")
Back to Journals » Journal of Inflammation Research » Volume 17
Estrogen Receptor β Alleviates Colitis by Inhibiting Ferroptosis in Intestinal Epithelial Cells
Authors Li J, Chen Y , Li S, Zhang X, Cheng Y, Fu X , Li J, Zhu L
Received 22 September 2024
Accepted for publication 15 November 2024
Published 10 December 2024 Volume 2024:17 Pages 10785—10805
DOI https://doi.org/10.2147/JIR.S492290
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Junrong Li,1,2,* Yidong Chen,1,* Shuang Li,1 Xiaopeng Zhang,1 Yiyu Cheng,1 Xiaoyu Fu,1 Jiamin Li,1 Liangru Zhu1
1Division of Gastroenterology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 2Division of Gastroenterology, Chongqing Hospital Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Chongqing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Liangru Zhu, Division of Gastroenterology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, No. 1277, Jiefang Avenue, Jianghan District, Wuhan, People’s Republic of China, Email [email protected]
Background: Ulcerative colitis (UC), a major type of inflammatory bowel disease, is characterized by chronic inflammation of the colonic mucosa and submucosa. Estrogen receptor β (ERβ) predominates in the colon and exerts anti-inflammatory effects. Ferroptosis, a recently discovered form of iron-dependent programmed cell death, is implicated in the pathogenesis of several diseases, including UC. However, the link between ferroptosis and the anti-inflammatory actions of ERβ in UC remains to be elucidated.
Methods: We analyzed colonic mucosal samples from inflammatory and non-inflammatory regions of UC patients to assess ferroptosis levels. Experimental colitis was induced in wild-type C57BL/6 mice and intestinal epithelial cell-specific ERβ knockout (ERβ−/−) mice using dextran sulfate sodium (DSS). We measured body weight, colon length, disease activity index (DAI), and histopathological scores. RNA sequencing was performed to identify differentially expressed genes and related signaling pathways, with additional ferroptosis assessment in vivo and in vitro through biochemical markers and cellular assays.
Results: In UC patients, ferroptosis was significantly elevated in inflammatory mucosal regions compared to non-inflammatory areas. Compared to the wild-type counterparts, ERβ−/− mice exacerbated DSS-induced experimental colitis, including reduced body weight, shortened colon length, and higher DAI scores. RNA sequencing showed enrichment of inflammatory and immune response pathways, with significant activation of JAK/STAT, NF-κB, and TNF signaling in ERβ−/− mice. ERβ deficiency induced ferroptosis in both in vitro and in vivo models. Ferroptosis indicators such as PTGS2 were upregulated, GPX4 expression was downregulated, and there were increases in malondialdehyde, iron content, reactive oxygen species, and mitochondrial damage.
Conclusion: Our findings demonstrate that ERβ deficiency exacerbates colitis and enhances ferroptosis in IECs. ERβ positively regulates GPX4 transcription, thereby inhibiting ferroptosis and alleviating colitis. These insights suggest that modulation of ERβ and its regulation of ferroptosis may represent a novel therapeutic strategy for UC.
Keywords: ulcerative colitis, estrogen receptor β, GPX4, ferroptosis
Introduction
Ulcerative colitis (UC), one of the principal types of inflammatory bowel disease (IBD), is a chronic and relapsing inflammatory disorder of the gastrointestinal tract.1 UC predominantly affects the mucosal and submucosal layers of the colon and rectum, starting at the rectum and extending proximally in a continuous fashion.2 To date, the total number of UC cases worldwide exceeds 5 million.3 Studies have shown that China is one of the countries with the highest incidence rates of IBD in Asia, with the incidence of UC being significantly higher than that of Crohn’s disease (CD), the other major type of IBD, with a ratio of 12.61.4 Despite ongoing research, the exact pathogenesis of UC remains incompletely understood; it is currently believed to involve a combination of genetic susceptibility, epithelial barrier dysfunction, dysregulation of immune responses and gut microbiome.5 Current treatments for UC include mesalamine, corticosteroids, immunosuppressants, and biologics.6 However, some patients do not respond adequately to these medications, experiencing poor symptom control or recurrent episodes of inflammation. Therefore, elucidating the pathogenesis of UC and identifying novel therapeutic targets is of utmost importance.
Intestinal epithelial cells (IECs) are a crucial component of the intestinal epithelium and play a central role in the pathogenesis of IBD.7,8 Beyond their digestive and absorptive functions, IECs serve as core regulators of intestinal immune homeostasis. Dysregulated death of IECs is a characteristic feature of UC, contributing to the disruption of the intestinal epithelial barrier and the perpetuation of inflammation, thus forming a vicious cycle that underlies the pathologic changes in UC.9,10
Cell death can be categorized into accidental cell death and programmed cell death (PCD), also known as regulated cell death. PCD is an autonomous and orderly form of cell death that can be modulated by various biological macromolecules. Ferroptosis is a recently discovered iron-dependent form of PCD that is distinct from other modes of cell death in terms of morphology, genetics, and biochemistry. Ferroptosis is initiated by the imbalance of cellular metabolism and redox homeostasis due to various factors (both endogenous and exogenous). Excessive iron ions and reactive oxygen species (ROS), along with polyunsaturated fatty acid-containing phospholipids, lead to uncontrolled lipid peroxidation, ultimately triggering ferroptosis.11 The regulation of ferroptosis is complex, with the glutathione/glutathione peroxidase 4 (GSH/GPX4) pathway playing a crucial role. GSH, a tripeptide composed of cysteine, glutamate, and glycine, is a major intracellular antioxidant.12 GSH serves as a key cofactor for GPX4, which facilitates the reaction between GPX4 and intracellular lipid peroxides, thereby suppressing ferroptosis.13 GPX4 is a selenoprotein and a member of the glutathione peroxidase family. Under physiological conditions, GPX4 uses GSH as a substrate to reduce cytotoxic lipid peroxides to non-toxic lipid alcohols. Inhibition of GPX4 leads to the accumulation of lipid peroxides and triggers ferroptosis.14,15 Prostaglandin-endoperoxide synthase 2 (PTGS2) is also associated with ferroptosis, commonly serving as a biomarker for this process due to its upregulation during lipid peroxidation and oxidative stress.16,17 Therefore, GPX4 acts as a primary regulator of ferroptosis, and factors that influence GPX4 expression or activity can modulate ferroptosis. Studies have shown that ferroptosis is involved in the pathogenesis of several diseases, including UC.18–20
Estrogens are lipophilic steroid hormones whose functions are primarily mediated by estrogen receptors (ERs), including ERα, ERβ, and the G protein-coupled estrogen receptor 1 (GPER1).21,22 ERβ is predominantly expressed in tissues such as the ovary, colon, kidney, cardiovascular system, and respiratory system.23 ERβ is the predominant subtype of ER in IECs, contributing to the maintenance of normal epithelial function and tissue architecture, and exerting anti-inflammatory effects.24,25 Studies have shown that compared to healthy controls, patients with UC exhibit significantly reduced expression levels of ERβ in peripheral blood T cells.4,26 ERβ has been proven to inhibit the JAK2/STAT3 signaling pathway in macrophages, reducing the release of interleukin-1β (IL-1β) and ameliorating experimental colitis.27 Activation of ERβ enhances the differentiation of regulatory T cells in CD4+ T cells and mitigates dextran sulfate sodium (DSS)-induced experimental colitis.26 However, the precise mechanisms by which ERβ exerts its anti-inflammatory effects in IECs remain unclear. Research has demonstrated that ERα and GPER1 regulate ferroptosis and participate in the pathogenesis of diseases such as osteoarthritis, hepatitis, breast cancer, and lung cancer.28–31 However, it remains unknown whether the anti-inflammatory effects of ERβ are associated with ferroptosis. Therefore, this study aims to explore whether ERβ participates in UC by regulating ferroptosis in IECs and to investigate the underlying mechanisms.
