cells can be modulated by lactate to produce either immunostimulatory or immunosuppressive effects.53,54 Lactate builds up as a result of metabolic reprogramming, which promotes effector T cell activation.55 Furthermore, as illustrated in Figure 3, activated T cells compete for nutrient consumption, which affects the glucose utilization by DCs. This competition leads to the inactivation of glucose-sensitive signaling pathways in DCs, resulting in increased DC synthesis. This process is accompanied by functional divergence of DCs and polarization towards pro-inflammatory phenotypes, which directly drives the inflammatory cascade through the release of inflammatory mediators and the disruption of immune tolerance.56 It has been shown that lactate inhibits the formation of glucose-derived serine and lowers the NAD+/NADH ratio, which in turn prevents effector T cell proliferation.57 It has been demonstrated that the accumulation of lactate from glycolysis triggered by the inflammatory response reduces the viability of activated T cells and controls their effector activities.58,59 Moreover, lactate inhibits glycolysis, a critical step in T cell migration, which in turn affects the motility of CD4+ and CD8+ T cells.60 Moreover, contradictory effects of lactate on the activity and cytokine production of various T-cell subsets have been reported. Lactate has been demonstrated to cause a functional change in CD4+ T cells in favor of the pro-inflammatory Th17 fraction.61,62 On the other hand, a different study found that lactate increases the activity of Foxp3-expressing Tregs while decreasing the functioning of effector T cells.53 Lactate, however, also aids in the differentiation and functionality of cytotoxic CD8+ T cells.63 Moreover, it enhances T cell plasticity by rerouting pro-inflammatory Th17 cells’ transcriptional program to produce a Foxp3-expressing T cell phenotype that is regulatory in nature. This implies that Th17 cell-driven inflammation and autoimmunity may be suppressed by extracellular lactate produced at inflammatory sites.64 As illustrated in Figure 4, lactate plays a dual role in immune regulation: it acts both as a metabolic byproduct that inhibits effector T cell function and through epigenetic modifications that reshape T cell fate. This metabolic-immune interaction is especially critical in the contexts of sepsis, the tumor microenvironment, and autoimmune diseases.
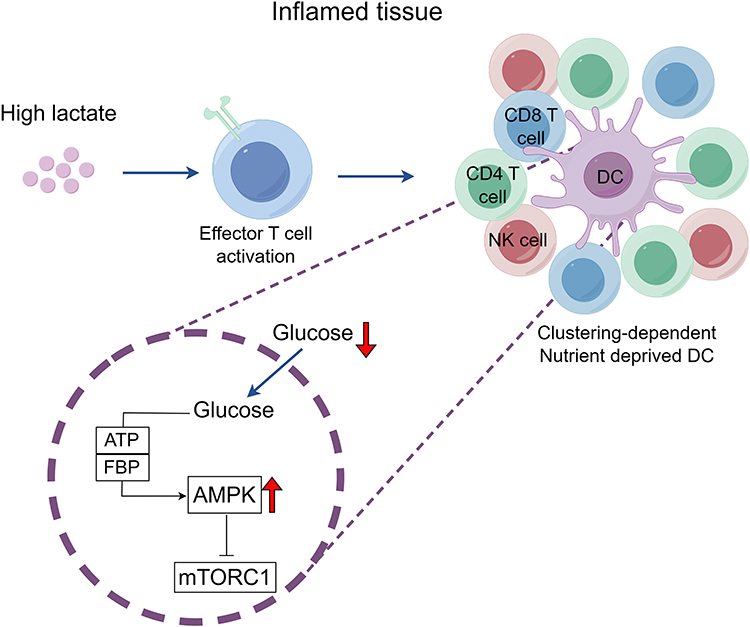 |
Figure 3 Antigen-presenting dendritic cells (DCs) are located at the center of cell clusters of activated immune cells. In an inflammatory environment, lactate increases and activates effector T cells, thus increasing the competition between immune cells for nutrients. The scarcity of glucose triggers the activation of AMP-activated protein kinase (AMPK), stemming from diminished concentrations of ATP and fructose-1,6-bisphosphate (FBP). This activation, in turn, represses the mTORC1 signaling cascade and elevates the inflammatory response produced by DCs. (by figdraw.com). |
 |
Figure 4 Nicotinamide adenine dinucleotide (NAD+) is reduced to NADH with the assistance of lactate dehydrogenase. NADH is not available for NAD+-dependent enzymatic reactions involving glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and 3-phosphoglycerate dehydrogenase (PGDH). The increase in lactate causes both the GAPDH and PGDH arrest and the depletion of the 3-phosphoglycerate-derived serine, which is known to be important for T cell proliferation. Lactate can impair T cell proliferation through reductive stress. (by figdraw.com). |
Notably, lactate not only directly influences immune cell function through metabolic reprogramming but also indirectly modulates immune responses via epigenetic modifications, such as lactylation. This dual regulatory mechanism may play a crucial role in the immune imbalance observed in sepsis, offering new insights into its pathological processes.
