")
Back to Journals » Journal of Inflammation Research » Volume 18
Identification and Analysis of Key Immune- and Inflammation-Related Genes in Idiopathic Pulmonary Fibrosis
Authors Tan Y, Qian B, Ma Q, Xiang K, Wang S
Received 11 September 2024
Accepted for publication 21 December 2024
Published 11 February 2025 Volume 2025:18 Pages 1993—2009
DOI https://doi.org/10.2147/JIR.S489210
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Yan Tan,1,* Baojiang Qian,1,* Qiurui Ma,2 Kun Xiang,1 Shenglan Wang1
1Department of Respiratory and Critical Care Medicine, the First People’s Hospital of Yunnan Province, Kunming, People’s Republic of China; 2Medical School of Kunming University of Science and Technolog, Kunming, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shenglan Wang, Email [email protected]
Background: Studies suggest that immune and inflammation processes may be involved in the development of idiopathic pulmonary fibrosis (IPF); however, their roles remain unclear. This study aims to identify key genes associated with immune response and inflammation in IPF using bioinformatics.
Methods: We identified differentially expressed genes (DEGs) in the GSE93606 dataset and GSE28042 dataset, then obtained differentially expressed immune- and inflammation-related genes (DE-IFRGs) by overlapping DEGs. Two machine learning algorithms were used to further screen key genes. Genes with an area under curve (AUC) of > 0.7 in receiver operating characteristic (ROC) curves, significant expression and consistent trends across datasets were considered key genes. Based on these key genes, we carried out nomogram construction, enrichment and immune analyses, regulatory network mapping, drug prediction, and expression verification.
Results: 27 DE-IFRGs were identified by intersecting 256 DEGs, 1793 immune-related genes, and 1019 inflammation-related genes. Three genes (RNASE3, S100A12, S100A8) were obtained by crossing two machine algorithms (Boruta and LASSO),which had good diagnostic performance with AUC values. These key genes were all enriched in the same pathways, such as GOCC_azurophil_granule, IL-12 signalling and production in macrophages is the pathway with the strongest role for key genes. Six distinct immune cells, including naive CD4 T cells, T cells CD4 memory resting, T cells regulatory (Tregs), Monocytes, Macrophages M2, Neutrophils were identified. Real-time quantitative polymerase chain reaction (RT-qPCR) results were consistent with the training and validation sets, and the expression of these key genes was significantly upregulated in the IPF samples.
Conclusion: This study identified three key genes (RNASE3, S100A12 and S100A8) associated with immune response and inflammation in IPF, providing valuable insights into the diagnosis and treatment of IPF.
Summary: This study focuses on cutting-edge research in IPF, with special attention to the development dynamics and application potential of IPF. By systematically reviewing the research results in recent years, this paper aims to provide a comprehensive perspective on the latest field. It is submitted to the journal not only because of its high international recognition, but also because of its emphasis on a progress in this field. The research scope covers a wide range of content from basic theory to practical cases, which has a guiding effect on clinical research.
This manuscript is closely related to the core purpose and readership of Journal of Inflammation Research, a journal known for its in-depth and comprehensive coverage of the immune system and encouragement of original research. The content of this study is highly aligned with the journal’s advocacy of pushing the boundaries of knowledge in the immune system and is expected to bring new insights to journal readers and positively influence the future direction of research in the field.
Keywords: bioinformatics, idiopathic pulmonary fibrosis, immunity, inflammation, machine learning
Introduction
IPF is a chronic interstitial lung disease marked by the deterioration of lung architecture and fibrosis.1 Approximately 3 million individuals globally are afflicted with IPF with an incidence rate of 2–9 per 100,000.2 The initial non-specific symptoms of IPF render early identification and treatment challenging. Diagnosis necessitates the exclusion of established causes of interstitial lung disease (ILD) and a thorough evaluation of high-resolution computed tomography (HRCT) findings alongside the UIP characteristics observed in lung biopsy, or a holistic assessment of HRCT and histological patterns.3 At present, pirfenidone and nintedanib are authorized for the management of IPF, demonstrating efficacy in decelerating functional deterioration and disease advancement; however, they do not provide a cure and are linked to tolerance complications.4 Ruaro B et al have motivated us to suggest IPF preventative techniques informed by IPF risk factors, including GERD, among others.5 Lung transplantation is the sole effective curative intervention for patients with IPF; however, due to age and comorbidities, it is appropriate for only a limited subset of patients.6 Consequently, prompt and precise identification of IPF is essential for developing successful treatment strategies and enhancing patient survival rates. Consequently, employing bioinformatics and next-generation sequencing data analysis to thoroughly investigate the fundamental gene markers in the diagnosis and treatment of IPF, as referenced in,7 has emerged as an imperative necessity in contemporary research to enhance diagnostic and therapeutic precision and efficacy.
Inflammation, as a subsequent occurrence following the activation of the innate and adaptive immune systems, manifests at various phases of IPF. Certain studies indicate that persistent inflammation contributes to the etiology of IPF.8 IPF is thought to be caused by certain molecular pathways that involve endoplasmic reticulum stress, TGF-β overactivation, and the release of growth factors, chemokines, or Wnt. These pathways affect epithelial-mesenchymal transition (EMT), fibroblast recruitment, and fibroblast differentiation. During the disease development phase, pathogenic interstitial cells release unusual types and amounts of matrix proteins, which changes the lungs even more and makes scars.9 Likewise, inflammation mediators are essential in IPF. In comparison to healthy controls, the expression levels of pro-inflammation factors (TNF-α and IL-8) in the lungs of patients with IPF are higher.10 Even though anti-inflammation drugs that target pro-inflammation factors in IPF patients did not show any significant effects in clinical trials,11 this does not mean that IPF is not immune-mediated. Recent studies suggest that immunological responses significantly contribute to IPF.12 IPF is caused by immune cells and fibroblasts not being able to tolerate T cells and B cells. This leads to autoimmune responses and interactions between these cells and fibroblasts.8 The inflammation response resulting from immunological dysregulation may contribute to the development of IPF.13 Regulating the pulmonary immune system can substantially enhance the severity of IPF.14 Numerous studies have shown a strong link between IPF and immune and inflammation processes. However, the exact mechanisms by which IPF and these factors interact are still not fully understood.