Materials and Methods
Chemicals and Reagents
DSS was purchased from MP Biomedicals (Solon, OH, USA). ERB041 was purchased from Tocris Biosciences (Bristol, UK). LPS was purchased from Sigma-Aldrich (St. Louis, MO). The siRNA was synthesized by GeneGene (Wuhan, China). The GAPDH primary antibody was purchased from AntGene Biotechnology (#ANT325, Wuhan, China). Primary antibodies against GPX4, PTGS2 were purchased from ABclonal Technology (#A1933, #A1253, Wuhan, China). ERβ primary antibody was obtained from GeneTex Biotechnology Company (#GTX70174, California, USA). Iron detection kits were purchased from Solebio (Beijing, China). Malondialdehyde (MDA) detection kits were obtained from Beyotime (Shanghai, China). Glutathione (GSH) detection kits were acquired from Jiancheng (Nanjing, China). Mitochondrial membrane potential assay kit with JC-1 were obtained from Yeasen (Shanghai, China). The chromatin immunoprecipitation (ChIP) assay kits were purchased from Beyotime (Shanghai, China). Dual-luciferase reporter assay kit was purchased from Promega (Madison, WI, USA). pRL Renilla luciferase control reporter vectors and PGL4.11 vectors were purchased from Promega (Madison, WI, USA). Reagents for quantitative real-time polymerase chain reaction (qPCR) were purchased from Vazyme (Nanjing, China).
Specimen Collection and Preservation
Clinical samples were collected from six patients with confirmed UC diagnosis during electronic colonoscopy procedures. The severity of intestinal inflammation in each patient was assessed using the Mayo endoscopic score. Biopsy specimens were obtained from histologically confirmed inflammatory and non-inflammatory regions. Specimens were flash-frozen in liquid nitrogen and stored at −80°C; samples were allocated for qPCR and Western blot analyses, while remaining portions were reserved for measurements of iron content, MDA, and GSH. Ethical approval was obtained from the Union Hospital Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology, with informed consent provided by each participant (Approval No. 2018-S425). This study complies with the principles of the Declaration of Helsinki.
Experimental Procedure for Colitis Induction
Villin-Cre recombinase transgenic C57BL/6 mice and ERβ flox/flox C57BL/6 mice were purchased from Cyagen (Jiangsu, China). The Villin-Cre and ERβ flox/flox mice were crossed to generate intestinal epithelial cell-specific ERβ knockout mice (ERβ−/− mice). Male ERβ−/− mice at 8 weeks were used for subsequent experiments. SPF-grade, 8-week-old male C57BL/6 mice were obtained from Biont (Hubei, China). Mice were housed under a 12-hour light/dark cycle with humidity between 60% and 70%, and food and water were provided ad libitum. After a one-week acclimatization period, the mice underwent modeling procedures. Mice were divided into three groups: Control, WT + DSS, and ERβ−/− + DSS, with 10 mice per group. The specific interventions were as follows: Control group: wild type (WT) mice fed a normal diet and provided with regular drinking water. WT + DSS group: WT mice fed a normal diet with drinking water replaced by a 3% DSS solution. ERβ−/− + DSS group: ERβ−/− mice fed a normal diet with drinking water replaced by a 3% DSS solution. The modeling protocol lasted for 7 days. At the end of the modeling period, mice were anesthetized with diethyl ether and euthanized by cervical dislocation. A segment of the colon, including a complete cross-section, was fixed in 4% paraformaldehyde for subsequent hematoxylin and eosin (H&E) staining and tissue immunofluorescence. Another small segment of the colon was preserved in electron microscopy fixative for transmission electron microscopy (TEM). A portion of the colon was immediately snap-frozen in liquid nitrogen and then transferred to a −80°C freezer for future RNA sequencing. The remaining colon tissue was processed for immediate isolation of IECs for further experimentation. All experimental procedures and protocols were conducted in strict accordance with relevant guidelines and regulations and were approved by the Animal Experiment Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology (Approval No. 2023–3389).
Disease Activity Index (DAI)
The DAI was assessed based on the average sum of scores for body weight loss, stool consistency, and presence of hematochezia. Scoring criteria were as follows: Body weight loss: 0 points for no weight loss, 1 point for 1~5% weight loss, 2 points for 6~10% weight loss, 3 points for 11~15% weight loss, 4 points for weight loss greater than 15%. Stool consistency: 0 points for normal stool, 2 points for unformed stool, 4 points for diarrhea. Presence of hematochezia: 0 points for no blood in stool, 2 points for occult blood positive, 4 points for gross blood in stool. The DAI was calculated as the mean of these three scores.
H&E Staining
Tissue samples were processed through fixation, dehydration, clearing, infiltration with wax, embedding, sectioning, and baking. Sections were deparaffinized and rehydrated. Hematoxylin was used to stain the cell nuclei, followed by eosin staining of the cytoplasm. The sections were then dehydrated through a series of graded alcohols and cleared in xylene. Finally, the slides were mounted using neutral resin and examined under a microscope.
Immunofluorescence
Cells were cultured to an appropriate density and then fixed with 4% paraformaldehyde for 20 min. Cells were washed 3 times with phosphate-buffered saline (PBS) for 5 min each time. Permeabilization was performed with 0.3% Triton X-100 at room temperature for 30 min, followed by three more washes with PBS for 5 min each. Cells were blocked with 5% normal donkey serum for 30 min to minimize non-specific binding. The blocking solution was removed, and the primary antibody against GPX4 (1:200 dilution) was added to the cells, which were incubated overnight at 4°C. Cells were washed three times with PBS for 5 min each time and then incubated with an appropriately diluted Alexa Fluor donkey anti-rabbit secondary antibody (1:200 dilution) for 1 hour at room temperature in the dark. Cells were washed again three times with PBS for 5 min each time. Nuclei were stained with DAPI for 5 min. Samples were visualized and imaged under a fluorescence microscope.
Cell Culture and Transfection
The human colon cancer cell line HT-29 was obtained from Pricella (Wuhan, China). Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin in a humidified incubator at 37°C with 5% CO2. For intervention experiments, cells were treated with 10 μg/mL lipopolysaccharide (LPS) or 100 nM ERB041 for 24 hours. When cells reached 60~70% confluence, small interfering RNA (siRNA) was transfected using Lipofectamine 3000 according to the manufacturer’s instructions. The efficiency of gene knockdown was confirmed by quantitative PCR (qPCR) 48 hours post-transfection.
Isolation of IECs
The colon was washed in cold Dulbecco’s PBS (DPBS) for three times, and then cut into 5-mm tissue segments. Then they were incubated in a 15 mL mixture containing ethylenediaminetetraacetic acid and dithiothreitol. The tissues were gently agitated for 75 min at 4°C. Post-incubation, the tissue samples were removed and thoroughly rinsed with cold DPBS, and then subjected to vigorous shaking to dislodge any remaining adherent cells. The resulting suspension was rapidly passed through a 70-μm filter and centrifuged at 800 g for 5 min at 4°C. The resultant sediment, enriched in IECs, was collected for further experiment.
qPCR
RNA was extracted using an RNA extraction kit. Following extraction, RNA was reverse-transcribed into cDNA using high-fidelity reverse transcriptase. qPCR was performed using the LightCycler 480 Real-Time PCR System (Roche Diagnostics, Basel, Switzerland). Each qPCR was performed in triplicate using gene-specific primers, with the sequences listed in Table 1. Gene expression levels were calculated using the ∆∆Ct method and normalized to the housekeeping gene GAPDH.
 |
Table 1 Primer Sequences |
Western Blot
Proteins were extracted using RIPA lysis buffer supplemented with a protease inhibitor cocktail. After centrifugation, the supernatants were harvested, and protein concentrations were determined using the BCA protein assay. Equal amounts of proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to PVDF membranes. Membranes were blocked with 5% non-fat milk to prevent nonspecific binding, and then incubated overnight at 4°C with primary antibodies. The membranes were incubated with horseradish peroxidase-conjugated secondary antibodies at room temperature. Protein bands were visualized using an ECL detection system, and densitometry analysis of these bands was performed using Fiji software.