Lactylation Modification and Sepsis
Lactylation modification, the newly discovered epigenetic modification induced by lactate, can regulate gene expression and signaling during inflammatory diseases such as sepsis. Lactylation modification links cellular metabolism to gene regulation and is closely related to the regulation of inflammatory response, immune regulation, and other processes. Lactylation modification can occur widely on histones and non-histone proteins and participate in many key cellular biological processes, revealing important mechanisms of action in the development of sepsis.
Histone Lactylation Modification
Lactate is an epigenetic regulator that can control the expression of important genes through epigenetic modification of histone lactylation, as demonstrated by recent research.9 Epigenetic regulation refers to the regulatory mechanism that controls gene expression but is not related to DNA sequence alteration, mainly including histone modifications, DNA methylation, and regulation of transcription by non-coding RNA, which alters gene expression in response to external stress. In eukaryotic cells, the basic unit of chromosomes is the nucleosome. The nucleosome core is an octamer of histones, consisting of dimers of histones H2A, H2B, H3, and H4. The DNA double helices are wrapped around the nucleosome. The structure of the histones includes a globular domain and an N-terminal tail. The globular domains are involved in histone-DNA interactions, and the N-terminal tail is affected by post-translational modifications (PTMs). PTMs of histones are performed by proteases known as “writers”, “erasers”, and “readers”. Histones can be acetylated, methylated, phosphorylated, citrullinated, etc., and these changes change how they interact with their DNA, regulating the chromatin’s opening and compaction to allow or suppress gene expression.65,66
The process of histone lactylation emerges as a pivotal regulator in the expression of genes within macrophages and other immune cells, thereby implying a profound association between this epigenetic modification and the pathogenesis of inflammatory diseases. Histone lactylation, a crucial mechanism of metabolic-epigenetic regulation, plays a pivotal role in the bidirectional modulation of immune responses during sepsis. When inflammation arises, macrophages recognize the presence of pathogen-associated molecular patterns (PAMPs) in bacteria through their surface-located pathogen recognition receptors, such as Toll-like receptors. This recognition promptly triggers the activation of M1 macrophages, also known as the pro-inflammatory phenotype. These activated M1 macrophages produce a substantial amount of pro-inflammatory mediators, leading to the expression of inflammatory genes like NOS2 and the generation of Warburg effectors. In the initial phases of inflammation, these processes play a critical role in the eradication of invasive infections and the activation of the adaptive immune response.67,68 As inflammation advances, sepsis and systemic inflammatory response syndrome (SIRS) can develop due to the uncontrolled activation of M1-type macrophages.69 However, macrophages must polarize into the M2 phenotype (an anti-inflammatory phenotype) later on, or after inflammation has been brought under control, to counteract the excessive inflammatory response and aid in the healing of collateral damage that results from inflammation. Lactification modification on the core histones occurs during this phase; this process is enriched in the promoter regions of steady-state genes within the chromatin structure. This modification directly stimulates chromatin gene transcription, resulting in the expression of steady-state genes, such as arginase 1 (Arg 1), which are involved in wound healing. Macrophages undergo this shift, which causes them to change from the M1 phenotype to the M2 phenotype.9 These macrophages secrete a multitude of anti-inflammatory chemicals when in the M2 state, which protects the host from excessive tissue damage and promotes wound healing.70 Nonetheless, there are times when macrophages experience an excessive amount of apoptosis, which may aid in the emergence of immunological suppression.71 It is noteworthy that the dynamic imbalance of lactylation during sepsis can result in the concurrent occurrence of both cytokine storms and immunosuppression.