In this study, we used a comprehensive bioinformatics approach to explore the links between immunity, inflammation and IPF. Leveraging machine learning algorithms, diagnostic analysis, and expression validation, we aim to identify key genes crucial in the diagnosis of IPF. In addition, a series of bioinformatics analyses were conducted to better understand the regulatory mechanisms and functions of immune- and inflammation-related genes in the occurrence and development of IPF. This integrated bioinformatics analysis provides valuable insights into the molecular pathways involved in IPF pathogenesis and highlights potential diagnostic and therapeutic implications of immune and inflammation genes in IPF.
Materials and Methods
Study Design and Data Collection
To investigate the role of immune- and inflammation-related genes in IPF, we used GSE93606 and GSE28042 datasets from the GEO database along with an existing list of known immune- and inflammation-related genes. This provided a theoretical foundation for exploring early IPF diagnosis and potential therapeutic targets.
First, we used the GSE93606 dataset to identify differentially expressed genes (DEGs), isolating immune- and inflammation-related genes. Next, intersections of inflammation-related genes, immune-related genes, and DEGs were identified. Significant genes were then validated in external datasets and further screened using multiple machine learning methods. We analyzed the function of key genes, developed a patient evaluation nomogram, and investigated the upstream regulation of key genes to identify potential therapeutic targets. A polymerase chain reaction (PCR) was conducted to confirm the expression of the key genes.
The technology roadmap is shown in Figure 1.
 |
Figure 1 The technology roadmap. Abbreviations: IPF, idiopathic pulmonary fibrosis; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, protein-protein interaction. |
Data Sources
The Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/gds) provided transcriptome datasets for IPF (GSE93606 and GSE28042). GSE93606 included 20 controls and 50 IPF peripheral blood samples, whereas GSE28042 included 19 controls and 75 IPF peripheral blood mononuclear cell (PBMC) samples. The ImmPort database (https://www.immport.org/home) showed 1793 immune-related genes, including those related to antigen-presenting cells, chemokines and receptors, cytokines and receptors, interferons, and interleukins.15 The GeneCards database (https://www.genecards.org/) provided 1019 inflammation-related genes (correlation score > 3).16
Identification and Analysis of Differentially Expressed Immune- and Inflammation-Related Genes (DE-IFRGs)
DEGs in the GSE93606 dataset (IPF vs control) were identified using the limma program (P < 0.05 and |log2 fold change (FC)| > 0.5)17 and visualized using the ggplot2 and pheatmap packages.18,19 DEGs, immune-related genes, and inflammation-related genes were intersected to identify DE-IFRGs. Enrichment analysis, based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) (Padjust < 0.05 and count ≥ 1), was performed using the clusterProfiler program.20 The STRING database (https://string-db.org/) was used to create the protein–protein interaction (PPI) network,21 and candidate genes were identified based on significant interactions within the PPI network.
Machine Learning
Feature genes were screened using the Boruta (Boruta package) and the LASSO (least absolute shrinkage and selection operator) (glmnet package) algorithms.22 The results of the Boruta and LASSO algorithms were intersected to obtain candidate genes. We then plotted the receiver operating characteristic (ROC) curve for candidate genes using the GSE93606 and GSE28042 datasets23 with the pROC software. Genes with an area under the curve (AUC) of > 0.7 were chosen for gene expression analysis, and those showing significant expression and consistent trends across both datasets were designated as key genes.
Construction of a Nomogram
Using the rms package, a nomogram comprising candidate genes was constructed for the GSE93606 and GSE28042 datasets.24 ROC and calibration curves were generated to evaluate the predictability of the model.
Gene Set Enrichment Analysis (GSEA) and Ingenuity Pathway Analysis (IPA)
Samples were divided into high- and low-expression groups based on the median expression values of candidate genes. GSEA was then carried out for both the groups using the clusterProfiler package and the org.Hs.eg.db package (|NES| > 1, NOM P < 0.05, q < 0.25) on the c5.go.v7.4.entrez.gmt and c2.cp.kegg.v7.4.entrez.gmt backgrounds.20 The IPA tool (www.ingenuity.com) was used to analyze signaling pathways associated with candidate genes, with a z-score of > 0 indicating pathway activation and a z-score of < 0 indicating pathway suppression.
Immune Analysis
The CIBERSORT method was used to determine the percentage abundance of infiltrating immune cells in the GSE93606 dataset. Using Pearson correlation analysis, the relationship between immune cells and candidate genes was examined.
