")
Back to Journals » Journal of Inflammation Research » Volume 17
High Translocation of High Mobility Group Box 1 in the Brain Tissue of Patients with Sturge-Weber Syndrome
Authors Cheng Z, Li X, Wang S , Sun W, Pan J, Wang X, Zhou J, Li T, Luan G, Guan Y
Received 11 April 2024
Accepted for publication 15 November 2024
Published 21 November 2024 Volume 2024:17 Pages 9347—9358
DOI https://doi.org/10.2147/JIR.S473377
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Zizhang Cheng,1,* Xiaoli Li,2,* Shu Wang,1 Weijin Sun,1 Junhong Pan,1 Xiongfei Wang,1 Jian Zhou,1 Tianfu Li,3– 5 Guoming Luan,1,4,5 Yuguang Guan1,4,5
1Department of Neurosurgery, SanBo Brain Hospital, Capital Medical University, Beijing, 100093, People’s Republic of China; 2Department of Neurology, Affiliated Zhong Da Hospital, Southeast University, Nanjing, People’s Republic of China; 3Department of Neurology, Sanbo Brain Hospital, Capital Medical University, Beijing, People’s Republic of China; 4Beijing Key Laboratory of Epilepsy, Beijing, 100093, People’s Republic of China; 5Center of Epilepsy, Beijing Institute of Brain Disorders, Collaborative Innovation Center for Brain Disorders, Laboratory for Clinical Medicine, Capital Medical University, Beijing, 100093, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yuguang Guan; Guoming Luan, Department of Neurosurgery, SanBo Brain Hospital Capital Medical University, Beijing, 100093, People’s Republic of China, Email [email protected]; [email protected]
Purpose: Sturge-Weber syndrome (SWS), a rare congenital neurological and skin disorder, is frequently associated with drug-resistant epilepsy. Translocation of high mobility group box 1 (HMGB1) protein from the nucleus to the cytoplasm or extracellular milieu has been implicated in neuroinflammatory processes contributing to the development of epileptogenesis. This study aimed to explore the expression and distribution of HMGB1 in brain tissue from SWS patients with drug-resistant epilepsy, with the goal of elucidating its potential involvement in the pathogenesis of epilepsy.
Patients and Methods: The study enrolled eight patients with drug-resistant epilepsy who underwent hemispherectomy. Brain tissue specimens were obtained and analyzed using immunofluorescence staining to detect HMGB1 distribution in microglia, astrocytes, or different neuronal subtypes. Correlation analyses were performed to investigate the potential relationship between HMGB1 translocation within cells and the clinical characteristics of SWS patients.
Results: In lesional tissues of SWS patients, we observed significantly higher cytoplasmic HMGB1 levels. Meanwhile, HMGB1 was widely distributed in the cytoplasm of microglia and neurons, while in astrocytes, it was primarily localized in the nucleus. This translocation occurred across many neuronal subtypes, including excitatory glutamatergic, inhibitory GABAergic, and cholinergic neurons. The lower proportion of HMGB1-translocated cholinergic neurons was seen compared to the other two neuronal subtypes. Furthermore, no correlation was found between cytoplasmic HMGB1 levels and clinical characteristics of SWS patients.
Conclusion: The results suggest the involvement of HMGB1 in the pathogenesis of drug-resistant epilepsy in SWS patients. Additional research is required to elucidate the precise mechanisms and potential therapeutic targets associated with HMGB1 that underlie the epilepsy linked to SWS.
Keywords: high mobility group box 1, translocation, Sturge Weber syndrome, drug-resistant epilepsy, cell subtype
Introduction
Sturge‒Weber syndrome (SWS) is a rare congenital neurocutaneous disorder, with an incidence ranging approximately from 0.19 to 3.08 per 100,000 people individuals annually across different regions.1 The typical clinical features of SWS encompass port-wine stains associated with cutaneous capillary or venous malformations, central nervous system abnormalities associated with leptomeningeal vascular malformations, and ocular abnormalities, including glaucoma and choroidal venous malformations.2–5 Among these, abnormalities of the central nervous system often manifest, and epilepsy is a common manifestation. More than 80% of patients with SWS are reported to experience concurrent epilepsy,6–8 and approximately 50% of these individuals develop drug-resistant epilepsy.9–11
Recently, considerable attention has been directed toward the potential pathogenic role of the High Mobility Group Box 1 (HMGB1) protein in the neuroinflammatory hypothesis of epileptogenesis.12,13 HMGB1, a highly conserved 215 amino acid protein with an affinity for DNA binding, plays a pivotal role in inflammation.14–16 Under physiological conditions, HMGB1 predominantly resides in the nucleus of most cells, where it is involved in DNA replication, repair, recombination, transcription, and genomic stability.17,18 However, under pathological conditions, HMGB1 is translocated from the nucleus to the cytoplasm or extracellular milieu.19,20 Upon relocation, HMGB1 interacts with various receptors, initiating the upregulation of cytokines released by pro-inflammatory cells, which in turn contributes to the development of drug-resistant epilepsy.15,16,20–24
The pro-inflammatory cascades triggered by HMGB1 may result in an imbalance between neuronal excitability and inhibition. Previous studies have suggested that HMGB1 contributes to neuronal excitotoxicity and may affect neurotransmitters dynamics, implicating its role in the pathogenesis of epilepsy.25–28 In the present study, we explored the expression and distribution of HMGB1 in abnormal brain tissues and the peripheral zone of lesion in SWS patients with drug-resistant epilepsy. A thorough investigation into the distribution of HMGB1 among various subtypes of glial cells and neurons, including excitatory, inhibitory, and cholinergic neurons, was conducted to delineate HMGB1’s differential impacts across these cell types. Furthermore, correlation analyses were undertaken to explore the potential association between HMGB1 translocation and the clinical manifestations of SWS patients, thereby providing novel insights and evidence towards elucidating the pathogenesis of drug-resistant epilepsy in SWS.