Lactylation modification is time-dependent. Histone lactylation increases during M1 macrophage polarization (within 24 h) in a closely time-dependent pattern, ie, the increase in histone lactylation at gene promoters is delayed after M1 macrophage stimulation. Within 16 to 24 hours, the particular H3K18la gene (lactylation on histone H3’s 23rd lysine residue) is active or reactivated, causing the expression of M2-like genes involved in the process of damage repair, such as Arg 1, which causes M1 macrophages to express M2. A “lactate timer” mechanism is comparable to this delayed temporal kinetic effect.9 This “lactate timer” in macrophages initiates the expression of homeostatic M2-like genes in an environment rich in histone Kla. As a result, it contributes to the eventual repair of collateral damage sustained by the host during infection. Additionally, the B cell adapter for phosphoinositide 3-kinase (BCAP) plays a role in promoting the expression of damage repair genes, such as Arg 1, in macrophages, by regulating glycolysis and histone lactate modification. Irizarry-Caro et al72 found that lactate production was impaired after macrophages lacked the BCAP gene, resulting in slow repair transformation. In sepsis, a dynamic imbalance in lactylation can lead to immune dysregulation. Insufficient early modification may impair pathogen clearance, while delayed late modification hinders the resolution of inflammation. Collectively, these factors exacerbate immunosuppression and contribute to organ damage.
The research stated above shows that histone lactylation has a beneficial effect on M2-like gene expression in macrophages, which is useful during the hyperimmune inflammatory phase. This process facilitates tissue repair and helps mitigate the spread of inflammation. However, in certain inflammatory diseases, such as sepsis, the anti-inflammatory effect of histone lactylation can be exacerbated by the presence of an immunosuppressive phase. Therefore, modulating the polarization state of macrophages by regulating histone lactylation levels at various inflammatory stages holds significant promise for improving the prognosis of inflammatory diseases. While several studies suggest that lactylation promotes M2 polarization, others indicate that its effects are context-dependent, necessitating further investigation. In a recent study, Susser et al73 emphasized the pivotal role of lactate modifications in regulating macrophage polarization. They found that increased lactate levels caused by mitochondrial fragmentation stimulate the expression of genes including Arg 1. Conversely, Dichtl et al74 challenged this view, arguing that lysine lactylation modifications and lactate do not typically correlate with macrophage activation or the expression of genes linked to tissue repair, including Arg 1. The current understanding of how histone lactylation affects the expression of macrophage homeostatic genes remains fragmented and requires further clarification. Additional studies are required to clarify these controversial findings.
The findings of Chu et al75 provide important insights into the prevalence and biological significance of lactylation. Their clinical study indicates that lactylation is not only present in histones but also prevalent in vivo proteins of both healthy individuals and those with sepsis. The significant differences observed between healthy and septic subjects suggest that lactylation may play a role in the pathogenesis of sepsis or related diseases. Patients with septic shock had the highest levels of H3K18la expression compared to non-septic shock patients, non-shocked critically ill patients, and healthy volunteers. H3K18la protein expression was positively correlated with serum lactate, levels of inflammatory molecules, and levels of transcription of the anti-inflammatory gene Arg 1. Therefore, lactated histone H3K18la can be used as a potential biomarker for the diagnosis and prediction of the severity of septic shock, which provides clues for further research on the application of histone lactylation in sepsis. Despite its potential as a diagnostic biomarker, the clinical application of lactylation is still in its infancy. Future studies must address challenges related to detection sensitivity, patient variability, and validation in large cohorts.
Non-Histone Lactylation Modification
Lactylation modifications are widely distributed in all types of proteins and are particularly highly enriched in energy metabolism pathways. Lactylation modifications are widely applicable, as they affect not just histones but also non-histone proteins, indicating the ubiquity of this modification process.76 High mobility group box protein 1 (HMGB1), which is released upon macrophage activation, participates in the inflammatory reaction.77 According to Yang et al,78 during sepsis, macrophages can take up extracellular lactate via a monocarboxylate transporter (MCT). This mechanism, which is dependent on p300/CBP, is shown to promote the lactylation and acetylation of the HMGB1 protein in the polymicrobial sepsis mouse model. Lactated or acetylated modified forms of HMGB1 are released from macrophages through the secretion of exosomes. This release process facilitates the onset and progression of sepsis by impairing the function of the endothelial cell barrier.78 It was discovered that in sepsis patients with sepsis, serum lactic acid and HMGB1 correlated positively.78,79 In conclusion, high blood lactate concentrations in sepsis patients can affect macrophage function through lactylation modifications of histones and non-histone proteins.