Construction of a Regulatory Network
Using the miRWalk database (http://mirwalk.umm.uni-heidelberg.de/), we identified miRNAs (energy < –30) of the candidate genes. We then used the miRTarBase database (https://mirtarbase.cuhk.edu.cn/) to predict the lncRNAs interacting with these miRNAs. Transcription factors (TFs) within 10 kb upstream of the candidate genes (TFRP_score > 0.5) were identified using the Cistrome database (http://cistrome.org/). The DrugBank database (https://go.drugbank.com/drugs/) provided information on potential therapeutic drugs targeting candidate genes. Finally, Cytoscape software was used to visualize the complete network.25
Expression Level Verification
Between February 2023 and March 2024, eight patients with IPF were recruited from the Respiratory and Critical Care Medicine Department of author contribution. The inclusion criterion was confirmed IPF and the exclusion criterion was other respiratory system diseases and tumors. Simultaneously, eight age- and sex-matched healthy volunteers were recruited. We collected 10 mL of venous blood samples from both patients and controls. Signed informed consent forms were obtained from all participants. This study was approved by the Medical Ethics Committee of The First People’s Hospital of Yunnan Province (no. KHLL2024-KY121).
PBMCs were isolated from the blood samples using a PBMC separation solution. RNAs were extracted using TRIzol solution (Ambion, Austin, Texas), and concentration was determined using NanoDrop. mRNAs were reverse transcribed into cDNA using SureScript First-Strand cDNA Synthesis Kit (Servicebio, Wuhan, China), as per instructions. Table 1 lists all the primers. A real-time fluorescence quantitative PCR was performed using CFX96 for 40 cycles, and Ct values were obtained. Relative gene expression was quantified using the 2−ΔΔCt method, and GraphPad Prism 5 was used to calculate the P value.
 |
Table 1 List of Primer Sequences |
Statistical Analysis
Data were processed and analyzed using the R package (v 4.1).Differences in gene expression levels between IPF patients and control samples in the dataset were compared using limma (v 3.48.3); functional enrichment analyses were performed using cluster Profiler (v 4.0.2); the diagnostic performance of the model was determined using pROC (v 1.18.0); the rms (v 6.1–0) package to construct column line plots and calibration curves for key genes; and correlation analysis with Pearson; the two groups were compared using the Wilcoxon rank sum test, with a P value of < 0.05 considered significant.
Results
Identification of 25 DE-IFRGs as Candidate Genes
In the GSE93606 dataset, 256 DEGs—including 192 upregulated and 64 downregulated—were identified between IPF and control samples (Figure 2a and 2b). By overlapping DEGs, immune-related genes, and inflammation-related genes, 27 DE-IFRGs were obtained (Figure 2c). A functional enrichment analysis was performed to investigate the biological activities and pathways of DE-IFRGs. In the GO biological process (BP) category, DE-IFRGs were primarily enriched in defense responses to bacteria, responses to lipopolysaccharides, and responses to bacterial molecules. In the GO cellular component (CC) category, DE-IFRGs were primarily linked to the lumen of secretory granules, cytoplasmic vesicles, and vesicles. In the GO molecular function (MF) category, DE-IFRGs were mostly associated with immune receptor activity, cytokine binding, and cytokine receptor activity (Figure 2d and Supplementary Material-Table Section). DE-IFRGs were also involved in Th17 cell differentiation, cytokine–cytokine receptor interactions, and T-cell receptor signaling pathways (Figure 2e and Table 2). In addition, the PPI network identified 25 DE-IFRGs with significant interactions as candidate genes (Figure 2f).
 |
Table 2 Results of KEGG Enrichment Analysis |
 |
Figure 2 Identification and of Functional enrichment analysis DE-IFRGs in the GSE93606 dataset. (a) Volcano plot of the DEGs. (b) Heatmap of the DEGs. (c) Venn diagram of DEGs, immunity-related genes, and inflammation-related genes. (d) The enriched GO terms of DE-IFRGs. (e) The KEGG enrichment results of DE-IFRGs. (f) The PPI network of DE-IFRG. Abbreviations: DEGs, differentially expressed gene; DE-IFRGs differentially expressed immunity-inflammation-related genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, Protein-protein interaction. |
Identification of Key Genes: RNASE3, S100A12, and S100A8
Two machine learning methods were used to screen key genes from candidate genes. Using the Boruta algorithm, 15 feature genes were identified, including S100A8, S100A12, ORM1, LCK, IL7R, CD247, SLPI, CX3CR1, CAMP, PF4, MMP9, RNASE3, PI3, PRKCA, and IL2RB (Figure 3a). The LASSO algorithm identified five feature genes: S100A12, ORM1, S100A8, RNASE3, and PF4 (Figure 3b and 3c). By combining the results of both algorithms, five genes (S100A12, ORM1, S100A8, RNASE3, and PF4) were selected for further analysis (Figure 3c). S100A12, S100A8, RNASE3, and PF4 showed an AUC of greater than 0.7 in both GSE93606 and GSE28042 datasets, suggesting their potential to distinguish between IPF and control samples and serving as effective diagnostic markers (Figure 3e and 3f). Of these, RNASE3, S100A12, and S100A8 were significantly upregulated in IPF samples across both datasets. Therefore, these three genes were chosen for further analysis (Figure 3g and 3h).
 |
Figure 3 Screening of characterized genes and ROC curves and expression analysis of characterized genes in control and IPF groups. (a) Boruta algorithm identifies 15 feature genes. (b and c) Screening 5 feature genes in LASSO analysis. (d) Intersection 5 key genes of the Boruta and LASSO algorithms. (e) ROC curves of candidate 5 key genes in GSE28042 dataset. (f) ROC curves of candidate 5 key genes in GSE93606 dataset. (g) Expression of 5 key genes in GSE28042 dataset. (h) Expression of 5 key genes in GSE93606 dataset. Abbreviations: ROC, receiver operating characteristic; LASSO, least absolute shrinkage and selection operator. |
Nomograms Constructed Based on Three Key Genes Showed High Predictive Accuracy
Nomograms based on the three key genes were created for both databases (Figure 4a and 4b). Calibration curves showed no significant difference between the expected and actual findings, with S.p=1 > 0.05 for both models (Figure 4c and 4d). AUCs for both models were above 0.8, suggesting a high predictive accuracy of the models (Figure 4e and 4f).