CCK-8
HT-29 cells (approximately 2000 cells per well) were seeded in 100 μL of suspension in a 96-well cell culture plate and subjected to the respective treatments as required by the experiment. CCK-8 (10 μL) solution were added to each well, and the plates were incubated for an additional 0.5 hours in the cell culture incubator. Absorbance was measured at 405 nm using a microplate reader for the blank wells, control wells, and test wells (Ablank, Acontrol, and Atest, respectively). Cell viability was calculated as follows: Cell viability (%) = (Atest - Ablank) / (Acontrol - Ablank) x 100%.
MDA Content
Tissue or cellular supernatants were extracted and their concentrations determined. TBA stock solution and MDA detection working solutions were prepared according to the manufacturer’s instructions. Standards were diluted to various final concentrations with PBS. MDA detection working solution (200μL) were added to test samples and standards of different concentrations. After thorough mixing, the samples were boiled for 15 min and then cooled to room temperature. They were centrifuged at 1000 g for 10 min at room temperature. The supernatants (200μL) were transferred to a 96-well plate, and absorbance was measured at 532 nm. The MDA content in the test samples was calculated from a standard curve generated using the different concentrations of the standards.
Iron Content
Tissue iron content: Tissue samples were prepared and iron detection standard solutions were diluted according to the manufacturer’s instructions. EP tubes were divided into blank, standard, and test groups. Samples were added and thoroughly mixed. The tubes were heated in a boiling water bath for 5 min and then cooled to room temperature. Chloroform (60μL) was added to each tube, followed by vigorous mixing. The tubes were centrifuged at 10000 rpm for 10 min at room temperature. The upper phase (200μL) was transferred to a 96-well plate, and absorbance was measured at 520 nm. The absorbance values were recorded as Ablank, Astandard, and Atest, and tissue iron content was calculated according to the manufacturer’s instructions.
Cellular iron content: Cell samples were prepared and iron detection standard solutions were diluted according to the manufacturer’s instructions. Cells were lysed by sonication and centrifuged at 8000 g for 10 min at 4°C. The supernatants were collected and their concentrations determined. EP tubes were divided into blank, standard, and test groups. Samples were added and thoroughly mixed. After 10 min, the mixture (200μL) was transferred to a 96-well plate, and absorbance was measured at 510 nm. The absorbance values were recorded as Ablank, Atest, and Astandard, and cellular iron content was calculated according to the manufacturer’s instructions.
GSH Content
Reagents and samples were prepared according to the manufacturer’s instructions. Homogenate (100 μL) was mixed with an equal volume of Reagent 1, followed by centrifugation at 3500 rpm for 10 min. The supernatant was collected. A 96-well plate was set up with blank, standard, and test wells, and the corresponding reagents were added to each well. After thorough mixing, absorbance was measured at 405 nm. GSH content was calculated according to the manufacturer’s instructions.
ROS Detection
DCFH-DA was diluted to a final concentration of 10 μmol/L (1:1000 dilution). The diluted DCFH-DA was added to the cells and incubated for 20 min in the cell culture incubator. Cells were then washed three times with serum-free medium. The cells were collected in a centrifuge tube. After centrifugation, the supernatant was discarded, and the cells were resuspended in serum-free medium and transferred to a flow cytometry tube. ROS levels were detected using a flow cytometer with excitation and emission wavelengths set at 488 nm and 525 nm, respectively. Data were analyzed using FlowJo v10.8.1 software.
Mitochondrial Membrane Potential Detection
JC-1 staining working solution was prepared according to the manufacturer’s instructions. Cells were collected, counted, and centrifuged for 5 min before being resuspended. Fifty to one hundred million cells were taken and 1 mL of JC-1 staining working solution was added. The cells were gently inverted several times to mix thoroughly and incubated for 20 min. After centrifugation, the supernatant was discarded. Cells were washed twice with JC-1 staining buffer (1×) by centrifugation and removal of the supernatant after each wash. Cells were resuspended in JC-1 staining buffer (1×) and transferred to a flow cytometry tube. Mitochondrial membrane potential was detected using a flow cytometer with excitation/emission wavelengths of 490/530 nm for JC-1 monomers and 525/590 nm for JC-1 aggregates. Data were analyzed using FlowJo v10.8.1 software.
ChIP
Cells were lysed and cross-linked protein-DNA complexes were sonicated to generate optimal chromatin fragments. After pre-clearance with protein A/G agarose beads, the fragmented chromatin was incubated overnight at 4°C with specific antibody, allowing for target DNA sequence binding. Antibody-bound complexes were isolated using protein A/G agarose beads. After removing nonspecifically bound materials via washing, the antibody-protein-DNA complexes were reversed, and DNA was extracted. The putative binding site was amplified using PCR with specifically designed primers (forward primer: 5’-GCTGAGGCTGGAGGATCACTTGA-3’; reverse primer: 5’-GACAACAGGCTCATCCACCATTCC-3’). qPCR was performed to confirm the binding of the transcription factor to the promoter region of the target gene.
Dual-Luciferase Reporter Gene Assay
HT-29 cells were transiently transfected with an overexpression plasmid or a corresponding control using Lipofectamine 3000. Reporter constructs were generated by integrating promoter sequence into the pGL4.11 luciferase reporter vector, thereby fabricating the wild-type (WT) reporter. An analogous procedure was followed to create a mutant (Mut) reporter by introducing an eight-base pair alteration into the promoter sequence within the pGL4.11 vector. After the initial transfection, the WT and Mut reporters, along with the Renilla luciferase vector (used as a transfection control), were transfected into HT-29 cells. After 48 h, a dual-luciferase reporter assay system was used to quantify the luciferase activity. The simultaneous utilization of both WT and Mut constructs, supplemented with a Renilla luciferase control, ensured robust and accurate evaluation of transcriptional activity.
TEM
Colon tissue samples (1 mm³ in size) were preserved in electron microscopy fixative. Post-fixation, room temperature dehydration, embedding, polymerization, sectioning, and staining were performed by Wuhan Servicebio Biotechnology Co., Ltd (Wuhan, China). Images were captured using a transmission electron microscope.
RNA Sequencing
Colon tissues from five randomly selected mice in both the ERβ−/− + DSS and WT + DSS groups were processed for RNA sequencing at Wuhan Frasergen Gene Information Co., Ltd., Wuhan, China. Following RNA extraction and quality validation, samples were sequenced on an Illumina NovaSeq 6000 platform. The resulting data have been deposited in the NCBI Sequence Read Archive under project number PRJNA1085055. Differential gene expression analysis was conducted using the DESeq2 package in R, with the Benjamini-Hochberg correction applied to control the false discovery rate. Genes with an absolute fold change greater than 1.5 and a p-value less than 0.05 were deemed significantly differentially expressed. Visualization of the data, including plots and heatmaps, was performed using the ggplot2 and ComplexHeatmap packages. Additionally, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses, and Gene Set Enrichment Analysis (GSEA) were executed, adopting a p-value threshold of less than 0.05 to establish statistical significance.
Statistical Analysis
A statistical analysis of the data was performed using GraphPad Prism 9.0 (GraphPad Software, CA, USA). Students’ t-tests were used to assess differences between two groups, while one-way ANOVA was used to compare multiple groups. Data are presented as mean (SD). The significance level was set at p < 0.05.
Results
Significant Ferroptosis in Inflammatory Regions of Colonic Mucosa in UC Patients
Compared to non-inflammatory regions, the mRNA and protein expression levels of GPX4 were significantly downregulated, while PTGS2 was significantly upregulated in the inflammatory regions of the colonic mucosa in UC patients (Figure 1A–C). Additionally, there was a significant increase in the levels of lipid peroxidation product (MDA) and iron content, along with a decrease in the level of the antioxidant GSH (Figure 1D–F). These findings indicated that significant ferroptosis occurred in the inflammatory regions of the colonic mucosa in UC patients.