Conclusion and Prospect
Although the study of lactylation in sepsis is still in its early stages, it has demonstrated significant clinical value as a key epigenetic mechanism linking immune regulation and lactate metabolism. The discovery of lactylation modification not only provides a novel molecular explanation for lactate-mediated immune dysregulation in sepsis but also reveals a profound connection between the metabolic, epigenetic, and immune regulatory networks. This paper systematically summarizes recent advances in the study of lactate-mediated immune dysfunction in sepsis and explores the complex interplay among metabolism, epigenetic regulation, and immune responses. However, several knowledge gaps still exist in this field, such as the relationship between histone lactylation modification and the expression of the Arg1 gene. Current studies have not fully elucidated this interaction, and these unresolved issues underscore the need for further investigation. Elucidating the molecular mechanisms and functional phenotypes of lactylation modification will fundamentally expand our understanding of the pathophysiology of sepsis and lay the groundwork for developing precise therapeutic strategies targeting the metabolic-epigenetic pathway. Systematic multi-omics studies and rigorously designed clinical trials are urgently needed to validate the feasibility of lactylation modification as diagnostic markers and therapeutic targets for sepsis. These efforts are expected not only to overcome the current bottlenecks in sepsis treatment but also to establish a new paradigm for critical care centered on metabolic reprogramming and epigenetic regulation.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from the National Natural Science Foundation of China (82004317), and the Science and Technology Planning Project of Guangdong Province (2023B1212060062).
Disclosure
The authors declare no conflicts of interest.
References
1. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801.
2. Joshi M, Ashrafian H, Arora S, et al. Digital alerting and outcomes in patients with sepsis: systematic review and meta-analysis. J Med Int Res. 2019;21(12):e15166–e15166. doi:10.2196/15166
3. Keskin A, Aci R. Procalcitonin to albumin ratio as a biomarker for predicting mortality in sepsis. J Coll Physicians Surg Pak. 2024;34(3):360–363.
4. Arı HF, Keskin A, Arı M, Aci R. Importance of lactate/albumin ratio in pediatric nosocomial infection and mortality at different times. Future Microbiol. 2024;19(1):51–59. doi:10.2217/fmb-2023-0125
5. Lehman KD. Update: surviving sepsis campaign recommends hour-1 bundle use. Nurse Pract. 2019;44(4):10. doi:10.1097/01.NPR.0000554123.08252.ae
6. Ferguson BS, Rogatzki MJ, Goodwin ML, et al. Lactate metabolism: historical context, prior misinterpretations, and current understanding. Eur J Appl Physiol. 2018;118(4):691–728. doi:10.1007/s00421-017-3795-6
7. Brooks GA. The science and translation of lactate shuttle theory. Cell Metab. 2018;27(4):757–785. doi:10.1016/j.cmet.2018.03.008
8. Brooks GA. Lactate shuttles in nature. Biochem Soc Trans. 2002;30(1):A10–A10. doi:10.1042/bst030a010
9. Zhang D, Tang Z, Huang H, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–580. doi:10.1038/s41586-019-1678-1
10. Du S, Zhang X, Jia Y, et al. Hepatocyte HSPA12A inhibits macrophage chemotaxis and activation to attenuate liver ischemia/reperfusion injury via suppressing glycolysis-mediated HMGB1 lactylation and secretion of hepatocytes. Theranostics. 2023;13(11):3856–3871. doi:10.7150/thno.82607
11. Wang Y, Chen L, Zhang M, et al. Exercise-induced endothelial Mecp2 lactylation suppresses atherosclerosis via the Ereg/MAPK signalling pathway. Atherosclerosis. 2023;375:45–58. doi:10.1016/j.atherosclerosis.2023.05.009