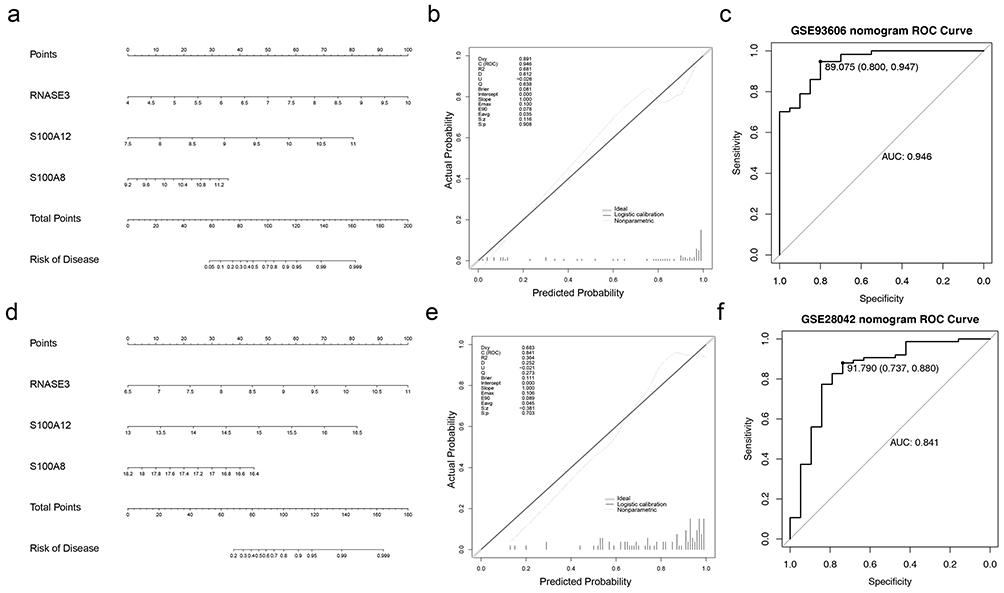 |
Figure 4 (a) Nomogram of overall survival based on the Cox model derived from dataset GSE36909. (b and c) Calibration curves versus ROC curves assessing the predictive value of the model for the prognosis of patients in the discovery cohort. (d) Cox model-based nomogram for overall survival derived from dataset GSE28042. (e and f) Calibration curves versus ROC curves assessing the predictive value of the model for the prognosis of patients in the validation cohort. Abbreviation: ROC, receiver operating characteristic. |
IL-12 Signalling and Production in Macrophages Is the Pathway With the Strongest Role for Key Genes
RNASE3, S100A12, and S100A8 were enriched in pathways associated with GOBP_antibacterial_humoral_responsea, GOCC_azurophil_granule, GOCC_specific_granule, GOCC_specific_granule_lumen, GOCC_tertiary_granule, and GOCC_tertiary_granule_lumen (Figure 5a, 5c and 5e). RNASE3 and S100A8 were enriched in pathways associated with KEGG_alzheimers_disease, KEGG_ribosome, and KEGG_parkinsons_disease pathways (Figure 5b and 5d), whereas S100A12 and S100A8 were associated with KEGG_allograft_rejection and KEGG_cell_adhesion_molecules_cams pathways (Figure 5d and 5f). In addition, IPA of the key genes identified involvement in six pathways, including the S100 family signaling pathway and LXR/RXR activation (Figure 6a), with the genes being most active in the IL-12 signaling and production in macrophages (Figure 6b). Supplementary pathways are shown in Supplementary Material-Figure section. This suggested that these genes might influence the immune function of macrophages by regulating IL-12 production.
 |
Figure 5 GSEA enrichment analysis of three key genes. (a and b) GSEA enrichment analysis of the RNASE3 gene: GO enrichment of the top 10 of significance and KEGG pathway mapping. (c and d) GSEA enrichment analysis of SA100A12 gene: GO enrichment of top 10 of significance and KEGG pathway mapping. (e and f) GSEA enrichment analysis of SA100A8 gene: top 10 significance GO enrichment and KEGG pathway mapping. |
 |
Figure 6 (a) IPA enrichment analysis. If z-score>0 is Orange, the path is activated. z-score<0, which is blue, indicates that it is suppressed 3 Key genes Key genes play a significant role in 6 pathways, mainly LXR/RXR activation, inflammation related, S100 family signaling pathways and other functions. (b) IL-12 Signaling and Production in Macrophages pathway process. |
S100A12 Correlated Significantly With Naive CD4 T Cells and Neutrophils
In the GSE93606 dataset, 22 infiltration immune cells were detected in each sample (Figure 7a). Correlation analysis showed interaction among certain immune cells, such as, a strong negative correlation between monocytes and neutrophils (Cor = –0.62; Figure 7b). Six immune cell types, including M2 macrophages and neutrophils, showed significant differences between the two groups (Figure 7c). The scatter plot showed that S100A12 had a strong negative correlation with naive CD4 T cells, whereas a significant positive correlation with neutrophils (Figure 7d). S100A12 may have been involved in processes such as differentiation, functional regulation or quantitative changes of immune cells.