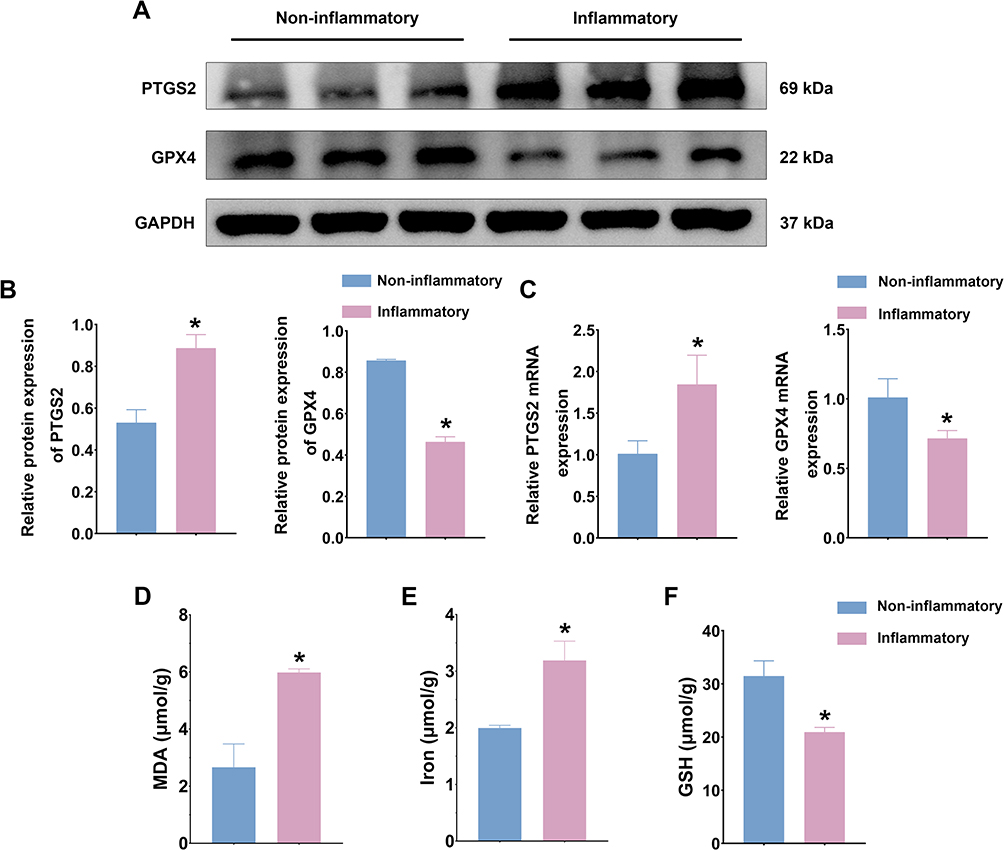 |
Figure 1 Significant ferroptosis in inflammatory regions of colonic mucosa in UC patients. (A). Western blot was used to assess PTGS2 and GPX4 protein expression levels in inflammatory and non-inflammatory colons from patients with UC. (B). Quantitative analysis of PTGS2 and GPX4 protein expression levels. (C). qPCR was used to assess PTGS2 and GPX4 mRNA expression levels in inflammatory and non-inflammatory colons from patients with UC. (D). Comparison of MDA content in inflammatory and non-inflammatory colons. (E). Comparison of iron content in inflammatory and non-inflammatory colons. (F). Comparison of GSH content in inflammatory and non-inflammatory colons. n = 3~6; * p < 0.05. |
Intestinal Epithelial Cell-Specific ERβ Gene Knockout Exacerbated Colitis
As shown in Figure 2A, mice in the control group exhibited stable weight gain, whereas mice in the WT + DSS group showed a decrease in body weight starting from day 3 of the modeling process. Compared with the WT + DSS group, mice in the ERβ−/− + DSS group had a further significant reduction in body weight. Compared with the control group, the colon length was significantly shortened, and the DAI and colon tissue histopathological scores were significantly increased in the WT + DSS group. Knockout of the ERβ gene in IECs exacerbated these changes: the colon was further shortened, and the DAI and histopathological scores were further increased (Figure 2B–E). Representative images of H&E staining of colon tissues from each group were shown in Figure 3F: compared with the Control group, the WT + DSS group displayed altered structural layers of the colonic wall, epithelial damage, and visible infiltration of inflammatory cells in the mucosal and submucosal layers, as well as swollen and deformed crypts. In the ERβ−/− + DSS group, epithelial damage was more pronounced, with disruption of continuity, extensive infiltration of inflammatory cells, and further aggravation of crypt destruction and loss. These results indicated that knockout of ERβ in IECs significantly exacerbated DSS-induced colitis in mice.
 |
Figure 2 Intestinal epithelial cell-specific ERβ gene knockout exacerbated colitis. (A). Percentage change in body weight for mice in the Control, WT + DSS, and ERβ−/− + DSS groups. (B). Representative colon images from mice in the three groups. (C). Quantification of mean colon lengths among the three groups. (D). DAI scores for mice in the three groups. (E). Histopathological scores of colon tissues from mice in the three groups. (F). Representative H&E-stained sections of colon tissues from mice in the three groups. n = 6; * p < 0.05. |
 |
Figure 3 Continued. |
 |
Figure 3 Intestinal epithelial cell-specific ERβ gene knockout impaired intestinal immune homeostasis. (A). PCA plot of RNA sequencing data from colon tissues of WT + DSS and ERβ−/− + DSS groups. (B). Volcano plot showing DEGs in colon tissues between the two groups. (C). Circular heatmap illustrating DEGs in colon tissues of the two groups. (D). GSEA. (E–G). GO enrichment analysis of DEGs in colon tissues from WT + DSS and ERβ−/− + DSS mice. (H). KEGG pathway enrichment analysis of DEGs. n = 5. |
Intestinal Epithelial Cell-Specific ERβ Gene Knockout Impaired Intestinal Immune Homeostasis
To further explore the regulatory role of ERβ in colitis, RNA sequencing was performed on colon tissues from five randomly selected mice from both the WT + DSS group and the ERβ−/− + DSS group. As shown in Figure 3A, the PCA plot demonstrated that the WT + DSS group and the ERβ−/− + DSS group were distinctly separated into two clusters, with each group displaying good internal consistency. A volcano plot based on all genes was presented in Figure 3B, showing a total of 3859 differentially expressed genes (DEGs). A circular heatmap based on the DEGs was depicted in Figure 3C. Gene set enrichment analysis (GSEA) indicated that, compared to the WT + DSS group, signaling pathways such as JAK/STAT, NF-κB, TNF, and IBD were significantly activated in the colon tissues of the ERβ−/− + DSS group (Figure 3D). Results of GO enrichment analysis revealed that the top ten enriched GO functions included regulation of inflammatory response, activation of immune response, signaling pathways involved in immune response regulation, Wnt signaling pathway, NF-κB signaling pathway (Figure 3E–G). KEGG pathway enrichment analysis demonstrated that, compared to the WT + DSS group, the ERβ−/− + DSS group was enriched for multiple signaling pathways related to inflammation and immunity, including cytokine-cytokine receptor interaction, TNF signaling pathway, PI3K-Akt signaling pathway, NF-κB signaling pathway, JAK-STAT signaling pathway, Toll-like receptor signaling pathway, and others, which were closely associated with the development of UC (Figure 3H). These findings suggested that knockout of ERβ in IECs exacerbated DSS-induced experimental colitis in mice and impairs intestinal immune homeostasis.
Intestinal Epithelial Cell-Specific ERβ Gene Knockout Exacerbated Ferroptosis
To further assess the ferroptosis in the IECs of the three groups of mice, we measured MDA and iron content, as well as GSH levels. As shown in Figure 4A–C, DSS treatment significantly increased MDA and iron content while decreasing GSH levels in IECs. qPCR and Western blot revealed that PTGS2 mRNA and protein expression levels were significantly upregulated, whereas GPX4 expression was significantly downregulated. The absence of ERβ in IECs exacerbated these changes (Figure 4D–F). Immunofluorescence indicated that GPX4 was abundantly expressed in the intestinal epithelium of control mice. After DSS treatment, GPX4 expression levels decreased. Compared with the WT + DSS group, GPX4 expression was further reduced in the ERβ−/− + DSS group, with statistical significance (Figure 4G). TEM images (Figure 4H) showed that in the control group, the structure of IECs was normal, with a columnar arrangement, intact cell membranes, and homogeneous cytoplasm. Mitochondria appeared normal, with a large number present, complete double membranes, homogenous matrix, parallel cristae, and normal structures. In the WT + DSS group, mitochondria were condensed and smaller in size, with some showing disrupted cristae and membrane damage. In the ERβ−/− + DSS group, mitochondrial damage was exacerbated: mitochondrial cristae were markedly reduced, mitochondrial membranes were severely damaged, the matrix was sparse, and the volume was decreased. These findings further suggested that the knockout of ERβ exacerbated ferroptosis in the IECs of mice with DSS-induced colitis.