12. Jia M, Yue X, Sun W, et al. ULK1-mediated metabolic reprogramming regulates Vps34 lipid kinase activity by its lactylation. Sci Adv. 2023;9(22):eadg4993. doi:10.1126/sciadv.adg4993
13. Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;10(2):172. doi:10.1016/j.ccr.2006.07.011
14. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519–530. doi:10.1085/jgp.8.6.519
15. Sharma NK, Pal JK. Metabolic ink lactate modulates epigenomic landscape: a concerted role of pro-tumor microenvironment and macroenvironment during carcinogenesis. Curr Mol Med. 2021;21(3):177–181. doi:10.2174/1566524020666200521075252
16. Brooks GA. Lactate as a fulcrum of metabolism. Redox Biol. 2020;35:101454. doi:10.1016/j.redox.2020.101454
17. Bennis Y, Bodeau S, Batteux B, et al. A study of associations between plasma metformin concentration, lactic acidosis, and mortality in an emergency hospitalization context. Crit Care Med. 2020;48(12):e1194–e1202. doi:10.1097/CCM.0000000000004589
18. Toffaletti JG. Blood lactate: biochemistry, laboratory methods, and clinical interpretation. Crit Rev Clin Lab Sci. 1991;28(4):253–268. doi:10.3109/10408369109106865
19. Garrabou G, Morén C, López S, et al. The effects of sepsis on mitochondria. J Infect Dis. 2012;205(3):392–400. doi:10.1093/infdis/jir764
20. Nolt B, Tu F, Wang X, et al. Lactate and immunosuppression in sepsis. Shock. 2018;49(2):120–125. doi:10.1097/SHK.0000000000000958
21. Mira JC, Gentile LF, Mathias BJ, et al. Sepsis pathophysiology, chronic critical illness, and persistent inflammation-immunosuppression and catabolism syndrome. Crit Care Med. 2017;45(2):253–262. doi:10.1097/CCM.0000000000002074
22. Kakihana Y, Ito T, Nakahara M, Yamaguchi K, Yasuda T. Sepsis-induced myocardial dysfunction: pathophysiology and management. J Intensive Care. 2016;4(1):22. doi:10.1186/s40560-016-0148-1
23. Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369(9):840–851. doi:10.1056/NEJMra1208623
24. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13(12):862–874. doi:10.1038/nri3552
25. Pena OM, Hancock DG, Lyle NH, et al. An endotoxin tolerance signature predicts sepsis and organ dysfunction at initial clinical presentation. EBioMedicine. 2014;1(1):64–71. doi:10.1016/j.ebiom.2014.10.003
26. Fan X, Liu Z, Jin H, Yan J, Liang HP. Alterations of dendritic cells in sepsis: featured role in immunoparalysis. Biomed Res Int. 2015;2015:903720. doi:10.1155/2015/903720
27. Cheng SC, Scicluna BP, Arts RJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. 2016;17(4):406–413. doi:10.1038/ni.3398
28. Sim WJ, Ahl PJ, Connolly JE. Metabolism is central to tolerogenic dendritic cell function. Mediators Inflamm. 2016;2016:2636701. doi:10.1155/2016/2636701
29. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5(10):987–995. doi:10.1038/ni1112
30. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1(2):135–145. doi:10.1038/35100529
31. Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. 2011;11(11):738–749. doi:10.1038/nri3071
32. Rodríguez-Prados JC, Través PG, Cuenca J, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185(1):605–614. doi:10.4049/jimmunol.0901698
33. Krawczyk CM, Holowka T, Sun J, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115(23):4742–4749. doi:10.1182/blood-2009-10-249540
34. Motwani MP, Gilroy DW. Macrophage development and polarization in chronic inflammation. Semin Immunol. 2015;27(4):257–266. doi:10.1016/j.smim.2015.07.002
35. Schultze JL, Schmieder A, Goerdt S. Macrophage activation in human diseases. Semin Immunol. 2015;27(4):249–256. doi:10.1016/j.smim.2015.07.003
36. Zhou D, Huang C, Lin Z, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. 2014;26(2):192–197. doi:10.1016/j.cellsig.2013.11.004