Material and Methods
Inclusion and Exclusion Criteria
This study was a retrospective analysis of patients with SWS complicated by epilepsy treated with focal resection between January 2020 and April 2022 at SanBo Brain Hospital of Capital Medical University (Beijing, China). All study procedures were performed in accordance with the guidelines outlined in the World Medical Association Code of Ethics, known as the Declaration of Helsinki. The study was approved by Ethics Committee of Sanbo Brain Hospital of Capital Medical University, and written informed consent was obtained from patient proxies. Patient proxies each agreed to the procedure and consented to the anonymous use of patient data for the purposes of the study. The investigators recorded clinical characteristics, including sex, the presence of facial hemangiomas, the presence of soft meningeal hemangiomas, and the presence of glaucoma. Various characteristics of epilepsy were also recorded, including the time of the first seizure, seizure type, seizure frequency, and location of epileptiform discharges observed on video electroencephalogram (VEEG).
Similar to our previous studies,2 for inclusion in the study, these patients were required to meet the following indications for surgery: (1) Patients met the clinical diagnosis of SWS, ie, imaging-confirmed soft meningeal hemangioma, often with typical imaging manifestations such as brain atrophy and foci of calcification, with or without typical clinical manifestations such as facial vascular nevus, glaucoma, cognitive developmental abnormalities, motor dysfunction, and postoperative pathology to aid in further confirmation of the diagnosis. (2) Patients were diagnosed with epilepsy by at least two experienced neurologists according to the International League Against Epilepsy 2017 classification. These patients had drug-resistant epilepsy, which is defined as failure to achieve sustained seizure freedom despite regular use of at least two antiepileptic drugs for 6 months. These patients also had motor deficits or cognitive decline. (3) The area at which surgery was scheduled to be performed could be determined by a detailed preoperative evaluation, such as a specific discharge site on VEEG and a clear lesion site on cranial MRI.
Exclusion criteria for this study were as follows: (1) the presence of a significant psychiatric disorder (eg, major depression or schizophrenia) prior to admission; (2) a clear history of central nervous system disease (eg, cerebral ischemia, cerebral hemorrhage, etc).; and (3) the presence of contraindications to anesthesia or surgery, such as severe underlying disease or other systemic disease (eg, severe diabetes mellitus, heart failure, etc).
Assessment
At least two independent and experienced neurologists were available for evaluation at the time of admission. Radiologically, each patient underwent a CT scan (2 mm, axial, Philips) and an MRI scan (1.5 T, Siemens or 3.0 T, GE). Each patient also underwent a minimum of 16 hours of 64-channel interictal and interictal scalp VEEG. In addition, all patients underwent assessments of cognitive function. Patients younger than 6 years of age were administered the Denver Developmental Screening Test-II. The results of this test were classified into three categories: “abnormal”, lagging behind peers in any area; “doubtful”, suspected abnormality in some areas; and normal, no abnormality in any area. Both “abnormal“ and ”doubtful” were considered to be cognitive decline. For individuals who are six years of age or older, the intelligence quotient (IQ) was evaluated using the Wechsler Children Intelligence Scale-IV (for individuals under the age of 16). Motor function was assessed by a professional neurologist on the basis of muscle strength, muscle tone, joint mobility, balance, coordination, and gait evaluations in neurological examinations.
Based on the different clinical presentations of SWS patients, SWS was classified as follows: type I patients had facial and soft meningeal hemangiomas, type II patients had isolated facial hemangiomas (without CNS involvement), and type III patients had isolated soft meningeal hemangiomas.3
Neurologists and neurosurgeons assessed epilepsy primarily through coordination and gait evaluations in neurological examinations.gh history, EEG findings, and clinical presentation. The experts based their diagnosis on the epilepsy classification scheme published by the International League Against Epilepsy (ILAE) in 2017, in which mainly seizure type and epilepsy type are recorded. In this study, the epilepsy of the patient for 3 months prior to admission was used as the baseline for observation. Assessment at admission was performed according to the patient’s condition.
Tissue Preparation
The surgical approach was determined based on the region of brain tissue affected, as detailed in our previous study.2 Tissue specimens were collected from the excised area, including from both the SWS lesion (SWS group) and the surrounding peri-lesion tissue (peri-SWS group), with each sample measuring 1 cubic centimeter. A portion of the tissue was stored at −80°C for protein extraction for Western Blot analysis. Another portion was fixed in 4% buffered formalin, processed into paraffin blocks, sectioned at 4 μm thickness, mounted on pre-coated glass slides, preparing them for immunohistochemical and immunofluorescence staining.
