")
Back to Journals » Clinical Ophthalmology » Volume 18
Expert Panel Consensus for Optimizing Outcomes in Neovascular Age-Related Macular Degeneration in the Context of Suboptimal Response to a Biosimilar: The Role of Aflibercept
Authors Narendran N, Bailey C, Downey L, Gale R, Kotagiri A, Pearce I, Rennie CA, Sivaprasad S , Talks J, Morgan-Warren P, Napier J, O’Neil C, Seeborne T
Received 10 June 2024
Accepted for publication 15 October 2024
Published 2 November 2024 Volume 2024:18 Pages 3133—3142
DOI https://doi.org/10.2147/OPTH.S481772
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Nirodhini Narendran,1,2 Clare Bailey,3 Louise Downey,4 Richard Gale,5 Ajay Kotagiri,6 Ian Pearce,7 Christina A Rennie,8 Sobha Sivaprasad,9 James Talks,10 Peter Morgan-Warren,11 Jackie Napier,11 Carolyn O’Neil,11 Timothy Seeborne11
1The Royal Wolverhampton NHS Trust, Wolverhampton, UK; 2School of Life & Health Sciences, Aston University, Birmingham, UK; 3University Hospitals Bristol and Weston NHS Foundation Trust, Bristol, UK; 4Hull University Teaching Hospitals NHS Trust, Hull, UK; 5York and Scarborough Teaching Hospitals NHS Foundation Trust, York, UK; 6South Tyneside and Sunderland NHS Foundation Trust, Sunderland, UK; 7Liverpool University Hospitals NHS Foundation Trust, Liverpool, UK; 8University Hospital Southampton NHS Foundation Trust, Southampton, UK; 9NIHR Moorfields Biomedical Research Centre, Moorfields Eye Hospital NHS Foundation Trust, London, UK; 10Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle, UK; 11Bayer plc, Reading, UK
Correspondence: Nirodhini Narendran, Ophthalmology Department, New Cross Hospital (Part of the Royal Wolverhampton NHS Trust), Wolverhampton Road, Wolverhampton, WV10 0QP, UK, Email [email protected]
Purpose: The inclusion of ranibizumab biosimilars into National Health Service England commissioning recommendations published in 2022 created a need for expert guidance to optimize treatment outcomes in patients with neovascular age-related macular degeneration (nAMD) who otherwise may not have received first-line ranibizumab. This article provides a consensus treatment pathway supporting timely identification and management of a suboptimal response to these therapies, thereby aiming to facilitate clinically meaningful outcomes and efficient management of service capacity under specific circumstances where ranibizumab biosimilars may be initiated as a first-line treatment.
Methods: Two structured round-table meetings of UK medical retina specialists were held in person and virtually on September 22 and November 3, 2022, respectively. These meetings were organized and funded by Bayer.
Results: The panel provided guidance on the implementation of an early treatment optimization pathway in cases where ranibizumab biosimilars are used as a first-line treatment, including recommendations on patient suitability and capacity requirements, and criteria for identification and strategies for management of a suboptimal response. The panel discussed the role of aflibercept treatment and its potential benefits and outlined recommendations on switching ranibizumab biosimilar suboptimal responders to an aflibercept treat-and-extend regimen, where appropriate.
Conclusion: Developed by a retinal expert panel, this early treatment optimization pathway provides guidance to facilitate optimal long-term patient outcomes while addressing capacity and resourcing constraints in circumstances of first-line ranibizumab biosimilar use for nAMD, including how aflibercept may be used in cases with a suboptimal response. Therefore, this fills an important gap in guidance on navigating the new treatment landscape.
Keywords: anti-VEGF agents, capacity, consensus pathway, ranibizumab
Introduction
Intravitreal anti–vascular endothelial growth factor (anti-VEGF) therapy is the current pharmacological standard of care for neovascular age-related macular degeneration (nAMD).1 Since its introduction, multiple treatment regimens have evolved, with differing potential impacts on treatment burden.2 Monthly fixed dosing and pro re nata (PRN) regimens with monthly monitoring both require visits every four weeks and thus place a high burden on patients and clinics alike.2 This can negatively affect the facilitation of and adherence to these strict monthly monitoring protocols, which may lead to poorer real-world visual outcomes than those observed in clinical trial settings.1,2 Fixed monthly dosing requires a high injection frequency, thereby increasing the associated costs and risk of overtreatment, whereas PRN is commonly associated with a risk of undertreatment.1–4 Proactive fixed regimens once every two months, which were first introduced with aflibercept 2 mg following its approval by the US Food and Drug Administration in 2011 and the European Medicines Agency in 2012, are associated with a lower treatment burden compared with monthly dosing or PRN with monthly monitoring; furthermore, data suggest real-world visual outcomes may be closer to those observed in randomized controlled trials compared with PRN.2,5,6 However, there is still a risk of undertreatment or overtreatment if the interval is suboptimal for the individual patient.1 Treat-and-extend (T&E) regimens, such as the aflibercept T&E pathway recommended by Bailey et al, provide a personalized approach designed to aid capacity planning while reducing unnecessary treatment burden for both patients and clinics compared with PRN and fixed monthly regimens.1,2,7 Evidence suggests that a proactive T&E approach is typically the regimen of choice for nAMD in clinical practice, with real-world studies indicating that long-term outcomes may be closer to those observed in clinical trials compared with PRN.2,8–10