37. Sica A, Erreni M, Allavena P, et al. Macrophage polarization in pathology. Cell Mol Life Sci. 2015;72(21):4111–4126. doi:10.1007/s00018-015-1995-y
38. Zhang A, Xu Y, Xu H, et al. Lactate-induced M2 polarization of tumor-associated macrophages promotes the invasion of pituitary adenoma by secreting CCL17. Theranostics. 2021;11(8):3839–3852. doi:10.7150/thno.53749
39. Zhang J, Muri J, Fitzgerald G, et al. Endothelial lactate controls muscle regeneration from ischemia by inducing M2-like macrophage polarization. Cell Metab. 2020;31(6):1136–1153.e7. doi:10.1016/j.cmet.2020.05.004
40. Wang C, Xue L, Zhu W, Liu L, Zhang S, Luo D. Lactate from glycolysis regulates inflammatory macrophage polarization in breast cancer. Cancer Immunol Immunother. 2023;72(6):1917–1932. doi:10.1007/s00262-023-03382-x
41. Ranganathan P, Shanmugam A, Swafford D, et al. GPR81, a cell-surface receptor for lactate, regulates intestinal homeostasis and protects mice from experimental colitis. J Immunol. 2018;200(5):1781–1789. doi:10.4049/jimmunol.1700604
42. Yang K, Xu J, Fan M, et al. Lactate suppresses macrophage pro-inflammatory response to LPS stimulation by inhibition of YAP and NF-κB activation via GPR81-mediated signaling. Front Immunol. 2020;11:587913. doi:10.3389/fimmu.2020.587913
43. Wang L, He HW, Xing ZQ, Tang B, Zhou X. Lactate induces alternative polarization (M2) of macrophages under lipopolysaccharide stimulation in vitro through G-protein coupled receptor 81. Chin Med J. 2020;133(14):1761–1763. doi:10.1097/CM9.0000000000000955
44. Hoque R, Farooq A, Ghani A, Gorelick F, Mehal WZ. Lactate reduces liver and pancreatic injury in toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology. 2014;146(7):1763–1774. doi:10.1053/j.gastro.2014.03.014
45. Arts RJ, Gresnigt MS, Joosten LA, Netea MG. Cellular metabolism of myeloid cells in sepsis. J Leukoc Biol. 2017;101(1):151–164. doi:10.1189/jlb.4MR0216-066R
46. Pena OM, Pistolic J, Raj D, Fjell CD, Hancock RE. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J Immunol. 2011;186(12):7243–7254. doi:10.4049/jimmunol.1001952
47. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. 2013;191(3):1486–1495. doi:10.4049/jimmunol.1202702
48. Mathias B, Delmas AL, Ozrazgat-Baslanti T, et al. Human myeloid-derived suppressor cells are associated with chronic immune suppression after severe sepsis/septic shock. Ann Surg. 2017;265(4):827–834. doi:10.1097/SLA.0000000000001783
49. Janols H, Bergenfelz C, Allaoui R, et al. A high frequency of MDSCs in sepsis patients, with the granulocytic subtype dominating in gram-positive cases. J Leukoc Biol. 2014;96(5):685–693. doi:10.1189/jlb.5HI0214-074R
50. Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212(9):1345–1360. doi:10.1084/jem.20151159
51. Chapman NM, Boothby MR, Chi H. Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol. 2020;20(1):55–70. doi:10.1038/s41577-019-0203-y
52. Shyer JA, Flavell RA, Bailis W. Metabolic signaling in T cells. Cell Res. 2020;30(8):649–659. doi:10.1038/s41422-020-0379-5
53. Angelin A, Gil-de-Gómez L, Dahiya S, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 2017;25(6):1282–1293.e7. doi:10.1016/j.cmet.2016.12.018
54. Pucino V, Certo M, Bulusu V, et al. Lactate buildup at the site of chronic inflammation promotes disease by inducing CD4+ T cell metabolic rewiring. Cell Metab. 2019;30(6):1055–1074.e8. doi:10.1016/j.cmet.2019.10.004
55. Kedia-Mehta N, Finlay DK. Competition for nutrients and its role in controlling immune responses. Nat Commun. 2019;10(1):2123. doi:10.1038/s41467-019-10015-4
56. Peng X, He Y, Huang J, Tao Y, Liu S. Metabolism of dendritic cells in tumor microenvironment: for immunotherapy. Front Immunol. 2021;12:613492. doi:10.3389/fimmu.2021.613492