Western Blotting
Western blotting was performed to assess the expression levels of target proteins in total and cytoplasmic extracts from tissue samples. Total proteins were extracted using RIPA lysis buffer containing protease inhibitors, whereas cytoplasmic proteins were isolated with a Nuclear-Cytosol Extraction Kit (P1200, APPLYGEN). Protein concentrations were determined using a BCA Protein Assay Kit. Equal amounts of protein were resolved on 12.5% SDS-PAGE gels and transferred to PVDF membranes. Membranes were blocked with 5% nonfat milk for 1.5 hours at room temperature, followed by overnight incubation at 4°C with specific primary antibodies, such as anti-beta actin (mouse monoclonal, #3700, CST) and anti-HMGB1 (mouse monoclonal, ab190377, Abcam), each at a 1:1,000 dilution, with beta actin serving as the loading control. The next day, membranes were incubated with HRP-conjugated secondary antibodies for 2 hours at room temperature. Protein bands were visualized using enhanced chemiluminescence. All experiments were independently performed three times to ensure reproducibility.
Immunofluorescence Staining
The obtained sections, after dewaxing to water, were subjected to antigen retrieval using citrate buffer (pH 6.0). The tissue sections were then blocked with 5% normal goat serum at room temperature for 1.5 hours. Afterward, sections were incubated overnight at 4°C with primary antibodies to the following proteins: HMGB1 (rabbit monoclonal, 1:400, ab79823, Abcam; mouse monoclonal, 1:200, ab190377, Abcam), Iba1 (mouse monoclonal, 1:300, MABN92, Millipore), GFAP (mouse monoclonal, 1:500, ab4648, Abcam), NeuN (mouse monoclonal, 1:400, ab104224, Abcam), CAMKIIa (mouse monoclonal, 1:300, ab22609, Abcam), CHAT (rabbit monoclonal, 1:300, ab178850, Abcam) and GAD65+GAD67 (rabbit monoclonal, 1:400, ab183999, Abcam). The next day, sections were incubated with Alexa Fluor 594 and Alexa Fluor 488 (anti-rabbit IgG or anti-mouse IgG; 1:200) for 2 hours at room temperature, then restained with DAPI for 2 minutes and covered with glass coverslips. Stained sections were observed using both a fluorescence microscope and a confocal fluorescence microscope. For each patient, 3–4 slices were analyzed, and from each slice, 5 high-magnification fields of view were randomly selected, resulting in a total of 15–20 fields per patient. HMGB1-positive cells, defined as cells showing HMGB1 translocation to the cytoplasm, were identified among different cell types (microglial cells, astrocytes, neurons, and specific neuronal subtypes). The percentage of these HMGB1-positive cells was calculated, ensuring that only cells with visible nuclei in the images were analyzed.
Statistical Analysis
Western Blotting band intensities and Immunofluorescence cell counts were analyzed using ImageJ, with continuous variables expressed as mean ± SD after testing for normality and homogeneity of variance. Paired-sample t-tests compared HMGB1 expression and translocation in SWS lesioned versus peri-lesional tissues, and Spearman correlation assessed relationships between cytoplasmic HMGB1 expression levels and clinical variables. All analyses were conducted using SPSS (version 26), considering P < 0.05 as statistically significant, and figures were generated with GraphPad PRISM (version 8).
Results
Baseline Characteristics
Eight patients (5 males and 3 females, mean age 41.500 ± 30.509 months) were included in this study. All patients were diagnosed with SWS by more than two neuroscientists, all had concurrent epilepsy, and all underwent hemispherectomy. Their mean age at first seizure was 15.125 ± 25.941 months, and the mean epilepsy history duration was 26.375 ± 25.404 months. The baseline characteristics and clinical presentation of these patients are shown in Table 1. All patients presented with focal epilepsy on EEG testing. All patients had leptomeningeal angiomas, while 62.5% (5/8) had clinical manifestations of facial hemangioma, meaning that 5 patients had type I SWS and 3 patients had type II SWS. In this study, the hemangiomas of type I patients were ipsilateral. Notably, one patient had bilateral hemangiomas. To further control variables and ensure consistency, we specifically selected the temporal and parietal lobes lesion tissue from each patient for our relevant studies and discussions. Regarding other characteristic clinical manifestations, 62.5% (5/8) of the patients had glaucoma, 62.5% (5/8) demonstrated cognitive decline, and 62.5% (5/8) had motor deficits.