Figure 4 Continued. Figure 4 Continued. Figure 4 Intestinal epithelial cell-specific ERβ gene knockout exacerbated ferroptosis. (A). MDA content in IECs from Control, WT + DSS, and ERβ−/− + DSS mice. (B). Iron content in IECs from the three groups. (C). GSH content in IECs from the three groups. (D). Western blot was used to assess PTGS2 and GPX4 protein expression levels in IECs from the three groups. (E). Quantitative analysis of PTGS2 and GPX4 protein expression levels in IECs from the three groups. (F). qPCR was used to assess PTGS2 and GPX4 mRNA expression levels in IECs from the three groups. (G). Immunofluorescence detection of GPX4 expression levels in colon tissues from the Control, WT + DSS, and ERβ−/− + DSS groups. (H). TEM analysis of mitochondrial morphology in IECs from the three groups. n = 3~6; * p < 0.05.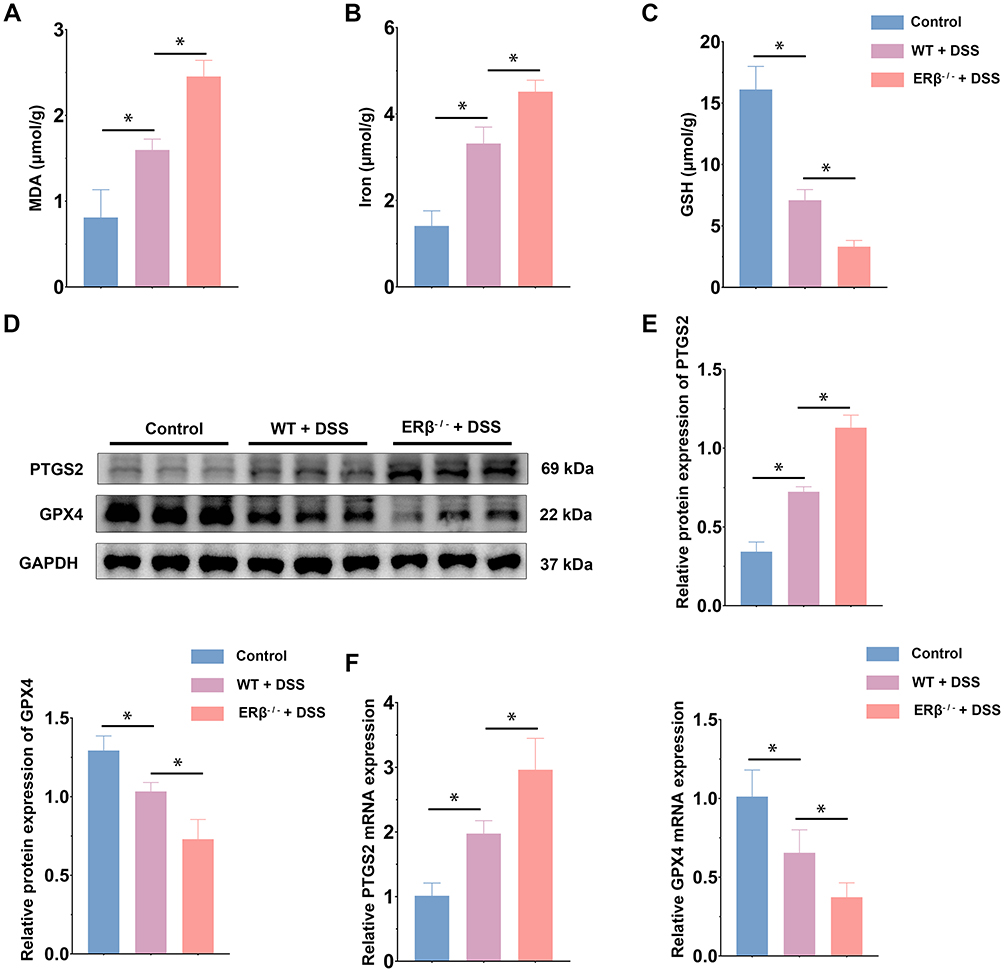


Activation of ERβ Attenuated LPS-Induced Ferroptosis in HT-29 Cells
In vitro, HT-29 cells were divided into three groups: Control, LPS, and LPS + ERB041. CCK-8 assay results showed that LPS inhibited the viability of HT-29 cells to 56% of normal levels, while ERB041 partially alleviated this inhibitory effect of LPS (Figure 5A). LPS treatment decreased GSH content and increased MDA and intracellular iron; activation of ERβ significantly increased GSH levels and reduced MDA and iron levels (Figure 5B–D). Similarly, Western blot demonstrated that activation of ERβ downregulated PTGS2 protein expression and upregulated GPX4, thereby mitigating ferroptosis in LPS-treated HT-29 cells (Figure 5E and F). ROS accumulation and mitochondrial damage are key features during the process of ferroptosis. We therefore measured intracellular ROS levels and changes in mitochondrial membrane potential. Results showed that LPS significantly increased ROS levels, and activation of ERβ reduced fluorescence intensity, indicating a decrease in ROS content after ERβ intervention (Figure 5G and H). Using the JC-1 fluorescent probe to detect mitochondrial membrane potential in the three groups of cells, we observed a decline in mitochondrial membrane potential in the LPS group, indicating depolarization and mitochondrial damage; activation of ERβ partially restored the decreased mitochondrial membrane potential, reducing mitochondrial damage (Figure 5I). These findings suggested that activation of ERβ attenuated LPS-induced ferroptosis in HT-29 cells.
Figure 5 Continued. Figure 5 Activation of ERβ attenuated LPS-induced ferroptosis in HT-29 cells. (A). CCK-8 assay was used to measure relative cell viability in the Control, LPS, and LPS + ERB041 groups. (B). GSH content in cells from the three groups. (C). Iron content in cells from the three groups. (D). MDA content in cells from the three groups. (E). Western blot was used to assess PTGS2 and GPX4 protein expression levels in cells from the three groups. (F). Quantitative analysis of PTGS2 and GPX4 protein expression levels in cells from the three groups. (G). Flow cytometry analysis of ROS level in cells from the three groups. (H). Mean fluorescence intensity of ROS in cells from the three groups. (I). Flow cytometry analysis of mitochondrial membrane potential in cells from the three groups. n = 3~6; * p < 0.05.

Knockdown of ERβ Exacerbated LPS-Induced Ferroptosis in HT-29 Cells
Following knockdown of ERβ using siRNA, HT-29 cells were divided into three groups: si-Con, si-Con + LPS, and si-ERβ + LPS. CCK-8 assay results showed that knockdown of ERβ expression suppressed the viability of HT-29 cells (Figure 6A). Consistent with the in vitro experimental results, knockdown of ERβ expression significantly decreased GSH content and increased MDA and intracellular iron levels (Figure 6B–D). Western blot demonstrated that, compared to the si-Con + LPS group, PTGS2 protein expression was significantly upregulated and GPX4 expression was significantly downregulated in the si-ERβ + LPS group, suggesting that knockdown of ERβ expression exacerbated LPS-induced ferroptosis in cells (Figure 6E and F). Additionally, compared to the si-Con + LPS group, the si-ERβ + LPS group showed significantly elevated ROS levels (Figure 6G and H). Knockdown of ERβ further decreased mitochondrial membrane potential, exacerbating mitochondrial damage (Figure 6I). These results indicated that knockdown of ERβ expression exacerbated LPS-induced ferroptosis in HT-29 cells.