57. Quinn WJ, Jiao J, TeSlaa T, et al. Lactate limits T cell proliferation via the NAD(H) Redox State. Cell Rep. 2020;33(11):108500. doi:10.1016/j.celrep.2020.108500
58. Lei J, Yang Y, Lu Z, et al. Taming metabolic competition via glycolysis inhibition for safe and potent tumor immunotherapy. Biochem Pharmacol. 2022;202:115153. doi:10.1016/j.bcp.2022.115153
59. Gu J, Zhou J, Chen Q, et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-β signaling in regulatory T cells. Cell Rep. 2022;39(12):110986. doi:10.1016/j.celrep.2022.110986
60. Haas R, Smith J, Rocher-Ros V, et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PLoS Biol. 2015;13(7):e1002202. doi:10.1371/journal.pbio.1002202
61. Certo M, Tsai CH, Pucino V, Ho PC, Mauro C. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat Rev Immunol. 2021;21(3):151–161. doi:10.1038/s41577-020-0406-2
62. Pucino V, Certo M, Varricchi G, et al. Metabolic checkpoints in rheumatoid arthritis. Front Physiol. 2020;11:347. doi:10.3389/fphys.2020.00347
63. Rundqvist H, Veliça P, Barbieri L, et al. Cytotoxic T-cells mediate exercise-induced reductions in tumor growth. Elife. 2020;9:e59996. doi:10.7554/eLife.59996
64. Lopez Krol A, Nehring HP, Krause FF, et al. Lactate induces metabolic and epigenetic reprogramming of pro-inflammatory Th17 cells. EMBO Rep. 2022;23(12):e54685. doi:10.15252/embr.202254685
65. García-Giménez JL, Garcés C, Romá-Mateo C, Pallardó FV. Oxidative stress-mediated alterations in histone post-translational modifications. Free Radic Biol Med. 2021;170:6–18. doi:10.1016/j.freeradbiomed.2021.02.027
66. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi:10.1016/j.cell.2007.02.005
67. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi:10.1126/science.123.3191.309
68. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. doi:10.1172/JCI59643
69. Ma J, Wei K, Liu J, et al. Glycogen metabolism regulates macrophage-mediated acute inflammatory responses. Nat Commun. 2020;11(1):1769. doi:10.1038/s41467-020-15636-8
70. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi:10.1038/nri3073
71. Luan YY, Yao YM, Xiao XZ, Sheng ZY. Insights into the apoptotic death of immune cells in sepsis. J Interferon Cytokine Res. 2015;35(1):17–22. doi:10.1089/jir.2014.0069
72. Irizarry-Caro RA, McDaniel MM, Overcast GR, Jain VG, Troutman TD, Pasare C. TLR signaling adapter BCAP regulates inflammatory to reparatory macrophage transition by promoting histone lactylation. Proc Natl Acad Sci U S A. 2020;117(48):30628–30638. doi:10.1073/pnas.2009778117
73. Susser LI, Nguyen MA, Geoffrion M, et al. Mitochondrial fragmentation promotes inflammation resolution responses in macrophages via histone lactylation. Mol Cell Biol. 2023;43(10):531–546. doi:10.1080/10985549.2023.2253131
74. Dichtl S, Lindenthal L, Zeitler L, et al. Lactate and IL6 define separable paths of inflammatory metabolic adaptation. Sci Adv. 2021;7(26):eabg3505. doi:10.1126/sciadv.abg3505
75. Chu X, Di C, Chang P, et al. Lactylated histone H3K18 as a potential biomarker for the diagnosis and predicting the severity of septic shock. Front Immunol. 2022;12:786666. doi:10.3389/fimmu.2021.786666
76. Fan H, Yang F, Xiao Z, et al. Lactylation: novel epigenetic regulatory and therapeutic opportunities. Am J Physiol Endocrinol Metab. 2023;324(4):E330–E338. doi:10.1152/ajpendo.00159.2022
77. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29(1):139–162. doi:10.1146/annurev-immunol-030409-101323
78. Yang K, Fan M, Wang X, et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022;29(1):133–146. doi:10.1038/s41418-021-00841-9
79. Charoensup J, Sermswan RW, Paeyao A, et al. High HMGB1 level is associated with poor outcome of septicemic melioidosis. Int J Infect Dis. 2014;28:111–116. doi:10.1016/j.ijid.2014.07.025
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.