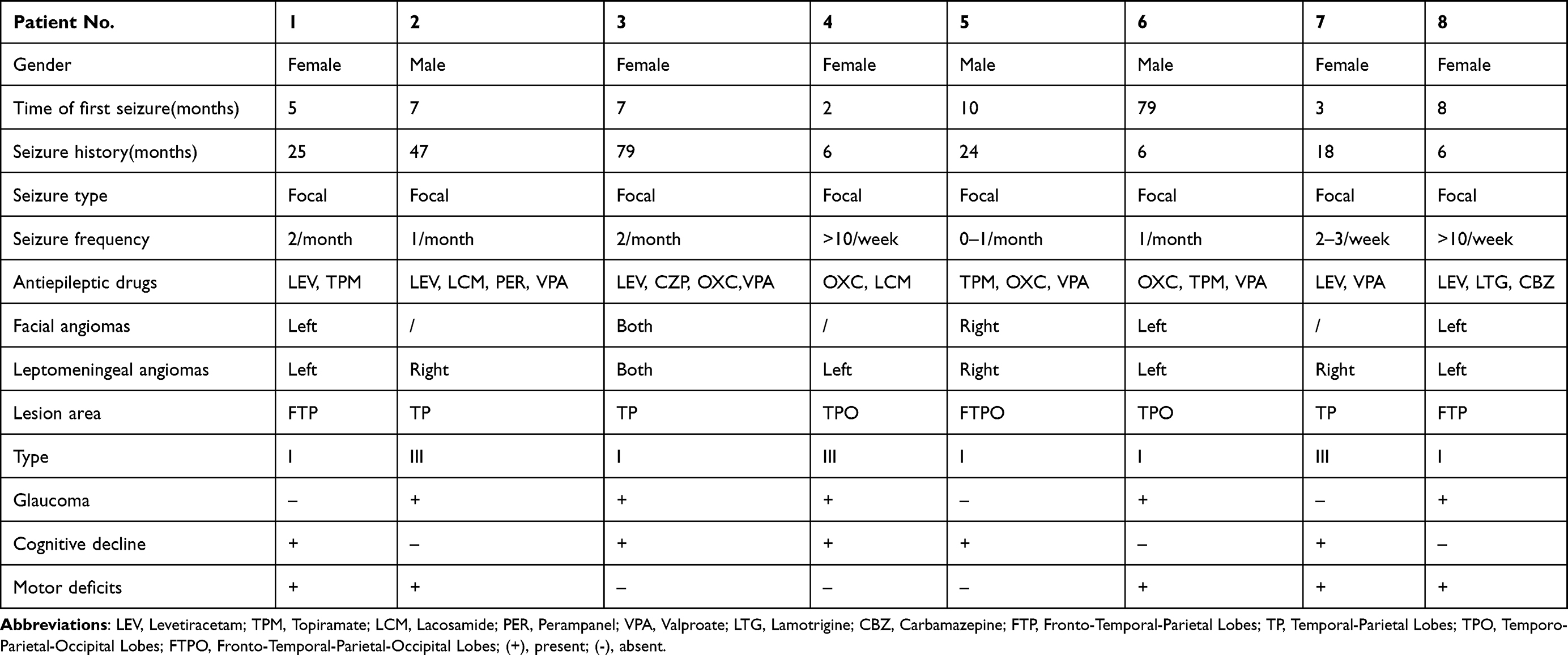 |
Table 1 Clinical Information of SWS Patients with Epilepsy |
Expression of HMGB1 in SWS Patients
HMGB1 levels in total protein showed no significant difference between lesional and perilesional tissues in SWS patients (t = −0.351, p = 0.736; Figure 1B). This suggests that the overall expression of HMGB1 remains consistent between these regions. However, in cytoplasmic protein, HMGB1 levels were markedly elevated in lesional tissue compared to the perilesional area (t = −3.395, p = 0.012; Figure 1D). This increase in cytoplasmic HMGB1 suggests a nuclear-to-cytoplasmic translocation of HMGB1 in lesional cells.
 |
Figure 1 Protein expression of HMGB1 in brain tissue of SWS patients. (A) Western blot results showing the total HMGB1 content in SWS lesion tissue and perilesional (peri-SWS) tissue. (B) Bar graph comparing the total HMGB1 levels between SWS lesion and peri-SWS tissues, revealing no significant difference. (C) Western blot results depicting the HMGB1 content specifically in the cytoplasmic fraction of SWS lesion tissue and peri-SWS tissue. (D) Bar graph illustrating the quantification of cytoplasmic HMGB1 levels, which were significantly higher in the lesion area compared to the perilesional tissue. Beta-actin was used as a loading control. (*P <0.05). |
Distribution of HMGB1 in Glial Cells in SWS Patients
HMGB1 was commonly detected in microglia (Figure 2). HMGB1 was detected in the nuclei of all microglia. In the SWS group of patients, HMGB1 was also distributed in the cytoplasm of microglia. However, little cytoplasmic staining of HMGB1 was observed in the microglia of the control group. That is, an increased proportion of HMGB1-translocated microglia was observed in SWS patients (t=11.26, p < 0.0001; Figure 2Q). In addition, a significant increase in the number of microglia was observed in the SWS group (t = 8.342, p < 0.0001; Figure 2R). In contrast, in astrocytes, HMGB1 was predominantly localized in the nuclei, with no significant distribution in the cytoplasm, regardless of whether it was in lesional or perilesional tissue.
 |
Figure 2 The expression and distribution of HMGB1 in glial cells of SWS lesions and perilesional tissue. HMGB1 immunoreactivity (green) was detected in glial cells, visualized by the glial marker Iba1 (red) (A–D) in SWS lesion tissue (as indicated by arrows in c, d) but not in perilesional tissue (E–H). In astrocytes, HMGB1 was predominantly localized in the nucleus (I–P, as indicated by arrows in k and I). The proportion of HMGB1-translocated glial cells was significantly higher in the SWS group than in the perilesional group (Q). The proportion of glial cells (ie, nuclei of Iba1-positive cells as a percentage of all nuclei) was significantly increased in SWS lesion tissue (R). (****P <0.0001). |
Distribution of HMGB1 in Cortical Neurons of SWS Patients
HMGB1 was ubiquitously detected in neurons in the cerebral cortex of SWS patients (Figure 3). HMGB1 was mainly localized in the nuclei of neurons in the control group. However, a widespread distribution of HMGB1 was observed in both the nuclei and the cytoplasm of neurons in the cortical tissue of SWS patients. The proportion of HMGB1-translocated neurons was increased significantly in the SWS group compared with that in the peri-SWS group (t=15.97, p < 0.0001, Figure 3I).