Figure 6 Continued. Figure 6 Knockdown of ERβ exacerbated LPS-induced ferroptosis in HT-29 cells. (A). CCK-8 assay was used to measure relative cell viability in the si-Con, si-Con + LPS, and si-ERβ + LPS groups. (B). GSH content in cells from the three groups. (C). Iron content in cells from the three groups. (D). MDA content in cells from the three groups. (E). Western blot was used to assess PTGS2 and GPX4 protein expression levels in cells from the three groups. (F). Quantitative analysis of PTGS2 and GPX4 protein expression levels in cells from the three groups. (G). Flow cytometry analysis of ROS content in cells from the three groups. (H). Mean fluorescence intensity of ROS in cells from the three groups. (I). Flow cytometry analysis of mitochondrial membrane potential in cells from the three groups. n = 3~6; * p < 0.05.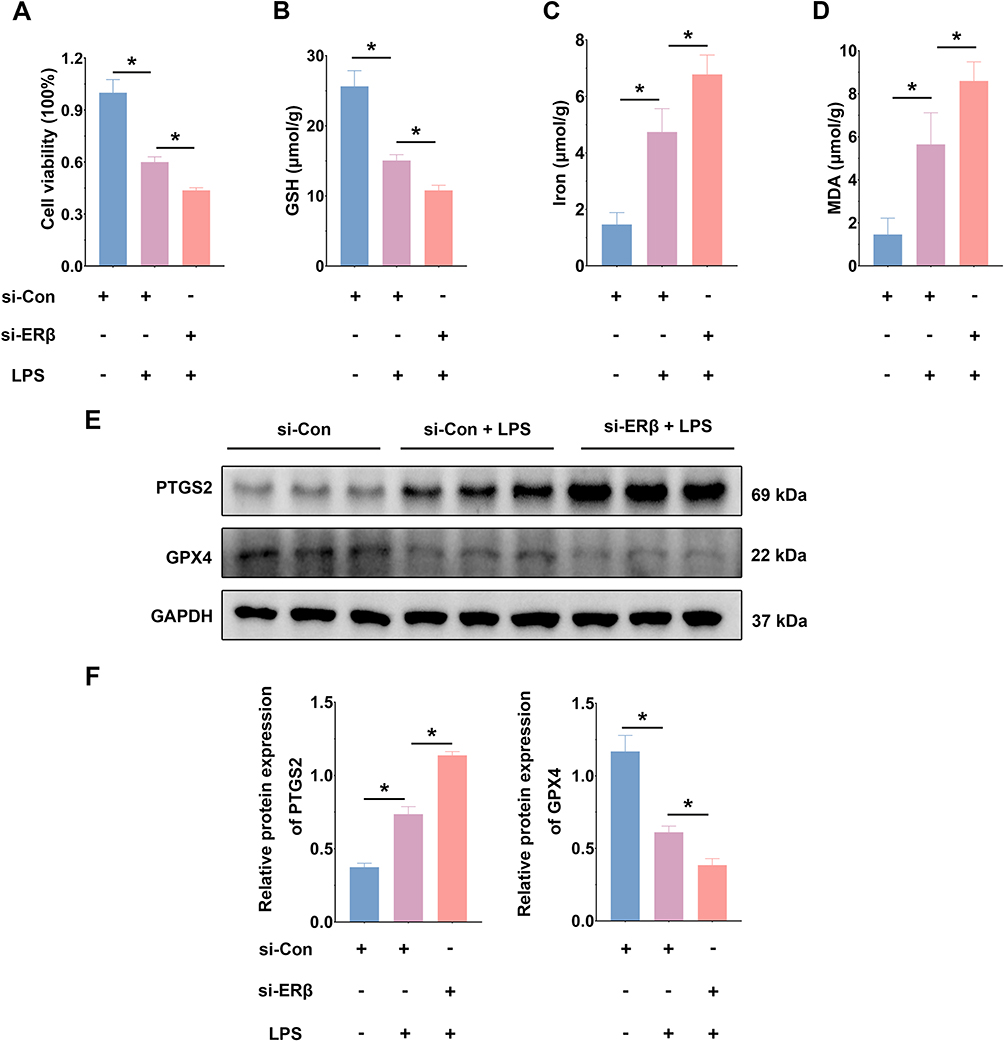

ERβ Regulated Ferroptosis Through GPX4
The above studies confirmed that ERβ alleviated colitis in vitro and in vivo by inhibiting ferroptosis. However, the specific mechanism by which ERβ inhibits ferroptosis is not fully understood. By predicting target genes of the transcription factor ERβ using the gene transcription regulation database (GTRD), we found that GPX4 was among them and played a critical regulatory role in ferroptosis. Therefore, we hypothesized that ERβ may participate in the regulation of ferroptosis via GPX4. As shown in Figure 7A, ChIP-qPCR confirmed that ERβ bound to the GPX4 promoter. The dual-luciferase reporter gene assay showed that overexpression of ERβ significantly increased luciferase activity in cells transfected with WT GPX4 promoter plasmids, whereas there was no significant difference in luciferase intensity in cells transfected with Mut plasmids (Figure 7B). These results suggested that ERβ bound to the promoter region of GPX4 and positively regulated its transcription. Next, we knocked down GPX4 expression using siRNA to induce ferroptosis and treated HT-29 cells with ERB041. CCK-8 assay results indicated that knockdown of GPX4 significantly suppressed the viability of HT-29 cells, decreased GSH content, and increased MDA and iron levels, as well as upregulated PTGS2 protein expression, suggesting significant ferroptosis occurred after GPX4 knockdown. However, under these conditions, activation of ERβ had no significant effect on cell viability, GSH, MDA, and iron content (Figure 7C–H). Flow cytometry results showed that the si-GPX4 group had a significant increase in intracellular ROS levels; mitochondrial membrane potential declined, indicating depolarization and mitochondrial damage. Compared to the si-GPX4 group, the si-GPX4 + ERB041 group showed no significant change in ROS levels or mitochondrial membrane potential (Figure 7I–K). These findings further confirmed that ERβ inhibited ferroptosis through the regulation of GPX4.
