")
Back to Journals » Journal of Inflammation Research » Volume 18
Identification of Diagnostic Biomarkers and Therapeutic Targets in Sepsis-Associated ARDS via Combining Bioinformatics with Machine Learning Analysis
Received 21 March 2025
Accepted for publication 10 July 2025
Published 18 July 2025 Volume 2025:18 Pages 9523—9536
DOI https://doi.org/10.2147/JIR.S529689
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Anh Ngo
Tingting Liu,1 Ling Gao,1 Xiaoyan Li2
1Department of Respiratory and Critical Care Medicine, Third Hospital of Shanxi Medical University, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Tongji Shanxi Hospital, Taiyuan, Shanxi, 030032, People’s Republic of China; 2Department of Pulmonary Critical Care Medicine, Shanghai Pudong New Area Zhoupu Hospital, Shanghai University of Medicine & Health Sciences Affiliated Zhoupu Hospital, Shanghai, 201318, People’s Republic of China
Correspondence: Xiaoyan Li, Department of Pulmonary Critical Care Medicine, Shanghai Pudong New Area Zhoupu Hospital, Shanghai University of Medicine & Health Sciences Affiliated Zhoupu Hospital, Shanghai, 201318, People’s Republic of China, Email [email protected]
Purpose: This study aims to identify key genes associated with Neutrophil Extracellular Traps (NETs) in sepsis-associated Acute Respiratory Distress Syndrome (ARDS) using bioinformatics and molecular docking for diagnostic and therapeutic purposes.
Methods: We obtained the GSE32707 datasets from the GEO database and selected the gene expression profiles of sepsis-associated ARDS patients and healthy controls. Differentially expressed genes (DEGs) were identified and subjected to functional enrichment analysis and immune infiltration analysis. Weighted Gene Co-expression Network Analysis (WGCNA) was performed to explore gene co-expression modules. The differential genes of the above screen were crossed with NETs gene sets to obtain the key NETs genes for sepsis-associated ARDS. Three machine learning algorithms were applied to refine the intersected genes. The expression of hub genes in clinical blood samples was verified by RT-qPCR. Molecular docking was conducted to predict small molecular compounds targeting hub genes.
Results: Analysis of the GSE32707 dataset using R software revealed 485 differential genes for sepsis-associated ARDS. WGCNA identified 332 common genes in the gene module associated with sepsis-associated ARDS. The differential genes of the above screen were crossed with NETs gene sets to obtain the key NETs genes for sepsis-associated ARDS. Further through machine learning, LTF and PRTN3 were identified as hub genes with excellent diagnostic potential. RT-qPCR analysis showed that PRTN3 and LTF expression were significantly upregulated in sepsis-associated ARDS patients as compared with healthy controls. Molecular docking results showed that nimesulide and minocycline were identified as potential therapeutic drugs for sepsis-associated ARDS.
Conclusion: LTF and PRTN3 are identified as key NETs genes in sepsis-associated ARDS and show promise as effective molecular markers for disease diagnosis and potential therapeutic targets.
Keywords: sepsis, ARDS, neutrophil extracellular traps, bioinformatics
Introduction
Sepsis, the prevalent acute medical condition in intensive care units (ICUs), is characterized as a systemic inflammatory response syndrome triggered by infections with pathogenic microorganisms, including bacteria and viruses.1 In 2016, sepsis was delineated as “life-threatening organ dysfunction due to a disproportionate host response to infection”2 Despite advances in the diagnosis, treatment, and management of sepsis in recent decades, the mortality rate of sepsis remains high (about 20–30%), particularly when it involves the lungs.3,4 Approximately 50% of sepsis patients will develop lung injury, which is the leading cause of sepsis-associated death.1 The case fatality rate for sepsis-induced ARDS is markedly higher than that for other ARDS etiologies, exceeding 40% in severe cases.5 Therefore, identifying the key molecules of sepsis-associated ARDS and discovering early diagnostic markers and potential therapeutic targets is crucial to reduce the case fatality rate of this disease.
In recent years, neutrophil extracellular traps (NETs) have emerged as significant biomarkers in scholarly research.6,7 NETs are structural frameworks composed of nuclear DNA, antimicrobial peptides, histones, and various bactericidal factors that immobilize, trap, and eliminate pathogens.8 The excessive formation or impaired clearance of NETs is, in fact, detrimental. They exacerbate the pathological process of sepsis-induced ARDS through mechanisms such as the release of damage-associated molecular patterns (DAMPs), which activate the inflammatory cascade, induce coagulation abnormalities, and directly damage lung tissue. Despite the potential of NETs as therapeutic targets, current NET-targeted therapies have several limitations. For example, DNases, which are used for NETs digestion, and therapeutics that capture circulating NETs have shown promise in preclinical studies. However, the clinical efficacy remains to be fully dissatisfied, and their use is also faced with challenges such as delivery efficiency and potential side effects.Therefore, there is an urgent need to identify NETs-related markers to better understand the pathophysiology of sepsis-associated ARDS and to develop more effective diagnostic and therapeutic strategies.