 |
Figure 3 The expression and distribution of HMGB1 in neurons of SWS lesions and perilesional tissue. HMGB1 immunoreactivity (green) was detected in neurons, which were visualized by the neuronal marker NeuN (red) (A–D) in SWS lesion tissue (as indicated by arrows in c, d) but not in peri-SWS tissue (E–H). The proportion of HMGB1-translocated cells was significantly higher in the SWS group than in the peri-SWS group (I). (****P <0.0001). |
Distribution of HMGB1 in Different Subtypes of Neurons of SWS Patients
HMGB1 was detected in all three subtypes of neurons in the cortex of SWS patients (Figures 4–6). Significant translocation of HMGB1 from the nucleus to the cytoplasm was observed in the abnormal brain tissues of SWS patients, while little HMGB1-translocated cytoplasm of neurons was observed in the perilesional tissue. Additionally, the proportion of HMGB1-translocated CaMKIIa-positive neurons or CHAT-positive neurons was significantly greater in the SWS group than in the peri-SWS group (t = 25.42 or 28.2, p < 0.0001, Figures 4I or 5I). The proportion of HMGB1-translocated GABAergic neurons was also increased significantly (t=27.19, p < 0.0001, Figure 6I).
 |
Figure 4 The expression and distribution of HMGB1 in excitatory neurons of SWS lesions and perilesional tissue. HMGB1 immunoreactivity (green) was detected in neurons visualized by the excitatory neuronal marker CaMKIIa (red) (A–D) in SWS lesion tissue (as indicated by arrows in c, d) but not in peri-SWS tissue (E–H). The proportion of HMGB1-translocated cells was significantly higher in the SWS group than in the peri-SWS group (I). (****P <0.0001). |
 |
Figure 5 The expression and distribution of HMGB1 in cholinergic neurons of SWS lesions and perilesional tissue. HMGB1 immunoreactivity (green) was detected in neurons visualized by the cholinergic neuronal marker CHAT (red) (A–D) in SWS lesion tissue (as indicated by arrows in c, d) but not in peri-SWS tissue (E–H). The proportion of HMGB1-translocated cells was significantly higher in the SWS group than in the peri-SWS group (I). (****P <0.0001). |
 |
Figure 6 The expression and distribution of HMGB1 in cholinergic neurons of SWS lesions and perilesional tissue. HMGB1 immunoreactivity (green) was detected in neurons that were visualized by the cholinergic neuronal marker GAD65+GAD67 (red) (A–D) in SWS lesion tissue (as indicated by arrows in c, d) but not in peri-SWS tissue (E–H). The proportion of HMGB1-translocated cells was significantly higher in the SWS group than in the peri-SWS group (I). (****P <0.0001). |
Furthermore, we explored the difference in the proportion of HMGB1 immunoreactive cells among different subtypes of neurons for each individual. The results showed that there was no significant difference in the HMGB1-translocated proportion of excitatory versus inhibitory neurons in any patient (t=−0.29, p=0.773). However, the proportion of HMGB1-translocated cholinergic neurons was smaller than that of excitatory or inhibitory neurons (t=2.96 or t=3.78, p=<0.05).
Correlation Analysis of Cytoplasmic HMGB1 in Lesional Tissues of SWS Patients
To assess whether cytoplasmic HMGB1 expression in lesional tissues correlates with clinical features in Sturge-Weber syndrome, we measured relative HMGB1 levels and analyzed their association with clinical variables. Using Spearman correlation analysis, we examined correlations between HMGB1 expressions and several clinical variables, including seizure history, seizure frequency, presence of facial and leptomeningeal angiomas, glaucoma, cognitive decline, and motor defects. The results indicated no significant associations between cytoplasmic HMGB1 expression and these clinical features (seizure history: r = 0.317, p > 0.05; seizure frequency: r = 0.352, p > 0.05; facial angiomas: r = 0.456, p > 0.05; leptomeningeal angiomas: r = 0.577, p > 0.05; glaucoma: r = −0.394, p > 0.05; cognitive decline: r = 0.218, p > 0.05; motor defects: r = 0.109, p > 0.05).
Discussion
In this study, we have observed that in patients with Sturge-Weber syndrome (SWS), although the total levels of HMGB1 do not significantly differ between lesional and perilesional tissues, there is a pronounced increase in cytoplasmic HMGB1 within the lesional regions. This finding implies a nuclear-to-cytoplasmic translocation, which could be associated with cellular stress or inflammatory responses. Consistently, several reports underscores HMGB1’s pivotal role as a mediator in neuroinflammatory pathways, notably in epilepsy and related disorders.12–14 After relocation, HMGB1 is capable of binding to receptors such as those for advanced glycation end products (RAGE) and Toll-like receptor 4 (TLR4). This binding activates signaling cascades like the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and mitogen-activated protein (MAP) kinase pathways, leading to the upregulation of cytokines from proinflammatory cells, a process involved in the pathogenesis of drug-resistant epilepsy.15,16,20–24 This body of evidence suggests that HMGB1 serves as a proinflammatory cytokine, contributing significantly to the pathophysiology of epilepsy.