 |
Figure 7 Immune infiltration analysis. (a) Immune cell stacking plots in samples: the CIBERSORT algorithm was used to determine the percentage abundance of infiltrating immune cells in each sample, along with the proportion of infiltrating immune cells in the disease and normal groups. (b) Immune Cell Correlation Plot: A Pearson correlation study was used to illustrate the interaction of immune cell infiltration in the immune environment. (c) Box plot of the proportion of immune cells in the disease group versus the normal group: the rank sum test was used to validate the difference in immune cell infiltration between the disease and normal. (d) Scatterplot of correlation between key genes and immune cells; Immune cell scores were compared between groups by Ryu yan, and immune cells that differed in the disease and normal groups were identified using rank and test analysis. **: P < 0.01; ***: P < 0.001; ****: P < 0.0001. Abbreviation: ns, not significant. |
Key Genes Were Significantly Upregulated in IPF and 11 Targeted Potential Therapeutic Agents Were Screened
Using miRWalk and miRTarBase databases, we predicted 61 miRNAs and 23 lncRNAs for the construction of ceRNA networks. C6orf223 regulated the expression of RNASE3 through hsa-miR-6769a-5p in the network. LINC00598 could simultaneously regulate RNASE3 and S100A12 through hsa-miR-4691-5p and hsa-miR-6779-5p, respectively (Figure 8a). Furthermore, a TF–mRNA network was constructed involving 69 TFs upstream of key genes, selected from the database. TCF7, PRKDC, and HDAC1 regulated S100A8; H2AZ and POLR2A regulated RNASE3; and DPF2, CEBPG, and HDAC2 regulated S100A12 (Figure 8b). These key genes may have been involved in the pathogenesis of IPF through multiple mechanisms. Using DrugBank database, 11 potential therapeutic drugs targeting key genes were identified; for example, S100A12 predicted DB00768 and DB01025 (Figure 8c). A real-time quantitative PCR (RT-qPCR) analysis confirmed the significant upregulation of the three key genes in IPF samples (Figure 9a–9c). This was consistent with the dataset results and further confirmed the reliability of our results.
 |
Figure 8 (a) Construction of ceRNA regulatory networks of key genes based on 61 miRNAs, 3 mRNAs and 23 lncrnas. (b) The upstream TFS of 69 important genes were selected for network visualization using Cytoscape software. (c) Discovered 11 therapeutic drugs targeting key IPF genes through the DrugBank database and created a table of drug-target interactions. |
 |
Figure 9 The expression of key genes was verified by qPCR. (a) Expression of S100A12 gene in IPF group and control group. (b) Expression of S100A8 gene in IPF group and control group. (c) Expression of RNASE3 gene in IPF group and control group. IPF: idiopathic pulmonary fibrosis; P < 0.05 indicates significant differences. |
Discussion
IPF is a chronic, progressive, and lethal lung illness of unclear etiology, marked by interstitial fibrosis. Researchers have found that immune and inflammation responses play a big role in the development of IPF. These responses hurt pulmonary epithelial cells, encourage the deposition of extracellular matrix (ECM) and activate fibroblasts.26 Immune cells exert therapeutic effects on IPF via innate immunity and the modulation of inflammation responses. Consequently, we assert that inflammation and immunological processes are pivotal in the initiation and advancement of IPF. This work utilized advanced machine learning techniques, Boruta and LASSO, to identify three significant genes: RNASE3, S100A12, and S100A8. These genes are essential in the immunological and inflammation mechanisms of IPF and effectively differentiate IPF patients from the control group. RT-qPCR technology has corroborated this discovery. Ding et al’s study27 demonstrates that S100A12, S100A8 and S100A9 serve as biomarkers for inflammation illnesses, with their serum levels markedly increased in patients with IPF.Qiu et al28 found a strong correlation between five genes (CXCL14, SLC40A1, RNASE3, CCR3, and RORA) and overall survival (OS) in patients with IPF, noting that patients with elevated RNASE3 expression have inferior survival results. These research findings align with our findings; however, by utilizing more advanced machine learning techniques, this study pinpoints essential genes that not only intricately link to inflammation but also significantly influence immunological systems. The nomogram model developed from these three pivotal genes demonstrates outstanding discriminatory capability, with an AUC value surpassing 0.9, reinforcing the significant diagnostic potential of these genes in IPF.
RNASE3 is a substantial particle found within eosinophilic granules, released upon activation by immunological stimuli. The protein molecular structure comprises a phycobiliprotein with a quaternary structure consisting of three subunits: α, β, and γ, which can interact with bacterial lipopolysaccharides and lipoteichoic acid.29 Studies indicate that RNASE3 exhibits significant cytotoxicity and can harm pulmonary epithelial cells.30 RNASE3 may facilitate pulmonary fibrosis by harming lung epithelial cells. This research validated the notable overexpression of this gene in IPF patients with RT-qPCR. S100 calcium-binding protein A12, a constituent of the S100 calcium-binding protein family, is situated on chromosome 1q21.3 and comprises 92 amino acids. It is predominantly situated in the cytoplasm, and upon an increase in intracellular calcium levels, and it translocates from the cytoplasm to the cytoskeleton and cell membrane. The activation of neutrophils correlates with the production of S100A12 via the microtubule-mediated alternative pathway. This investigation confirmed a substantial positive correlation between S100A12 expression and neutrophil levels.S100A12 interacts with calcyclin in a calcium-dependent way and has been recognized as a predictive serum biomarker for IPF.31S100A12 expression is markedly elevated in the blood and bronchoalveolar lavage fluid (BALF) of patients with IPF, particularly in those with unfavorable prognoses.32S100A12 suppresses lung fibroblast migration via the RAGE-p38 MAPK signaling pathway, contributing to the healing of defective lung tissue. This pathway is anticipated to serve as a therapeutic target for the repair and remodeling of lung tissue.33 The protein expressed by the S100 calcium-binding protein A8 gene belongs to the S100 protein family, characterized by its calcium-binding zinc finger domain and its role in cell cycle regulation. This family comprises over 13 members, predominantly situated in the cytoplasm and nucleus, impacting many cellular processes. S100A8 enhances leukocyte recruitment in the inflammation milieu of bleomycin-induced lung injury via exacerbating acute lung injury. This mechanism relies on the activation of AECs via Toll-like receptor 4 and is measured by the release of interleukin-6, cytokines, and monocyte chemoattractant protein-1.34 This aligns with our findings that S100A8 levels are considerably positively linked with neutrophil levels. Studies indicate that blood S100A8 levels in patients experiencing acute exacerbation of IPF are markedly elevated compared to age-matched controls, and those with elevated S100A8 levels exhibit reduced short-term survival rates. This study corroborates this finding. Consequently, the blood concentration of S100A8 serves as a significant predictive biomarker for individuals with qualitative pneumonia during acute exacerbation; however, the precise mechanism requires additional investigation.