Even with the introduction of T&E regimens with aflibercept and ranibizumab, long-term economic burden and lack of clinic capacity remain key unmet needs for National Health Service (NHS) medical retina services in the UK.11 These challenges are anticipated to worsen as the prevalence of nAMD, and thus the demand for intravitreal anti-VEGF injections, continues to grow, with a 64% rise in nAMD cases predicted from 2015 to 2035 in the UK.11,12
Following the expiration of the European patent for Lucentis® (reference ranibizumab) in 2022, ranibizumab biosimilar agents have started to become available.13,14 These biosimilars provide potential cost savings compared with reference ranibizumab.14 Several ranibizumab biosimilars are licensed in the European Union and Great Britain for the treatment of nAMD; Byooviz™ (ranibizumab biosimilar), Ongavia® (ranibizumab biosimilar), and Ximluci® (ranibizumab biosimilar) were approved in the UK in August 2021, May 2022, and January 2023, respectively.15–20
In anticipation of the introduction of ranibizumab biosimilars, an NHS England (NHSE) national procurement exercise for medical retinal vascular medicines was conducted.11 The purpose of this exercise was to support the delivery of the NHS Pathway Improvement Programme through reducing unwarranted variation in treatment, maintaining clinical choice, and making the best use of NHS resources.11 Following its conclusion, updated NHSE commissioning recommendations were published in August 2022, with minor updates released in July 2023.11 These recommendations state that, in consultation with the patient, the lowest cost treatment option should be used where clinically appropriate.11 Additionally, NHSE propose that ranibizumab biosimilars should be considered in patients commencing treatment with nAMD where clinically appropriate and there is capacity to do so.11
Although NHSE does not mandate use of ranibizumab biosimilars as first-line treatment, it is expected that, following the introduction of these recommendations, clinicians are likely to be encouraged to offer their patients the choice of ranibizumab first-line for newly diagnosed nAMD, even where previously another product may have been offered preferentially.11 However, NHSE recommends consideration of several factors when determining the most appropriate treatment option, including clinic capacity and individual patient circumstances.11
NHSE emphasizes the importance of reviewing treatment in patients with a suboptimal response.11 In such cases, NHSE guidelines state that switching to an alternative anti-VEGF agent or ceasing treatment should be considered.11 In cases of suboptimal response to ranibizumab biosimilars, NHSE guidelines state that the clinician should consider switching to aflibercept, brolucizumab, or faricimab.11 However, no guidance is provided on how to implement this within existing treatment pathways or what a suboptimal response may look like in practice.
The objective of this article is to fill this important knowledge gap in existing guidance through providing expert consensus recommendations for optimizing patient outcomes in treatment-naive eyes with nAMD initiated on ranibizumab biosimilars in a UK NHS clinic setting, in cases where use of reference ranibizumab may not have previously been considered, and in services where its use will not adversely affect the ability to treat all patients in the anti-VEGF service in a timely manner. To help clinicians navigate the new treatment landscape and support evidence-based rational clinical decision-making, this article introduces an early treatment optimization pathway to guide timely identification of suboptimal response and facilitate switching to alternative anti-VEGF agents, such as aflibercept with a T&E regimen, in appropriate cases.
Methods
A panel of UK medical retina specialists was convened to explore the practical guidance required to optimize outcomes in eyes with treatment-naive nAMD in cases where ranibizumab biosimilar use is initiated within a standard UK NHS clinic and develop a consensus pathway for the management of suboptimal responders. Two structured round-table meetings were held in person and virtually on September 22 and November 3, 2022, respectively. Both meetings were organized and funded by Bayer. A series of predefined questions was used alongside a review of the latest available evidence to generate the consensus pathway and expert recommendations. Ethics committee approval is not required for this type of consensus document.
Guidance from a UK Expert Panel
Overview
The early treatment optimization pathway for nAMD, where switching to a more durable anti-VEGF agent such as aflibercept is recommended for patients with a suboptimal response to ranibizumab biosimilars, is outlined in Figure 1. This pathway is designed for use in individual cases where ranibizumab biosimilars have been selected as first-line treatment for treatment-naive eyes with nAMD in which the use of reference ranibizumab may not have previously been considered, and in services where clinic capacity will not adversely affect the ability to treat all patients requiring anti-VEGF injections in a timely manner. The consensus pathway is not official NHS guidance; it is intended to serve only as advice for clinicians.