In our study, we particularly focused on the translocation of HMGB1 from the nucleus due to the diverse array of HMGB1 receptors. In the resected abnormal brain tissue of the SWS individuals, HMGB1 was significantly translocated in microglia and neurons compared to the relatively normal tissue in the periphery of the lesion. However, this relocation was not observed in astrocytes. Interestingly, several studies have reported the discovery of the colocalization of HMGB1 with GFAP in patients with epilepsy. HMGB1 translocation in astrocytes has been observed in patients with focal cortical dysplasia (FCD) and epileptic patients with depression.14,23,29 Astrocytes may play a role in the development of epileptogenesis in these patients, as suggested by these researchers. However, our findings indicate that neurons and microglia, rather than astrocytes, are more closely associated with HMGB1-related neuroinflammatory responses in SWS patients with drug-resistant epilepsy.
Microglia play a crucial role in neuroinflammation associated with epilepsy.30 They can produce pro-inflammatory Microglia can produce pro-inflammatory mediators early in the onset of epilepsy, well before morphological cell activation is detectable.31 Previous animal experiments have proven that these pro-inflammatory molecules lower the seizure threshold, and the intensity of the expression correlates with seizure frequency.32–34 In the present experiment, there was an elevated number of microglia in the lesional tissues of SWS patients. Additionally, there was a significant increase in the number of HMGB1-translocated microglia, which to some extent can reflect their role in neuroinflammation in SWS.
Neuronal excitation/inhibition imbalance is a known cause of seizures.35 However, the previous studies did not determine the relocation of HMGB1 in different subtypes of neurons. Therefore, we further examined the distribution of HMGB1 in several subtypes of neurons, including glutamatergic neurons (positive for CaMKIIa), GABAergic neurons (positive for GAD65+GAD67), and cholinergic neurons (positive for ChAT), in SWS patients. In the excised lesion brain tissue of SWS patients, all types of neurons, including excitatory glutamatergic neurons, inhibitory GABAergic neurons, and cholinergic neurons, were HMGB1-translocated cells, demonstrating HMGB1 translocation. There was no difference in the proportion of cells with HMGB1 translocation between excitatory and inhibitory neurons. Therefore, we speculate that there may be a cell-selective activation process downstream of the HMGB1-related pathway, that requires further exploration in a progressive study. Furthermore, the number of excitatory neurons in the cerebral cortex exceeds that of inhibitory neurons, which may contribute to the heightened neuronal excitatory activity during epileptogenesis. However, the precise function of these neuron subtypes in the context of neuroinflammation requires further investigation.
Abnormalities in the functioning of the cholinergic system play a crucial role in the modulation of epilepsy, in addition to the influence of excitatory and inhibitory neurotransmitters on epileptogenesis.36,37 We detected the relocation of HMGB1 in cholinergic neurons. However, the proportion of HMGB1-translocated cholinergic neurons was lower than that in the other two subtypes of neurons. The cholinergic anti-inflammatory pathway (CAP) is a neural-immunomodulatory pathway. When stimulated by immune signals, the efferent vagus nerve releases acetylcholine, which binds to α7 nicotinic acetylcholine receptors. This regulates immune cell function and inhibits inflammatory responses. The CAP helps regulate the body’s immune response to inflammation through acetylcholine. ACh, which is released by cholinergic neurons can regulate immune function in an autocrine/paracrine manner by acting on its receptors.38 From this, we propose that cholinergic neurons may be involved in epileptogenesis in SWS patients and have a lower proportion of HMGB1-translocated cells due to the CAP.
An increasing body of research suggests that HMGB1 may serve as a potential biomarker for epilepsy. Several studies have indicated elevated levels of HMGB1 in the serum and cerebrospinal fluid of epilepsy patients.39,40 Notably, some researchers have also identified a correlation between HMGB1 levels and the severity of epilepsy.39,41 However, in our study, we did not observe a significant association between the relative expression of translocated HMGB1 and the clinical characteristics of patients with SWS. This discrepancy might be attributed to the limited sample size and the cross-sectional nature of our study, which may not adequately capture the dynamic changes in HMGB1 distribution throughout the course of epilepsy. Given the experimental constraints, further research utilizing larger, well-characterized cohorts is necessary to clarify the role of HMGB1 in the pathophysiology and clinical progression of epilepsy in SWS patients.
Conclusion
The objective of this study was to investigate the expression and distribution of HMGB1 in abnormal brain tissues and perilesional areas in individuals with drug-resistant epilepsy and Sturge-Weber Syndrome. Our findings indicate an augmented presence of HMGB1-translocated cells, predominantly in microglia and neurons, but not in astrocytes. Notably, HMGB1 translocation was evident across various neuronal subtypes, encompassing excitatory glutamatergic, inhibitory GABAergic, and cholinergic neurons. There was no significant difference in the proportions of HMGB1-translocated cells between excitatory and inhibitory neurons, but both were higher than in cholinergic neurons. Building upon our findings and previous studies, we hypothesize that the translocation of HMGB1 from the nucleus to the cytoplasm is likely to be involved in the onset and development of drug-resistant epilepsy in SWS patients. In future studies, we will further explore the relationship between HMGB1 and epilepsy in SWS patients.