The IPA enrichment analysis shows that the three main genes that were looked at are strongly linked to macrophages making and signaling IL-12. Monocytes, macrophages, and dendritic cells predominantly synthesize IL-12, a pivotal cytokine. You can make more interferon-γ (IFN-γ) when you take it. This helps helper T cells 1 (Th1) become more specific.35 IPF is a condition mostly caused by Th2, and the lack of IFN-γ is linked to the disease getting worse over time due to fibrosis.36 A rise in prostaglandin E2 (PGE2) may be linked to a rise in IL-13 and a fall in IL-12 in people with IPF.37 This suggests that blocking the IL-12 signaling pathway may play a major role in the development of IPF. Finally, these important genes may have an indirect effect on the Th1/Th2 balance by stopping the production of IL-12 or the expression and function of its receptors, which can make IPF worse. The precise methods of action for these three pivotal genes in IPF necessitate additional experimental investigation for clarification.
The subsequent stage is to assess the immune cell infiltration between the diseased cohort and the healthy cohort. There are big differences in the numbers of six types of immune cells: naive CD4 T cells, resting CD4 memory T cells, regulatory T cells (Tregs), monocytes, M2 macrophages, neutrophils. Simultaneously, we identified a substantial link between essential genes and varying immune cells. IPF is made worse by M2 macrophages, Th17 cells, CD8+ T cells, and Tregs. On the other hand, Th1 and tissue-resident memory (TRM) CD4+ T cells seem to protect against it. Also, studies show that CD4+ T cells help IPF get worse by increasing CD4(+)IL-21(+)T cells and CD4(+)21R(+)T cells and also by encouraging the growth of Th17 cells through IL-2138. The production of macrophage inflammation protein 1 by CD4+ T lymphocytes helps pulmonary fibrosis cells move to the lungs, which ultimately speeds up the progression of pulmonary fibrosis.39 CD4+ T cells that release TGF-β can increase the expression of PD-1 in people with fibrotic lung disease. Inhibiting the PD-1 pathway can similarly diminish bleomycin-induced lung fibrosis. Consequently, CD4+ T lymphocytes may be associated with pulmonary fibrosis via the PD-1 pathway.40 Quiescent M2 macrophages can suppress inflammation responses and facilitate the resolution of advanced fibrosis. They can also mitigate lung fibrosis by secreting inhibitory chemokines and cytokines.41 People with very different types of IPF and their related groups can use peripheral blood monocyte counts to guess how likely they are to die from any cause. Higher levels of monocytes are linked to higher risks of hospitalization, death and IPF getting worse. This means that the number of monocytes may be a simple and inexpensive way to predict IPF.42 Neutrophil elastase (NE) levels are higher in the BALF of people with IPF. This activates the transforming growth factor-ß pathway and encourages fibroblast participation in the pathophysiological mechanisms of IPF, among other things.43 In conclusion, further research is required to comprehensively comprehend IPF.
The DrugBank database ultimately led to the identification of potential therapeutic agents targeting 11 pivotal genes associated with IPF. The drugs include Calcium, Copper, Zinc, Zinc acetate, Zinc chloride, Zinc sulfate, Pranlukast, Adenosine-2’-5’-Diphosphate, Citric acid, Amlexanox, and Olopatadine. Calcium-activated potassium channel targets have demonstrated potential for modulating the pathogenesis of IPF and therefore impeding disease progression.44 In IPFAEC2 cells, zinc metabolism is out of whack, and the zinc transporter SLC39A8/ZIP8 has been identified as a key pathogenic factor contributing to IPF fibrosis. Zinc supplementation in the diet may mitigate lung fibrosis.45 Precisely predicting the essential genes targeted by pharmaceuticals could serve as potential biomarkers for the clinical diagnosis and management of IPF.
This study undoubtedly possesses numerous limitations. The database’s sample lacks comprehensive diagnostic and treatment data, which could potentially affect the subsequent analysis of key genes in the study of IPF. Secondly, this work is a GEO retrospective investigation characterized by a relatively limited sample size and an absence of data from multicenter clinical trials. Third, the independent expression analysis of each gene in the sample leaves the potential synergistic effect among the genes ambiguous; fourth, the function of essential genes in IPF remains unelucidated. Ultimately, this study does not do enough functional studies to figure out how the key genes in IPF work and how much of the related biomolecules are expressed. We did not evaluate the expression of these genes in other interstitial lung disorders. In order to get around these problems, we plan to do bigger prospective studies, collect a lot of clinical samples for validation, and use animal models and functional experiments to learn more about how key genes in IPF work. We will also look at how these genes are similar and different in other ILDs to get a fuller picture.