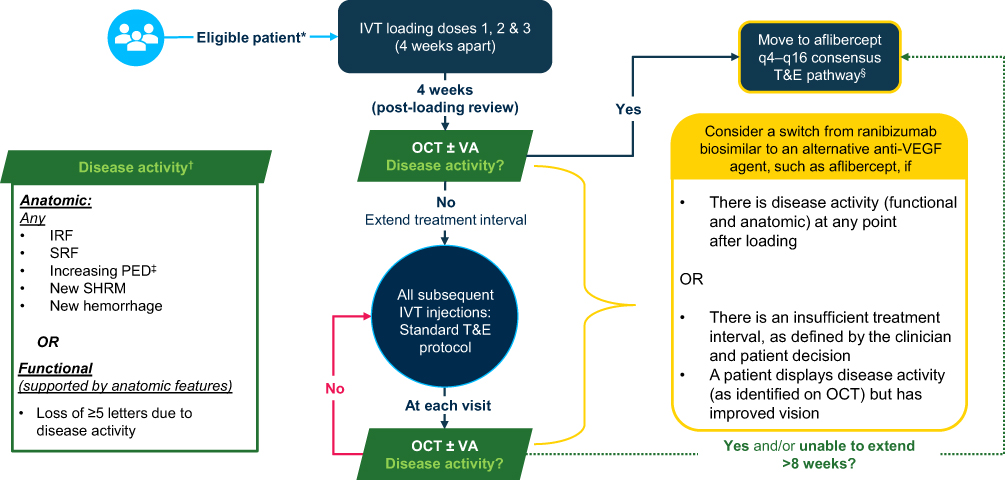 |
Figure 1 Early treatment optimization pathway for nAMD, in which suboptimal responders to a ranibizumab biosimilar are switched to aflibercept. Abbreviations: IRF, intraretinal fluid; IVT, intravitreal; nAMD, neovascular age-related macular degeneration; NHS; National Health Service; OCT, optical coherence tomography; PED, pigment epithelial detachment; q4, every 4 weeks; q16, every 16 weeks; SHRM, subretinal hyperreflective material; SRF, subretinal fluid; T&E, treat-and-extend; VA, visual acuity; VEGF, vascular endothelial growth factor. Notes: The consensus pathway is not official NHS guidance; it is intended to serve only as advice for clinicians. *As per the predefined suitability factors as outlined in Table 1. †According to clinician discretion. ‡With reference to OCT scans acquired since commencing treatment. §Three 4-weekly loading doses should be administered, followed by an injection 8 weeks later prior to commencing T&E, as per the licensed posology;21 the impact on clinic capacity should be considered. |
 |
Table 1 Clinical Factors to Consider When Determining the Suitability of Patients with nAMD for First-Line Treatment with a Ranibizumab Biosimilar |
In such cases, where there is capacity and following fully informed consent, ranibizumab biosimilar treatment would be initiated in eligible patients with a loading phase of a minimum of three 4-weekly doses followed by a standard T&E regimen for injections thereafter, with the option for a 2-week interval extension according to the licensed posology.18–20
At Week 12 (four weeks after the third loading dose) and at all subsequent injection visits, disease activity should be assessed to determine if the response to treatment is suboptimal. If, at any assessment, the clinician determines that disease activity is present or the treatment interval cannot be extended sufficiently, switching from a ranibizumab biosimilar to an alternative anti-VEGF agent should be considered. This consensus outlines how and when to switch to aflibercept by commencing treatment on the aflibercept T&E pathway recommended by Bailey et al (Figure 2).7
 |
Figure 2 The aflibercept T&E pathway for the treatment of patients with nAMD. Abbreviations: IVT, intravitreal injection; nAMD, neovascular age-related macular degeneration; OCT, optical coherence tomography; q4, every 4 weeks; q16, every 16 weeks; RPE, retinal pigment epithelium; T&E, treat-and-extend; VA, visual acuity. Notes: Figure reproduced under a Creative Commons BY 4.0 license (http://creativecommons.org/licenses/by/4.0/) from Bailey C, Cackett P, Kotagiri A, et al. Practical implementation of a q4-q16 aflibercept treat-and-extend pathway for the treatment of neovascular age-related macular degeneration: updated guidance from a UK expert panel. Eye. 2023;37(9):1916–1921.7 |
Initiating Patients on the Early Treatment Optimization Pathway
Determining Patient Suitability
The NHSE commissioning recommendations include factors for consideration when commencing first-line treatment with ranibizumab biosimilars. Alternative agents such as aflibercept, brolucizumab, or faricimab should be considered (subject to relevant National Institute for Health and Care Excellence [NICE] technology appraisal guidance) where there are contraindications to ranibizumab biosimilars or in circumstances where ranibizumab biosimilars are not deemed clinically appropriate for the individual patient, such as non-response to ranibizumab in the fellow eye previously, subretinal bleed >50% of lesion, and/or diagnosis of idiopathic polypoidal choroidal vasculopathy (PCV).11
Of note, PCV is typically more prevalent in Asian populations than in white populations.22 Indocyanine green angiography, the gold standard for PCV diagnosis, is not commonly used in many centers when diagnosing patients with nAMD and thus underdiagnosis of PCV may be common; however, findings using this imaging technique suggest that up to 9% of Caucasian patients with nAMD in the UK may have PCV (NB the reference uses the term “Caucasian”, which we have followed here despite the term being considered outdated).22,23 Since the presence of idiopathic PCV is only one of several factors listed by NHSE when considering treatment with ranibizumab biosimilars, it is likely that a further proportion of the patient population may not be suitable for this treatment option in UK clinical practice.11
Treatment with ranibizumab biosimilars should only proceed if no contraindications to drug usage are identified, as detailed in the ranibizumab summaries of product characteristics.18–20,24
Recommendations: In line with previous recommendations, several factors should be considered when determining if a patient is suitable for commencing treatment with a ranibizumab biosimilar (Table 1). Notably, the fellow eye is an important factor; patients may be unsuitable for a ranibizumab biosimilar if they have experienced a suboptimal or no response to prior ranibizumab treatment in the fellow eye.