Disclosure
The authors disclose that the research, writing, and/or publication of this article received the following financial support: This work was supported by the Beijing Municipal Natural Science Foundation (grant No. 7222098) and by the UCB Foundation of China Association Against Epilepsy (UC-2023-061).
References
1. Ha A, Hwan Kim SH, Uk Baek SU, Kim J-S, Yoon H-J, Kook Kim YK. Incidence of Sturge-Weber Syndrome and Risk of Secondary Glaucoma: a Nationwide Population-based Study Using a Rare Disease Registry. Am J Ophthalmol. 2023;247:121–126. doi:10.1016/j.ajo.2022.11.009
2. Wang S, Pan J, Zhao M, et al. Characteristics, surgical outcomes, and influential factors of epilepsy in Sturge-Weber syndrome. Brain. 2021. doi:10.1093/brain/awab470
3. Bianchi F, Maria Auricchio A, Immacolata Battaglia D, Rosaria Pia Chieffo D, Massimi L. Sturge-Weber syndrome: an update on the relevant issues for neurosurgeons. Child’s Nerv Syst. 2020;36(10):2553–2570. doi:10.1007/s00381-020-04695-3
4. Javaid U, Hassaan Ali M, Jamal S, Hafeez Butt N. Pathophysiology, diagnosis, and management of glaucoma associated with Sturge–Weber syndrome. Intl Ophthalmol. 2017. doi:10.1007/s10792-016-0412-3
5. Shirley MD, Tang H, Gallione CJ, et al. Sturge–Weber Syndrome and Port-Wine Stains Caused by Somatic Mutation in GNAQ. N Engl J Med. 2013;368(21):1971–1979. doi:10.1056/nejmoa1213507
6. la Torre D, Alejandro J, Luat AF, et al. A Multidisciplinary Consensus for Clinical Care and Research Needs for Sturge-Weber Syndrome. Pediatr Neurol. 2018;84:11–20. doi:10.1016/j.pediatrneurol.2018.04.005
7. Sujansky E, Conradi S. Sturge-Weber Syndrome: age of Onset of Seizures and Glaucoma and the Prognosis for Affected Children. J Child Neurol. 1995;10(1):49–58. doi:10.1177/088307389501000113
8. Powell S, Fosi T, Sloneem J, Hawkins C, Richardson H, Aylett S. Neurological presentations and cognitive outcome in Sturge-Weber syndrome. Eur J Paediatr Neurol. 2021;34:21–32. doi:10.1016/j.ejpn.2021.07.005
9. Pascual-Castroviejo I, Pascual-Pascual S-I, Velazquez-Fragua R, Viaño J. Sturge-Weber Syndrome. Study of 55 Patients. Canad J Neurological Sci. 2008;35(3):301–307. doi:10.1017/s0317167100008878
10. Maton B, Kršek P, Jayakar P, et al. Medically intractable epilepsy in Sturge-Weber syndrome is associated with cortical malformation: implications for surgical therapy. Epilepsia. 2010;51(2):257–267. doi:10.1111/j.1528-1167.2009.02304.x
11. Annapurna S, Ardern-Holmes SL. Sturge–Weber syndrome: from the past to the present. Eur J Paediatr Neurol. 2014;18(3):257–266. doi:10.1016/j.ejpn.2013.10.003
12. Amna R, Musto AE. The role of inflammation in the development of epilepsy. J Neuroinflammation. 2018;15(1). doi:10.1186/s12974-018-1192-7
13. Kiarash R, Galic MA, Pittman QJ. Contributions of peripheral inflammation to seizure susceptibility: cytokines and brain excitability. Epilepsy Res. 2010;89(1):34–42. doi:10.1016/j.eplepsyres.2009.09.004
14. Xiao-Li L, Wang S, Tang C-Y, et al. Translocation of High Mobility Group Box 1 From the Nucleus to the Cytoplasm in Depressed Patients With Epilepsy. ASN Neuro. 2022;14:175909142211366. doi:10.1177/17590914221136662
15. Mao D, Zheng Y, Fenfen X, Han X, Zhao H. HMGB1 in nervous system diseases: a common biomarker and potential therapeutic target. Front Neurol. 2022;13:1029891. doi:10.3389/fneur.2022.1029891
16. Zhang S, Chen F, Zhai F, Liang S. Role of HMGB1/TLR4 and IL-1β/IL-1R1 Signaling Pathways in Epilepsy. Front Neurol. 2022;13:904225. doi:10.3389/fneur.2022.904225
17. Shi Y, Zhang L, Teng J, Miao W. HMGB1 mediates microglia activation via the TLR4/NF-κB pathway in coriaria lactone induced epilepsy. Mol Med Rep. 2018;17(4):5125–5131. doi:10.3892/mmr.2018.8485
18. Wang S, Guan Y-G, Zhu Y-H, Wang M-Z. Role of high mobility group box protein 1 in depression: a mechanistic and therapeutic perspective. World J Psychiatry. 2022;12(6):779–786. doi:10.5498/wjp.v12.i6.779
19. Zhao L, Liu P, Kepp O, Kroemer G. Methods for measuring HMGB1 release during immunogenic cell death. In: Methods in Enzymology. Elsevier; 2019:177–193. doi:10.1016/bs.mie.2019.05.001