Figure 7 Continued. Figure 7 ERβ regulated ferroptosis through GPX4.(A). ChIP-qPCR was used to confirm the binding of ERβ to the GPX4 promoter. (B). Dual-luciferase reporter assay was used to detect the transcriptional regulation of GPX4 by ERβ. (C). CCK-8 assay was used to measure relative cell viability in the si-Con, si-GPX4, and si-GPX4 + ERB041 groups. (D). GSH content in cells from the three groups. E. Iron content in cells from the three groups. (F). MDA content in cells from the three groups. (G). Western blot was used to assess PTGS2 and GPX4 protein expression levels in cells from the three groups. (H). Quantitative analysis of PTGS2 and GPX4 protein expression levels in cells from the three groups. (I). Flow cytometry analysis of ROS content in cells from the three groups. (J). Mean fluorescence intensity of ROS in cells from the three groups. (K). Flow cytometry analysis of mitochondrial membrane potential in cells from the three groups. n = 3~6; * p < 0.05; ns: p > 0.05.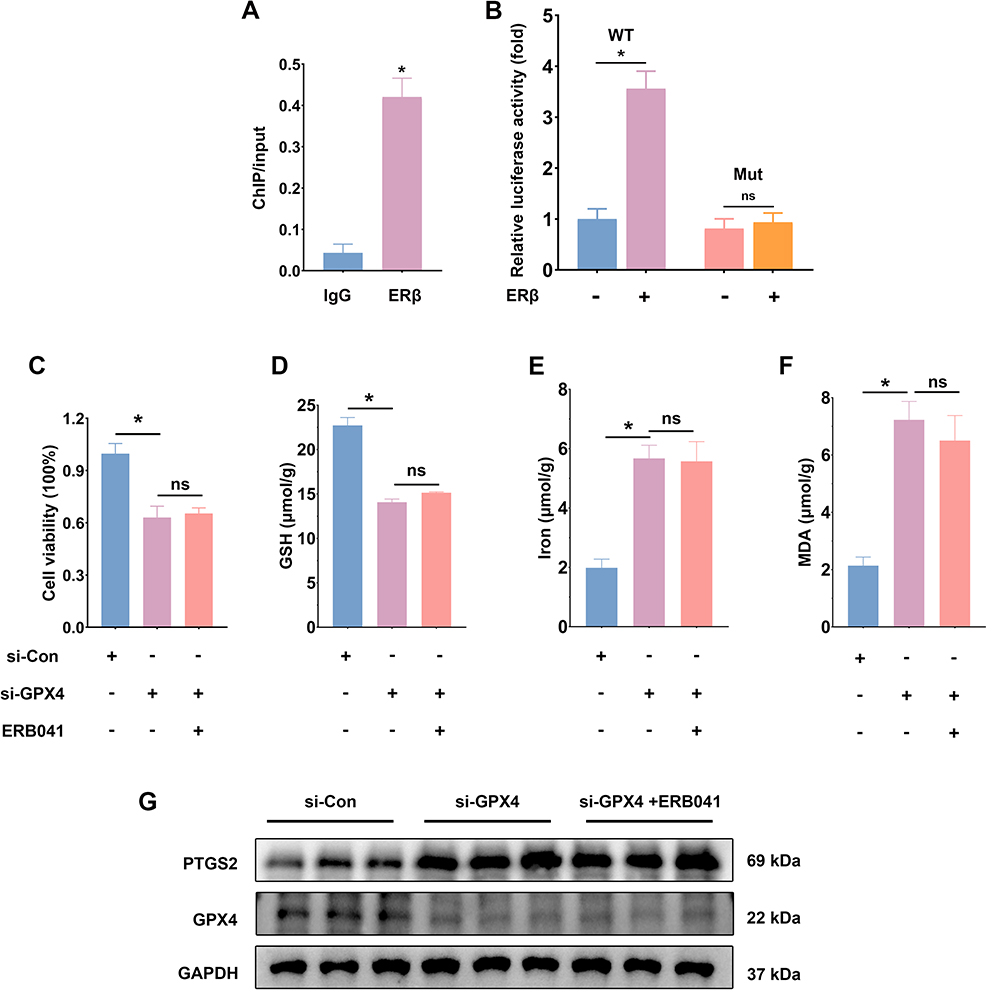

Discussion
Since 1990, the incidence of UC has been increasing in emerging industrialized countries in Asia and Latin America, including China.32 The typical clinical manifestations include abdominal pain, diarrhea, and mucopurulent bloody stools, with extraintestinal manifestations occurring in 20% to 35% of patients.33 Ultimately, more than 10% of UC patients require surgical treatment, which not only affects quality of life but also imposes a significant economic burden on patients and society.34 Given the crucial role of IECs in the pathogenesis of UC, finding therapeutic agents that act on IECs is an urgent issue that needs to be addressed.
The anti-inflammatory effects of ERβ in the colon have been preliminarily established, with research showing that ERβ deficiency impairs the normal gut microbiota, leading to more severe colitis.35 In this study, we found that compared to wild-type mice, mice with intestinal epithelial cell-specific ERβ knockout exhibited more severe weight loss, shortened colon length, higher DAI, and higher colon tissue histopathological scores after DSS treatment. Furthermore, the JAK/STAT, NF-κB, TNF, and IBD signaling pathways were significantly activated in the colon tissues. This further confirmed that ERβ deficiency in IECs exacerbated DSS-induced experimental colitis.
As previously mentioned, ferroptosis is an iron-dependent form of cell death. The regulatory pathway of ferroptosis is complex, with GPX4 being a key regulator. Factors affecting GPX4 expression or activity can modulate cellular ferroptosis. Iron ions play a critical role in cellular metabolic processes such as energy metabolism and oxygen transport; however, excessive iron ions can generate large amounts of ROS via enzymatic and non-enzymatic pathways, inducing ferroptosis.36 MDA is a product of lipid peroxidation and directly reflects the degree of oxidative damage in cells, with its levels positively correlated with the level of ferroptosis.37 GSH is one of the main intracellular antioxidants and a critical substrate for GPX4, with its levels decreasing during ferroptosis.38 PTGS2 is a marker gene of ferroptosis, with increased expression observed during ferroptosis.39 ROS production occurs through enzymatic and non-enzymatic pathways; under normal conditions, ROS can participate in physiological processes such as mitochondrial signaling and transcription factor activation. However, when the balance between ROS generation and degradation is disrupted, excess ROS attack the cellular membrane, leading to structural and functional disruption and triggering ferroptosis.40 Additionally, during ferroptosis, mitochondria exhibit morphological changes, decreased membrane potential, and increased permeability.41 Recent studies have identified the involvement of ferroptosis in the pathogenesis of UC.42 DSS-induced colitis in mice shows significant activation of ferroptosis in the colon, with ferroptosis inhibitors able to significantly restore colon length, decrease DAI, protect the intestinal epithelial barrier, and reduce the expression levels of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α, improving colitis.43–45 In this study, consistent with other studies, DSS induced ferroptosis in IECs, and ERβ deficiency exacerbated cellular ferroptosis, characterized by increased MDA and iron content, decreased GSH content, downregulated GPX4 expression, upregulated PTGS2 expression, and aggravated mitochondrial morphology disruption. In vitro experiments showed that LPS induced ferroptosis in HT-29 cells, and silencing ERβ expression using siRNA led to results consistent with in vivo experiments, with further exacerbation of ferroptosis, manifested as reduced cell viability, increased ROS content, and decreased mitochondrial membrane potential. Conversely, ERβ activation with ERB041 inhibited ferroptosis. Therefore, we concluded that activation of ERβ inhibited ferroptosis. Given that GPX4 is a target gene of ERβ and plays a critical role in resistance to ferroptosis, we hypothesize that ERβ regulates cellular ferroptosis through GPX4, exerting an anti-inflammatory effect. ChIP-qPCR confirmed the binding of ERβ to the GPX4 promoter, and dual luciferase reporter assays demonstrated that ERβ positively regulated GPX4 transcription. Knockdown of GPX4 expression using siRNA, followed by ERβ activation with ERB041, did not significantly alter the level of ferroptosis. Based on these experimental results, we concluded that ERβ inhibited ferroptosis in IECs through positive regulation of GPX4 transcription, thereby alleviating DSS-induced experimental colitis. This study not only confirms the anti-inflammatory effect of ERβ in IECs but also further explores its regulatory role in ferroptosis and the underlying molecular mechanisms, underscoring the significance of ERβ in improving colitis.
Data Sharing Statement
Data is available on request from the corresponding author.
Author Contributions
All authors made a significant contribution to the work reported, whether in conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The study was supported by National Key R&D Program of China (2023YFC2507300) and the National Natural Science Foundation of China (No.82470562; No. 82170547).
Disclosure
The authors declare that there is no conflict of interest in this study.