Informed Consent
Guidance from the Royal College of Ophthalmologists highlights the importance of providing sufficient information to help patients reach an informed decision and provide informed consent, including details on disease pathology and progression; available treatment options; and the likelihood of repeated injections and their frequency.25 UK guidance on biosimilar licensing also notes that all biosimilars should be prescribed by the specific brand name, with both the prescriber and the patient aware of the brand name of the product received.26,27
Recommendations: Consent from the patient should be documented at pathway outset and include the name of the prescribed drug and proprietary names of any reference and biosimilar products. The patient should be given detailed information regarding the injection procedure and its associated risks, as well as the possibility of switching agents, even after a short period of treatment. Prior counsel may facilitate switching treatment; however, re-consenting will be required at the time of the switch. Patients should be made aware of the available licensed anti-VEGF agents and their individual risks and benefits prior to treatment initiation.
Capacity Limitations
Clinic capacity must be assessed when considering the use of ranibizumab biosimilars. The NHSE commissioning recommendations state that ranibizumab biosimilars should only be considered where there is capacity to do so, but no further information on capacity requirements is provided.11
Inadequate clinic capacity to either assess or treat patients delays the provision of care and can lead to patient harm. Multiple factors may negatively affect capacity when implementing ranibizumab biosimilars according to the early treatment optimization pathway. Treatment according to the pathway is associated with a minimum of eight clinic visits in Year 1, including the requirement for optical coherence tomography (OCT) assessment four weeks after the third loading dose to determine initial treatment response. Switching agents in cases of suboptimal response to a ranibizumab biosimilar also has capacity implications, although the additional resource requirements (eg to obtain mandatory re-consent) should not hinder a timely switch in the interest of achieving the best possible outcomes for the patient.
Recommendations: Capacity limitations should determine whether clinics are able to implement ranibizumab biosimilars in clinical practice and must, therefore, be considered prior to treatment initiation. Only clinics that have adequate capacity to monitor patients using OCT at four weeks after the third loading dose with a ranibizumab biosimilar should consider using the early treatment optimization pathway.
Managing Patients with a Suboptimal Response to Ranibizumab Biosimilars
To optimize both long-term clinical outcomes for patients and service efficiency and capacity for clinics, suboptimal responses to ranibizumab and its biosimilars should be determined at the earliest possible opportunity. This allows patients to be switched in a timely fashion to an alternative anti-VEGF agent. Criteria for determining suboptimal response to treatment are therefore required.
The licensed posology in nAMD for ranibizumab and its biosimilars states that one injection per month should be administered until maximum visual acuity is achieved and/or there are no signs of disease activity as assessed by visual and/or anatomic parameters, with the option to extend intervals with T&E thereafter.18–20,24 Several factors have been identified as potential disease activity markers in nAMD, including retinal fluid, subretinal hyperreflective material, pigment epithelial detachment, and hemorrhage.28
In a previous UK consensus, Amoaku et al recommended that anti-VEGF responsiveness in nAMD is assessed using visual acuity and macular morphology on spectral domain OCT at any time after the loading phase, noting that morphological changes precede loss of vision.29 Good visual response was defined as a gain in visual acuity of >5 Early Treatment Diabetic Retinopathy Study letters from baseline; good anatomic response was defined as an absence of subretinal fluid, intraretinal fluid, or intraretinal cysts, or a reduction in central retinal thickness of >75% from baseline, although tolerance of some persistent subretinal fluid in nAMD is a debated topic in medical retina.29–31 Amoaku et al concluded that primary response to anti-VEGF treatment is best determined one month after the final loading dose, and maintained treatment response and late response can be identified from Months 4 to 11 and Month 12 onward, respectively.29
NHSE states that in cases of suboptimal response, clinicians should consider stopping treatment or switching to an alternative anti-VEGF agent; if ranibizumab biosimilars were used initially, switching to aflibercept, brolucizumab, or faricimab should be considered.11 Additionally, in line with NICE guidance, continuing treatment is only recommended for patients with an adequate response to therapy.11,32
Recommendations: Clinicians should monitor for functional and anatomic changes in response to ranibizumab biosimilar at all visits after the final loading dose, including the post-loading review at Week 12. Suboptimal response should be determined at the discretion of the clinician using the disease activity criteria listed in Figure 1. Notably, response may be considered suboptimal if any anatomic disease activity criteria are met, even if vision improves. Response may also be considered suboptimal if the treatment interval cannot be extended sufficiently, as defined by clinician and patient decision. In cases of suboptimal response at any visit after the loading phase, including the post-loading review at Week 12, clinicians should consider switching from a ranibizumab biosimilar to an alternative anti-VEGF agent; however, if a patient displays a suboptimal response at a treatment interval of >8 weeks, clinicians may first consider shortening the treatment interval (patient factors and patient preference may affect this decision) (Figure 1). Cases in which there is no response or a dramatic decline in response to an anti-VEGF agent warrant investigation of differential diagnoses or treatment-refractory nAMD.