20. Ulf A, Tracey KJ, Yang H. Post-Translational Modification of HMGB1 Disulfide Bonds in Stimulating and Inhibiting Inflammation. Cells. 2021;10(12):3323. doi:10.3390/cells10123323
21. Paudel YN, Semple BD, Jones NC, Othman I, Farooq Shaikh M. High mobility group box 1 (HMGB 1) as a novel frontier in epileptogenesis: from pathogenesis to therapeutic approaches. J Neurochem. 2019;151(5):542–557. doi:10.1111/jnc.14663
22. Paudel YN, Farooq Shaikh M, Chakraborti A, et al. HMGB1: a Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front Neurosci. 2018;12:12. doi:10.3389/fnins.2018.00628
23. Zhang Z, Liu Q, Ming L, et al. Upregulation of HMGB1-TLR4 inflammatory pathway in focal cortical dysplasia type II. J Neuroinflammation. 2018;15(1). doi:10.1186/s12974-018-1078-8
24. Pauletti A, Terrone G, Shekh-Ahmad T, et al. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain. 2019;142(7):e39–e39. doi:10.1093/brain/awz130
25. Shuxian H, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. Cytokine Effects on Glutamate Uptake by Human Astrocytes. Neuroimmunomodulation. 2000;7(3):153–159. doi:10.1159/000026433
26. Bezzi P, Domercq M, Brambilla L, et al. CXCR4-activated astrocyte glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4(7):702–710. doi:10.1038/89490
27. Viviani B, Bartesaghi S, Gardoni F, et al. Interleukin-1β Enhances NMDA Receptor-Mediated Intracellular Calcium Increase through Activation of the Src Family of Kinases. J Neurosci. 2003;23(25):8692–8700. doi:10.1523/jneurosci.23-25-08692.2003
28. Vezzani A. Epilepsy and Inflammation in the Brain: overview and Pathophysiology. Epilepsy Currents. 2014;14(2_suppl):3–7. doi:10.5698/1535-7511-14.s2.3
29. Maroso M, Balosso S, Ravizza T, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16(4):413–419. doi:10.1038/nm.2127
30. Orrin D, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013;36(3):174–184. doi:10.1016/j.tins.2012.11.008
31. Avignone E, Ulmann L, Levavasseur F, Rassendren F, Audinat E. Status Epilepticus Induces a Particular Microglial Activation State Characterized by Enhanced Purinergic Signaling. J Neurosci. 2008;28(37):9133–9144. doi:10.1523/jneurosci.1820-08.2008
32. Annamaria V, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2010;7(1):31–40. doi:10.1038/nrneurol.2010.178
33. Galic MA, Riazi K, Pittman QJ. Cytokines and brain excitability. Front Neuroendocrinol. 2012;33(1):116–125. doi:10.1016/j.yfrne.2011.12.002
34. Ravizza T, Gagliardi B, Noé F, Boer K, Aronica E, Vezzani A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29(1):142–160. doi:10.1016/j.nbd.2007.08.012
35. Niemeyer JE, Gadamsetty P, Chun C, et al. Seizures initiate in zones of relative hyperexcitation in a zebrafish epilepsy model. Brain. 2022;145(7):2347–2360. doi:10.1093/brain/awac073
36. Akyuz E, Kristina Polat A, Eroglu E, Kullu I, Angelopoulou E, Nath Paudel Y. Revisiting the role of neurotransmitters in epilepsy: an updated review. Life Sci. 2021;265:118826. doi:10.1016/j.lfs.2020.118826
37. Wang Y, Tan B, Wang Y, Chen Z. Cholinergic Signaling, Neural Excitability, and Epilepsy. Molecules. 2021;26(8):2258. doi:10.3390/molecules26082258
38. Han B, Xiuping L, Hao J. The cholinergic anti-inflammatory pathway: an innovative treatment strategy for neurological diseases. Neurosci Biobehav Rev. 2017;77:358–368. doi:10.1016/j.neubiorev.2017.04.002
39. Zhu M, Chen J, Guo H, Ding L, Zhang Y, Yun X. High Mobility Group Protein B1 (HMGB1) and Interleukin-1β as Prognostic Biomarkers of Epilepsy in Children. J Child Neurol. 2018;33(14):909–917. doi:10.1177/0883073818801654
40. Chen Y, Chen X, Liang Y. Meta-analysis of HMGB1 levels in the cerebrospinal fluid and serum of patients with epilepsy. Neurological Sci. 2023;44(7):2329–2337. doi:10.1007/s10072-023-06720-0
41. Kan M, Song L, Zhang X, Zhang J, Fang P. Circulating high mobility group box-1 and toll-like receptor 4 expressions increase the risk and severity of epilepsy. Braz J Med Biol Res. 2019;52(7). doi:10.1590/1414-431x20197374
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.