References
1. Du L, Ha CEpidemiology and pathogenesis of ulcerative colitis. Gastroenterol Clin North Am. 2020;49. doi:10.1016/j.gtc.2020.07.005
2. Krugliak Cleveland N, Torres J, Rubin DT. What does disease progression look like in ulcerative colitis, and how might it be prevented? Gastroenterology. 2022;162(5):1396–1408. doi:10.1053/j.gastro.2022.01.023
3. Le Berre C, Honap S, Peyrin-Biroulet L. Ulcerative colitis. Lancet. 2023;402. doi:10.1016/S0140-6736(23)00966-2
4. Xu L, He B, Sun Y, et al. Incidence of inflammatory bowel disease in urban China: a nationwide population-based study. Clin Gastroenterol Hepatol. 2023;21(13):3379–3386.e29. doi:10.1016/j.cgh.2023.08.013
5. Nakase H, Sato N, Mizuno N, et al. The influence of cytokines on the complex pathology of ulcerative colitis. Autoimmun Rev. 2022;21(3):103017. doi:10.1016/j.autrev.2021.103017
6. Hirten RP, Sands BE. New therapeutics for ulcerative colitis. Annu Rev Med. 2021;72(1):199–213. doi:10.1146/annurev-med-052919-120048
7. Allaire JM, Crowley SM, Law HT, et al. The intestinal epithelium: central coordinator of mucosal immunity. Trends Immunol. 2018;39(9):677–696. doi:10.1016/j.it.2018.04.002
8. Jv P, B C. Cell death in the gut epithelium and implications for chronic inflammation. Nat Rev Gastroenterol Hepatol. 2020;17. doi:10.1038/s41575-020-0326-4
9. Günther C, Neumann H, Neurath MF, et al. Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut. 2013;62(7):1062–1071. doi:10.1136/gutjnl-2011-301364
10. Odenwald MA, Turner JR. The intestinal epithelial barrier: a therapeutic target? Nat Rev Gastroenterol Hepatol. 2017;14. doi:10.1038/nrgastro.2016.169
11. Romani P, Valcarcel-Jimenez L, Frezza C, Dupont S. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(1):22. doi:10.1038/s41580-020-00324-8
12. Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. doi:10.1038/s41422-020-00441-1
13. Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med. 2019;133:130–143. doi:10.1016/j.freeradbiomed.2018.09.043
14. Liu Y, Wan Y, Jiang Y, et al. GPX4: the hub of lipid oxidation, ferroptosis, disease and treatment. Biochim Biophys Acta Rev Cancer. 2023;1878(3):188890. doi:10.1016/j.bbcan.2023.188890
15. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144–152. doi:10.1016/j.freeradbiomed.2018.09.014
16. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi:10.1016/j.cell.2017.09.021
17. Fang X, Cai Z, Wang H, et al. Loss of cardiac ferritin h facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ Res. 2020;127(4):486–501. doi:10.1161/CIRCRESAHA.120.316509
18. Xu C, Liu Z, Xiao J. Ferroptosis: a double-edged sword in gastrointestinal disease. Int J Mol Sci. 2021;22(22):12403. doi:10.3390/ijms222212403
19. Xu S, He Y, Lin L, et al. The emerging role of ferroptosis in intestinal disease. Cell Death Dis. 2021;12(4):289. doi:10.1038/s41419-021-03559-1
20. Xiong Z, Chen P, Yuan M. The induction mechanism of ferroptosis, necroptosis, and pyroptosis in inflammatory bowel disease, colorectal cancer, and intestinal injury. Biomolecules. 2023;14(1):13. doi:10.3390/biom13050820
21. Chakraborty B, Byemerwa J, Krebs T, et al. Estrogen receptor signaling in the immune system. Endocr Rev. 2023;44(1):117–141. doi:10.1210/endrev/bnac017
22. Chen P, Li B, Ou-Yang L. Role of estrogen receptors in health and disease. Front Endocrinol. 2022;13. doi:10.3389/fendo.2022.839005
23. Chen C, Gong X, Yang X. The roles of estrogen and estrogen receptors in gastrointestinal disease. Oncol Lett. 2019;18. doi:10.3892/ol.2019.10983
24. Garcia-Villatoro EL, Allred CD. Estrogen receptor actions in colitis. Essays Biochem. 2021;65(6):1003–1013. doi:10.1042/EBC20210010
25. Looijer-van LM, Hotte N, Dieleman LA, et al. Estrogen receptor-β signaling modulates epithelial barrier function. Am J Physiol Gastrointest Liver Physiol. 2011;300(4):G621–626. doi:10.1152/ajpgi.00274.2010
26. Guo D, Liu X, Zeng C, et al. Estrogen receptor β activation ameliorates DSS-induced chronic colitis by inhibiting inflammation and promoting Treg differentiation. Int Immunopharmacol. 2019;77:105971. doi:10.1016/j.intimp.2019.105971
27. Jiang Q, Li W, Zhu X, et al. Estrogen receptor β alleviates inflammatory lesions in a rat model of inflammatory bowel disease via down-regulating P2X7R expression in macrophages. Int J Biochem Cell Biol. 2021;139:106068. doi:10.1016/j.biocel.2021.106068
28. Zhao Z, Niu S, Chen J, et al. G protein-coupled receptor 30 activation inhibits ferroptosis and protects chondrocytes against osteoarthritis. J Orthop Translat. 2024;44:125–138. doi:10.1016/j.jot.2023.12.003
29. He W, Gao Z, Liu S, et al. G protein-coupled estrogen receptor activation by bisphenol-A disrupts lipid metabolism and induces ferroptosis in the liver. Environ Pollut. 2023;334:122211. doi:10.1016/j.envpol.2023.122211
30. Liu L, Zhang C, Qu S, et al. ESR1 inhibits ionizing radiation-induced ferroptosis in breast cancer cells via the NEDD4L/CD71 pathway. Arch Biochem Biophys. 2022;725:109299. doi:10.1016/j.abb.2022.109299
31. Chen J, Zhao R, Wang Y, et al. G protein-coupled estrogen receptor activates PI3K/AKT/mTOR signaling to suppress ferroptosis via SREBP1/SCD1-mediated lipogenesis. Mol Med. 2024;30(1):28. doi:10.1186/s10020-023-00763-x
32. Burri E, Maillard MH, Schoepfer AM, et al. Treatment algorithm for mild and moderate-to-severe ulcerative colitis: an update. Digestion. 2020;101 Suppl 1(Suppl. 1):2–15. doi:10.1159/000504092
33. Feuerstein JD, Moss AC, Farraye FA. Ulcerative Colitis. Mayo Clin Proc. 2019;94(7):1357–1373. doi:10.1016/j.mayocp.2019.01.018
34. Gros B, Kaplan GG. Ulcerative colitis in adults: a review. JAMA. 2023;330(10):951–965. doi:10.1001/jama.2023.15389
35. Ma Y, Liu T, Li X, et al. Estrogen receptor β deficiency impairs gut microbiota: a possible mechanism of IBD-induced anxiety-like behavior. Microbiome. 2022;10(1):160. doi:10.1186/s40168-022-01356-2
36. Rochette L, Dogon G, Rigal E, et al. Lipid peroxidation and iron metabolism: two corner stones in the homeostasis control of ferroptosis. Int J Mol Sci. 2022;24(1):449. doi:10.3390/ijms24010449
37. Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–379. doi:10.1038/cdd.2015.158
38. Deng L, He S, Guo N, et al. Molecular mechanisms of ferroptosis and relevance to inflammation. Inflamm Res. 2023;72(2):281–299. doi:10.1007/s00011-022-01672-1
39. Sm C, Oh M-C, M M, et al. Targeting lipid peroxidation for cancer treatment. Molecules. 2020;25. doi:10.3390/molecules25215144
40. Zeng F, Nijiati S, Tang L, et al. Ferroptosis detection: from approaches to applications. Angew Chem Int Ed Engl. 2023:62:e202300379. doi:10.1002/anie.202300379
41. Chen Z, Lin H, Wang X, et al. The application of approaches in detecting ferroptosis. Heliyon. 2024;10(1):e23507. doi:10.1016/j.heliyon.2023.e23507
42. Wang X, Zhou Y, Min J, Wang F. Zooming in and out of ferroptosis in human disease. Front med. 2023;17. doi:10.1007/s11684-023-0992-z
43. Gao S, Sun C, Kong J. Vitamin D attenuates ulcerative colitis by inhibiting ACSL4-mediated ferroptosis. Nutrients. 2023;15. doi:10.3390/nu15224845
44. Wu Y, Ran L, Yang Y, et al. Deferasirox alleviates DSS-induced ulcerative colitis in mice by inhibiting ferroptosis and improving intestinal microbiota. Life Sci. 2023;314:121312. doi:10.1016/j.lfs.2022.121312
45. Millar AD, Rampton DS, Blake DR. Effects of iron and iron chelation in vitro on mucosal oxidant activity in ulcerative colitis. Aliment Pharmacol Ther. 2000;14(9):1163–1168. doi:10.1046/j.1365-2036.2000.00828.x
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.