Switching Patients from Ranibizumab Biosimilars to Aflibercept 2 Mg in Clinical Practice
At the time these consensus recommendations were developed, there was no published real-world evidence of ranibizumab biosimilars nor any head-to-head studies comparing ranibizumab biosimilars with aflibercept 2 mg. However, several comparison studies suggest that favorable outcomes may be achieved with aflibercept compared with reference ranibizumab in patients with nAMD.33–37
In the pivotal Phase III VIEW trials, patients were randomized to receive aflibercept 0.5 mg every 4 weeks (q4), aflibercept 2 mg q4, aflibercept 2 mg every 8 weeks (q8), or ranibizumab 0.5 mg q4 following the loading phase. Compared with ranibizumab, all aflibercept arms were clinically equivalent and met the primary endpoint of non-inferiority in the proportion of patients maintaining vision at Week 52, with similar anatomic improvements across all treatment arms.38 However, a post hoc analysis found that 29.8% and 33.5% of eyes treated with aflibercept 2 mg q4 and q8, respectively, had retinal fluid at Week 12 compared with 41.5% of eyes treated with ranibizumab q4, which may suggest a numerically greater drying effect with aflibercept versus ranibizumab.33
Aflibercept may also be associated with a reduction in treatment burden compared with ranibizumab.34,35 A post hoc analysis of the VIEW trials found that, following use of a modified extended dosing regimen in Year 2, a numerically greater proportion of patients received ≤3 injections from Week 52 to Week 96 with aflibercept 2 mg than with ranibizumab (54%, 48%, and 43% in the aflibercept q4 arm, aflibercept q8 arm, and ranibizumab q4 arm, respectively) while maintaining vision gains.34 Furthermore, a network meta-analysis of major clinical trials noted comparable vision gains with significantly fewer injections over two years with aflibercept T&E when indirectly compared with ranibizumab T&E.35
Functional outcomes with ranibizumab may vary significantly in clinical practice, with vision gains typically smaller than those reported in the pivotal ranibizumab trials.36 In contrast, although undertreatment with anti-VEGF therapies continues to be a challenge in clinical practice, real-world vision gains with aflibercept are generally closer to those reported in the pivotal aflibercept trials.36,39 Moreover, despite some previous evidence suggesting similar outcomes with aflibercept and ranibizumab, a 2020 meta-analysis of 23 real-world studies found that, compared with ranibizumab, aflibercept achieved greater vision gains, with fewer injections required to achieve clinically meaningful functional improvements in clinical practice.36,40 A real-world study comparing switch and non-switch cohorts found that switching from ranibizumab to aflibercept improved retinal morphology and prolonged planned treatment intervals, irrespective of previous anti-VEGF response.37
Recommendations: Given the potential for favorable outcomes with aflibercept over reference ranibizumab in clinical practice, and that biosimilars must be clinically equivalent to the reference medicine for approval, aflibercept is a recommended alternative for patients receiving ranibizumab biosimilars who experience a suboptimal response at any visit after the loading phase.
Initiating Aflibercept T&E in Switch Patients
When switching from ranibizumab to an alternative agent such as aflibercept 2 mg, the impact on treatment capacity should continue to be considered.
Both the ALTAIR and ARIES clinical trials of aflibercept T&E found that a notable proportion of patients can be extended to dosing every 16 weeks (q16) while maintaining vision gains; 41.5%–46.3% of patients were on q16 dosing at Week 96 in ALTAIR and 26.9%–30.2% of patients were on ≥q16 dosing at Week 104 in ARIES.41,42 Taking this into account, Bailey et al developed a consensus aflibercept T&E pathway (Figure 2) with guidance for UK clinics, including those with capacity limitations, to help alleviate capacity challenges while improving patient outcomes and experience.7
Despite the need to re-consent patients and reinitiate the loading phase, which may initially contribute to treatment burden, switching from ranibizumab biosimilars to aflibercept via the aflibercept T&E pathway (Figure 2) in cases of suboptimal response has the potential to improve long-term outcomes for patients with extended treatment intervals up to 16 weeks. This potential reduction in treatment burden may not only benefit patients but could also improve service capacity.
Recommendations: When switching patients from ranibizumab biosimilars to aflibercept 2 mg, clinicians should consider implementing the aflibercept T&E pathway recommended by Bailey et al (Figure 2).7 Three 4-weekly loading doses should be administered, followed by an injection 8 weeks later prior to commencing T&E, as per the licensed posology.21 OCT should be assessed at baseline, at the fourth injection visit (8 weeks after the third loading dose), and at all subsequent treatment visits. Clinicians should be cautious when deciding to extend aflibercept treatment intervals by 2 or 4 weeks, as patients requiring a switch from a ranibizumab biosimilar are likely to demonstrate signs of disease activity.
Closing Comments
The increasing prevalence of nAMD and accompanying rise in economic burden of treatment are anticipated to increase pressure to use the lowest cost treatment option where appropriate. However, when exploring alternative treatment options within a service, the wider costs of treatment beyond the drug itself must be considered. These include, but are not limited to, the burden of particular agents and their impact on different treatment pathways in capacity-challenged settings, as well as patient-specific factors that may contribute to drug choice.