Conclusion
RT-qPCR further verified the identification of three key genes (RNASE3, S100A12, and S100A8) that effectively distinguish health status from IPF in this work, using two machine learning algorithms. It is important to emphasize that these three genes are not only associated with immune control but also intricately linked to the inflammation response. Key genes exhibiting a singular characteristic may demonstrate low diagnostic accuracy for diseases; however, the integration of key genes with many disease traits might enhance diagnostic precision. This work developed a nomogram model utilizing these three pivotal genes, which showed a robust capacity for differentiation. A thorough investigation also shows that the three key genes in the IL-12 production and signaling pathway in macrophages are very important. This gives a strong theoretical basis for more mechanistic studies. Most importantly, we were able to find 11 possible therapeutic agents by using these three unique genes. This gives us a new perspective and a solid base for future clinical research on IPF treatment.
Data Sharing Statement
The datasets [GSE93606 and GSE28042] for this study can be found in the [Gene Expression Omnibus (GEO)] [https://www.ncbi.nlm.nih.gov/gds]. The Immune-related gene datasets for this study can be found in the ImmPort database (https://www.immport.org/home). The Inflammation related genes datasets for this study can be found in the GeneCards database (https://www.genecards.org/).
Institutional Review Board Statement
This study was performed inline with the principles of the Declaration of Helsinki. Approval was granted by Medical Ethics Committee of the First People’s Hospital of Yunnan Province (KHLL2024-KY121).
Acknowledgments
I thank all the faculty members of Respirad Critical Care Medicine in the First People’s Hospital of Yunnan Province, for making this Research Topic possible.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The work was supported by the Yunnan Provincial Clinical Medical Center Open project (2022LCZXKFHX-08) and Academician Zhong Nanshan Workstation(2019IC032-1).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. doi:10.1164/rccm.2009-040GL
2. Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Rres J. 2015;46(3):795–806. doi:10.1183/09031936.00185114
3. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: an Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205(9):e18–e47. doi:10.1164/rccm.202202-0399ST
4. Spagnolo P, Kropski JA, Jones MG, et al. Idiopathic pulmonary fibrosis: disease mechanisms and drug development. Pharmacol Ther. 2021;222:107798. doi:10.1016/j.pharmthera.2020.107798
5. Ruaro B, Pozzan R, Confalonieri P, et al. Gastroesophageal Reflux Disease in Idiopathic Pulmonary Fibrosis: viewer or Actor? To Treat or Not to Treat? Pharmaceuticals. 2022;15(8):1033. doi:10.3390/ph15081033
6. George PM, Patterson CM, Reed AK, Thillai M. Lung transplantation for idiopathic pulmonary fibrosis. Lancet Respir Med. 2019;7(3):271–282. doi:10.1016/S2213-2600(18)30502-2
7. Giriyappagoudar M, Vastrad B, Horakeri R, Vastrad C. Study on Potential Differentially Expressed Genes in Idiopathic Pulmonary Fibrosis by Bioinformatics and Next-Generation Sequencing Data Analysis. Biomedicines. 2023;11(12):3109. doi:10.3390/biomedicines11123109
8. Heukels P, Moor CC, von der Thüsen JH, Wijsenbeek MS, Kool M. Inflammation and immunity in IPF pathogenesis and treatment. Respir Med. 2019;147:79–91. doi:10.1016/j.rmed.2018.12.015
9. Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9(1):157–179. doi:10.1146/annurev-pathol-012513-104706
10. Parra ER, Kairalla RA, de Carvalho RCR, Eher E, Capelozzi VL. Inflammatory cell phenotyping of the pulmonary interstitium in idiopathic interstitial pneumonia. Respiration. 2007;74(2):159–169. doi:10.1159/000097133
11. Raghu G, Brown KK, Costabel U, et al. Treatment of idiopathic pulmonary fibrosis with etanercept: an exploratory, placebo-controlled trial. Am J Respir Crit Care Med. 2008;178(9):948–955. doi:10.1164/rccm.200709-1446OC
12. O’Dwyer DN, Armstrong ME, Trujillo G, et al. The Toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2013;188(12):1442–1450. doi:10.1164/rccm.201304-0760OC
13. Harrell CR, Sadikot R, Pascual J, et al. Mesenchymal Stem Cell-Based Therapy of Inflammatory Lung Diseases: current Understanding and Future Perspectives. Stem Cells Int. 2019;2019:4236973. doi:10.1155/2019/4236973
14. Xu Y, Lan P, Wang T. The Role of Immune Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina. 2023;59(11):1984. doi:10.3390/medicina59111984
15. Nie H, Yan C, Zhou W, Li T-S, Moreira H. Analysis of Immune and Inflammation Characteristics of Atherosclerosis from Different Sample Sources. Oxid Med Cell Longev. 2022;2022:5491038. doi:10.1155/2022/5491038
16. Xing M, Li J. A New Inflammation-Related Risk Model for Predicting Hepatocellular Carcinoma Prognosis. Biomed Res Int. 2022;2022(1):5396128. doi:10.1155/2022/5396128
17. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
18. Gustavsson EK, Zhang D, Reynolds RH, Garcia-Ruiz S, Ryten M. ggtranscript: an R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics. 2022;38(15):3844–3846. doi:10.1093/bioinformatics/btac409
19. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–2849. doi:10.1093/bioinformatics/btw313
20. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
21. Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–d8. doi:10.1093/nar/gkw937
22. Li Y, Lu F, Yin Y. Applying logistic LASSO regression for the diagnosis of atypical Crohn’s disease. Sci Rep. 2022;12(1):11340. doi:10.1038/s41598-022-15609-5
23. Yan P, Ke B, Song J, Fang X. Identification of immune-related molecular clusters and diagnostic markers in chronic kidney disease based on cluster analysis. FronT Genet. 2023;14:1111976. doi:10.3389/fgene.2023.1111976
24. Sachs MC. plotROC: a Tool for Plotting ROC Curves. Journal of Statistical Software. 2017;79. doi:10.18637/jss.v079.c02
25. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
26. Lee JW, Chun W, Lee HJ, et al. The Role of Macrophages in the Development of Acute and Chronic Inflammatory Lung Diseases. Cells. 2021;10(4):897. doi:10.3390/cells10040897
27. Ding D, Luan R, Xue Q, Yang J. Prognostic significance of peripheral blood S100A12, S100A8, and S100A9 concentrations in idiopathic pulmonary fibrosis. Cytokine. 2023;172:156387. doi:10.1016/j.cyto.2023.156387
28. Qiu L, Gong G, Wu W, et al. A novel prognostic signature for idiopathic pulmonary fibrosis based on five-immune-related genes. Ann Transl Med. 2021;9(20):1570. doi:10.21037/atm-21-4545
29. Richards TJ, Kaminski N, Baribaud F, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;185(1):67–76. doi:10.1164/rccm.201101-0058OC
30. Zagai U, Dadfar E, Lundahl J, Venge P, Sköld CM. Eosinophil cationic protein stimulates TGF-beta1 release by human lung fibroblasts in vitro. Inflammation. 2007;30(5):153–160. doi:10.1007/s10753-007-9032-4
31. Xia P, Ji X, Yan L, Lian S, Chen Z, Luo Y. Roles of S100A8, S100A9 and S100A12 in infection, inflammation and immunity. Immunology. 2024;171(3):365–376. doi:10.1111/imm.13722
32. Li Y, He Y, Chen S, et al. S100A12 as Biomarker of Disease Severity and Prognosis in Patients With Idiopathic Pulmonary Fibrosis. Front Immunol. 2022;13:810338. doi:10.3389/fimmu.2022.810338
33. Pruenster M, Vogl T, Roth J, Sperandio M. S100A8/A9: from basic science to clinical application. Pharmacol Ther. 2016;167:120–131. doi:10.1016/j.pharmthera.2016.07.015
34. Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in Inflammation. Front Immunol. 2018;9:1298. doi:10.3389/fimmu.2018.01298
35. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3(2):133–146. doi:10.1038/nri1001
36. Latsi P, Pantelidis P, Vassilakis D, Sato H, Welsh KI, du Bois RM. Analysis of IL-12 p40 subunit gene and IFN-gamma G5644A polymorphisms in Idiopathic Pulmonary Fibrosis. Respir Res. 2003;4(1):6. doi:10.1186/1465-9921-4-6
37. Li JZ, Li ZH, Kang J, Hou XM, Yu RJ. Change of prostaglandin E2 and interleukin-12, interleukin-13 in the bronchoalveolar lavage fluid and the serum of the patients with idiopathic pulmonary fibrosis. Zhonghua Jie He He Hu Xi Za Zhi. 2004;27(6):378–380.
38. Lei L, Zhong XN, He ZY, Zhao C, Sun XJ. IL-21 induction of CD4+ T cell differentiation into Th17 cells contributes to bleomycin-induced fibrosis in mice. Cell Biol Int. 2015;39(4):388–399. doi:10.1002/cbin.10410
39. Luzina IG, Todd NW, Iacono AT, Atamas SP. Roles of T lymphocytes in pulmonary fibrosis. J Leukoc Biol. 2008;83(2):237–244. doi:10.1189/jlb.0707504
40. Celada LJ, Kropski JA, Herazo-Maya JD, et al. PD-1 up-regulation on CD4+ T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-β1 production. Sci Transl Med. 2018;10(460):eaar8356. doi:10.1126/scitranslmed.aar8356
41. Yang HZ, Cui B, Liu HZ, et al. Targeting TLR2 attenuates pulmonary inflammation and fibrosis by reversion of suppressive immune microenvironment. J Immunol. 2009;182(1):692–702. doi:10.4049/jimmunol.182.1.692
42. Kreuter M, Lee JS, Tzouvelekis A, et al. Monocyte Count as a Prognostic Biomarker in Patients with Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2021;204(1):74–81. doi:10.1164/rccm.202003-0669OC
43. Achaiah A, Fraser E, Saunders P, Hoyles RK, Benamore R, Ho LP. Neutrophil levels correlate with quantitative extent and progression of fibrosis in IPF: results of a single-centre cohort study. BMJ Open Respir Res. 2023;10(1):e001801. doi:10.1136/bmjresp-2023-001801
44. Vaidya B, Patel R, Muth A, Gupta V. Exploitation of Novel Molecular Targets to Treat Idiopathic Pulmonary Fibrosis: a Drug Discovery Perspective. Curr Med Chem. 2017;24(22):2439–2458. doi:10.2174/0929867324666170526123607
45. Foster PS, Tay HL, Oliver BG. Deficiency in the zinc transporter ZIP8 impairs epithelia renewal and enhances lung fibrosis. J Clin Invest. 2022;132(11):e160595. doi:10.1172/JCI160595
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.