As further ranibizumab biosimilars are launched, a clear understanding of how best to implement their use in clinical practice is required; this is especially relevant for UK services (particularly those with capacity challenges) that are considering the use of ranibizumab biosimilars as a first-line treatment in cases where they may not have considered use of reference ranibizumab previously.
As outlined in this consensus, many factors must be considered to determine if ranibizumab biosimilars can be used; these include patient suitability (including fellow eye considerations), capacity considerations, and the recommendation to obtain agent-specific patient consent before treatment initiation. In cases where patients are given ranibizumab biosimilars as a first-line treatment, this consensus treatment pathway provides guidance on optimizing patient outcomes in treatment-naive eyes with nAMD. This guidance includes how to identify suboptimal responders at an early stage and how to ensure a timely switch to alternative agents, such as aflibercept via the T&E pathway recommended by Bailey et al.7 As such, this guidance supports evidence-based clinical decision-making through providing a framework for the efficient management of service capacity and optimal long-term clinical response for patients with nAMD.
Abbreviations
nAMD, neovascular age-related macular degeneration; NHS, National Health Service; NHSE, NHS England; NICE, National Institute for Health and Care Excellence; OCT, optical coherence tomography; PCV, polypoidal choroidal vasculopathy; PRN, pro re nata; q4, every 4 weeks; q8, every 8 weeks; q16, every 16 weeks; T&E, treat-and-extend; VEGF, vascular endothelial growth factor.
Acknowledgments
Medical writing support was provided by Catherine McEnhill of Porterhouse Medical Group, UK, and funded by Bayer plc, UK, in accordance with Good Publication Practice (GPP3) guidelines.
Bayer-affiliated authors were included based on their relevant clinical experience and standing as ophthalmology experts in their own right and to provide a compliance perspective. Bayer checked that the content was factually accurate, balanced, and compliant with the Association of the British Pharmaceutical Industry Code of Practice.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
All authors except PM-W, JN, CO’N, and TS received honoraria from Bayer plc, UK, for their contributions to the round-table meetings. The round-table meetings were organized and funded by Bayer. The production of this manuscript was facilitated through medical writing support funded by Bayer.
Disclosure
NN: Advisory activity for Bayer, Novartis, and Roche; speaker fees from Bayer, Novartis, and Roche; travel grants from Bayer, Novartis, and Roche. CB: Advisor / travel fees: Bayer, Novartis, Alimera Sciences, Roche, Janssen, Apellis, Boehringer Ingelheim; speaker fees: Apellis, Alimera Sciences, Bayer, Roche. LD: Advisory board and/or speaker fees: Alimera, Allergan (AbbVie), Bausch and Lomb, Bayer, Biogen, Novartis, Roche; travel grants: Allergan, Bayer, Novartis, Roche. RG: Fees from: Apellis, AbbVie, Alimera, Amgen, Astellas, Bayer, Biogen, Boehringer Ingelheim, Heidelberg, Novartis, Notal Vision, Roche, Santen. AK: Advisory board attendances for AbbVie, Bayer, Novartis, and Roche; speaker fees from AbbVie and Bayer; travel grants from AbbVie, Bayer, and Novartis. IP: Advisor fees: Apellis, Bayer, Biogen, Boehringer Ingelheim, Novartis, Roche; lecture/travel fees: Apellis, Bayer, Biogen, Roche. CR: Consultant for Allergan (a subsidiary of AbbVie Ltd) and Alimera; advisory board attendances for Alimera Sciences, Allergan (a subsidiary of AbbVie Ltd), Bayer, Novartis, and Roche; travel grants from Bayer; sponsored research with Allergan (a subsidiary of AbbVie Ltd). SS: Financial support from AbbVie, Amgen, Apellis, Bayer, Biogen, Boehringer Ingelheim, Novartis, Eyebiotech, Janssen Pharmaceuticals, Novo Nordisk, Optos, Ocular Therapeutix, OcuTerra, Roche; Editor-in-Chief for Eye. JT: Personal fees/grants for clinical trials from Bayer, Novartis, and Roche; sponsored research with Alexion, Bayer, Janssen, and Roche. PM-W, JN, CO’N, and TS: Employees of Bayer plc, UK. The authors report no other conflicts of interest in this work.
References
1. Lanzetta P, Loewenstein A. Vision Academy Steering Committee. Fundamental principles of an anti-VEGF treatment regimen: optimal application of intravitreal anti–vascular endothelial growth factor therapy of macular diseases. Graefes Arch Clin Exp Ophthalmol. 2017;255(7):1259–1273. doi:10.1007/s00417-017-3647-4
2. Daien V, Finger RP, Talks JS, et al. Evolution of treatment paradigms in neovascular age-related macular degeneration: a review of real-world evidence. Br J Ophthalmol. 2021;105(11):1475–1479. doi:10.1136/bjophthalmol-2020-317434
3. Victor AA, Putri YM. Pro re nata versus fixed aflibercept regimen for neovascular age-related macular degeneration: a systematic review and meta-analysis. Int J Retina Vitreous. 2022;8(1):67. doi:10.1186/s40942-022-00416-x
4. Li E, Donati S, Lindsley KB, Krzystolik MG, Virgili G. Treatment regimens for administration of anti-vascular endothelial growth factor agents for neovascular age-related macular degeneration. Cochrane Database Syst Rev. 2020;5(5):CD012208. doi:10.1002/14651858.CD012208.pub2
5. Bayer AG. Eylea® (Aflibercept) 40 Mg/Ml Solution for Injection [Summary of Product Characteristics]. Leverkusen: Bayer AG; 2024.
6. Regeneron Pharmaceuticals, Inc. Eylea® (Aflibercept) Injection, for Intravitreal Use [Highlights of Prescribing Information]. Tarrytown: Regeneron Pharmaceuticals, Inc.; 2023.
7. Bailey C, Cackett P, Kotagiri A, et al. Practical implementation of a q4-q16 aflibercept treat-and-extend pathway for the treatment of neovascular age-related macular degeneration: updated guidance from a UK expert panel. Eye. 2023;37(9):1916–1921. doi:10.1038/s41433-022-02264-3
8. ASRS Global Trends in Retina. 2019 global trends in retina survey; 2019. Available from: https://www.asrs.org/content/documents/2019-global-trends-survey-for-website.pdf.
9. Lukic M, Eleftheriadou M, Hamilton RD, Rajendram R, Bucan K, Patel PJ. Four-year outcomes of aflibercept treatment for neovascular age-related macular degeneration: results from real-life setting. Eur J Ophthalmol. 2021;31(4):1940–1944. doi:10.1177/1120672120938565
10. Traine PG, Pfister IB, Zandi S, Spindler J, Garweg JG. Long-term outcome of intravitreal aflibercept treatment for neovascular age-related macular degeneration using a “treat-and-extend” regimen. Ophthalmol Retina. 2019;3(5):393–399. doi:10.1016/j.oret.2019.01.018
11. National Health Service England. Operational note: updated commissioning recommendations for medical retinal vascular medicines following the national procurement for ranibizumab biosimilars; 2023. Available from: https://www.england.nhs.uk/long-read/operational-note-updated-commissioning-recommendations-for-medical-retinal-vascular-medicines-following-The-national-procurement-for-ranibizumab-biosimilars/.
12. Buchan JC, Norman P, Shickle D, Cassels-Brown A, MacEwen C. Failing to plan and planning to fail. Can we predict the future growth of demand on UK eye care services? Eye. 2019;33(7):1029–1031. doi:10.1038/s41433-019-0383-5
13. Derbyshire M, Shina S. Patent expiry dates for biologicals: 2018 update. GaBI J. 2019;8(1):24–31. doi:10.5639/gabij.2019.0801.003
14. Kaiser PK, Schmitz-Valckenberg MS, Holz FG. Anti–vascular endothelial growth factor biosimilars in ophthalmology. Retina. 2022;42(12):2243–2250. doi:10.1097/IAE.0000000000003626
15. Midas Pharma GmbH. Ranivisio® (Ranibizumab) 10 Mg/Ml Solution for Injection [Summary of Product Characteristics]. Ingelheim: Midas Pharma GmbH; 2023.
16. Samsung Bioepis NL B.V. Byooviz™ (Ranibizumab) 10 Mg/Ml Solution for Injection [Summary of Product Characteristics]. Delft: Samsung Bioepis NL B.V.; 2024.
17. STADA Arzneimittel AG. Ximluci® (Ranibizumab) 10 Mg/Ml Solution for Injection [Summary of Product Characteristics]. Bad Vilbel: STADA Arzneimittel AG.; 2024.
18. Samsung Bioepis UK Limited. Byooviz™ (Ranibizumab) 10 Mg/Ml Solution for Injection [Summary of Product Characteristics]. Middlesex: Samsung Bioepis UK Limited; 2023.
19. Midas Pharma GmbH. Ongavia® (Ranibizumab) 10 Mg/Ml Solution for Injection [Summary of Product Characteristics]. Ingelheim: Midas Pharma GmbH; 2023.
20. Genus Pharmaceuticals Ltd. Ximluci® (Ranibizumab) 10 Mg/Ml Solution for Injection [Summary of Product Characteristics]. Huddersfield: Genus Pharmaceuticals Ltd.; 2023.
21. Bayer plc. Eylea® (Aflibercept) 40mg/Ml Solution for Injection in a Vial [Summary of Product Characteristics]. Reading: Bayer plc; 2023.
22. Kokame GT, deCarlo TE, Kaneko KN, Omizo JN, Lian R. Anti-vascular endothelial growth factor resistance in exudative macular degeneration and polypoidal choroidal vasculopathy. Ophthalmol Retina. 2019;3(9):744–752. doi:10.1016/j.oret.2019.04.018
23. Yadav S, Parry DG, Beare NAV, Pearce IA. Polypoidal choroidal vasculopathy: a common type of neovascular age-related macular degeneration in Caucasians. Br J Ophthalmol. 2017;101(10):1377–1380. doi:10.1136/bjophthalmol-2016-310074
24. Novartis Pharmaceuticals UK Limited. Lucentis® (Ranibizumab) 10 Mg/Ml Solution for Injection [Summary of Product Characteristics]. London: Novartis Pharmaceuticals UK Limited; 2023.
25. The Royal College of Ophthalmologists. Commissioning guidance: age related macular degeneration services; 2021. Available from: https://curriculum.rcophth.ac.uk/wp-content/uploads/2021/07/AMD-Commissioning-Guidance-Full-June-2021.pdf.
26. Medicines & Healthcare products Regulatory Agency. Guidance on the licensing of biosimilar products; 2022. Available from: https://www.gov.uk/government/publications/guidance-on-the-licensing-of-biosimilar-products/guidance-on-the-licensing-of-biosimilar-products.
27. National Health Service England. What is a biosimilar medicine?; 2023. Available from: https://www.england.nhs.uk/long-read/what-is-A-biosimilar-medicine/.
28. Kodjikian L, Parravano M, Clemens A, et al. Fluid as a critical biomarker in neovascular age-related macular degeneration management: literature review and consensus recommendations. Eye. 2021;35(8):2119–2135. doi:10.1038/s41433-021-01487-0
29. Amoaku WM, Chakravarthy U, Gale R, et al. Defining response to anti-VEGF therapies in neovascular AMD. Eye. 2015;29(6):721–731. doi:10.1038/eye.2015.48
30. Holekamp NM, Sadda S, Sarraf D, et al. Effect of residual retinal fluid on visual function in ranibizumab-treated neovascular age-related macular degeneration. Am J Ophthalmol. 2022;233:8–17. doi:10.1016/j.ajo.2021.06.029
31. Jaffe GJ, Ying G-S, Toth CA, et al. Macular morphology and visual acuity in year five of the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2019;126(2):252–260. doi:10.1016/j.ophtha.2018.08.035
32. National Institute for Health and Care Excellence. Age-related macular degeneration: NICE guideline [NG82]; 2018. Available from: https://www.nice.org.uk/guidance/ng82.
33. Moshfeghi DM, Hariprasad SM, Marx JL, et al. Effect of fluid status at week 12 on visual and anatomic outcomes at week 52 in the VIEW 1 and 2 trials. Ophthalmic Surg Lasers Imaging Retina. 2016;47(3):238–244. doi:10.3928/23258160-20160229-06
34. Richard G, Monés J, Wolf S, et al. Scheduled versus pro re nata dosing in the VIEW trials. Ophthalmology. 2015;122(12):2497–2503. doi:10.1016/j.ophtha.2015.08.014
35. Ohji M, Lanzetta P, Korobelnik J-F, et al. Efficacy and treatment burden of intravitreal aflibercept versus intravitreal ranibizumab treat-and-extend regimens at 2 years: network meta-analysis incorporating individual patient data meta-regression and matching-adjusted indirect comparison. Adv Ther. 2020;37(5):2184–2198. doi:10.1007/s12325-020-01298-x
36. Carrasco J, Pietsch G-A, Nicolas M-P, Koerber C, Bennison C, Yoon J. Real-world effectiveness and real-world cost-effectiveness of intravitreal aflibercept and intravitreal ranibizumab in neovascular age-related macular degeneration: systematic review and meta-analysis of real-world studies. Adv Ther. 2020;37(1):300–315. doi:10.1007/s12325-019-01147-6
37. Granstam E, Aurell S, Sjövall K, Paul A. Switching anti-VEGF agent for wet AMD: evaluation of impact on visual acuity, treatment frequency and retinal morphology in a real-world clinical setting. Graefes Arch Clin Exp Ophthalmol. 2021;259(8):2085–2093. doi:10.1007/s00417-020-05059-y
38. Heier JS, Brown DM, Chong V, et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology. 2012;119(12):2537–2548. doi:10.1016/j.ophtha.2012.09.006
39. Monés J, Singh RP, Bandello F, Souied E, Liu X, Gale R. Undertreatment of neovascular age-related macular degeneration after 10 years of anti-vascular endothelial growth factor therapy in the real world: the need for a change of mindset. Ophthalmologica. 2020;243(1):1–8. doi:10.1159/000502747
40. Gillies MC, Nguyen V, Daien V, Arnold JJ, Morlet N, Barthelmes D. Twelve-month outcomes of ranibizumab vs. aflibercept for neovascular age-related macular degeneration: data from an observational study. Ophthalmology. 2016;123(12):2545–2553. doi:10.1016/j.ophtha.2016.08.016
41. Ohji M, Takahashi K, Okada AA, et al. Efficacy and safety of intravitreal aflibercept treat-and-extend regimens in exudative age-related macular degeneration: 52- and 96-week findings from ALTAIR; a randomized controlled trial. Adv Ther. 2020;37(3):1173–1187. doi:10.1007/s12325-020-01236-x
42. Mitchell P, Holz FG, Hykin P, et al. Efficacy and safety of intravitreal aflibercept using a treat-and-extend regimen for neovascular age-related macular degeneration: the ARIES study: a randomized clinical trial. Retina. 2021;41(9):1911–1920. doi:10.1097/IAE.0000000000003128
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.