Bioinformatics analysis have currently become an indispensable part of biomedical research, and machine learning is a relatively new study way. Accordingly, we plan to present novelly the NETs-related molecular biomarker by combining bioinformatics with machine learning analysis. This study aims to identify key genes associated with NETs in sepsis-associated ARDS using bioinformatics analysis, thereby offering new insights into potential molecular mechanisms and providing promising novel targets for diagnostic and therapeutic strategies.
Materials and Methods
Data Download and Pre-Processing
We downloaded the GSE32707 dataset from the NCBI Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) database. From these, 65 samples were selected, including 31 from patients with sepsis-associated ARDS and 34 from normal healthy controls. The platform and grouping information of the relevant data set for this study is provided in Supplementary Table 1.
Screening for Differentially Expressed Genes
Screening for differentially expressed genes (DEGs) between the sepsis-associated ARDS group and the healthy control group in the GSE32707 dataset using the limma package in R software. Gene expression values were normalized with limma, and DEGs were identified with criteria of |log2FC| > 1 and adjusted P < 0.05.9 The GEOquery package in R version 4.1.1 (http://rproject.org/) was applied to visualize the DEGs.10
Enrichment Analysis of Differentially Expressed Genes
The identified DEGs underwent gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses using the R software. During the enrichment analysis, “org.Hs.eg.db”, “clusterProfiler”, “enrichplot” and “ggplot2” packages were employed in R software. The analyses focused on biological processes (BP), cellular components (CC), and molecular functions (MF), with a significance threshold of P < 0.05. Meanwhile, KEGG provided the enriched signaling pathways demonstrated by the DEGs. Afterward, the results were visualized using the bar plot regarded the threshold of p-value < 0.05.
Weighted Gene Co-Expression Network Analysis (WGCNA)
The WGCNA package in R was employed to cluster specimens from the sepsis-associated ARDS and healthy control groups within the GSE32707 dataset. The soft threshold (β) for network construction was determined using the dynamic minimum tree cutting algorithm, and co-expression modules significantly associated with sepsis-associated ARDS were identified.11
Key Gene Screen for NETs in Sepsis-Associated ARDS
The intersection of DEGs, WGCNA gene modules, and NETs gene sets was utilized to identify key genes associated with NETs in sepsis-associated ARDS.
Machine Learning
For further refining the filtered candidate genes correlated with sepsis-associated ARDS and NETs, we utilized three machine learning algorithms. The Least Absolute Shrinkage and Selection Operator (LASSO),12 in detail, the “glmnet” package was employed to operate LASSO. cv.glmnet was exploited to optimize lambda. For the parameters, the scale of “λ” was set ranging from 0 to 100 with “binomial” and “class”. Under the minimum λ, glmnet was processed to the LASSO with alpha and a “binomial” method. Afterward, we use the support vector machine-recursive feature elimination (SVM-RFE) algorithm. SVM was implemented using the “e1071” package. tune.svm was adopted to improve the settings parameter with the kernel of “linear”, and the cost ranging from 1 to 20. Following, compliance with the optimal number of support vectors, the SVM model was completed. RF was carried out with the “randomForest” package. Firstly, the tuneRF function was employed to optimize 0–700 trees with one step size. RF was managed according to the minimum error rate to acquire the optimal tree number. The selection of these three algorithms was based on their complementary strengths and the ability to provide a comprehensive assessment of the most relevant genes. LASSO regression excels in feature selection for linear models, SVM-RFE provides robust feature ranking through recursive elimination, and Random Forest offers a powerful approach for handling non-linear interactions and assessing feature importance. By combining the results of these algorithms, we aimed to enhance the reliability and accuracy of our biomarker identification process.
Collection of Clinical Samples
The clinical trial was implemented strictly complying with the Declaration of Helsinki and obtained approval from the Institute Research Ethics Committee at Shanxi Bethune Hospital to make sure there were no ethical issues.Twenty sepsis-associated ARDS patients hospitalized between September 2023 and May 2024, and twenty healthy adults who underwent physical examinations during the same period, were selected as control groups. Inclusion criteria for both groups included age ≥18 years and informed consent. Inclusion criteria: ①Test group: I meet the diagnostic criteria of sepsis-associated ARDS; II age ≥18 years old; III informed consent. ② Healthy control group: I prompted by the physical examination center of the hospital with good health status; II age ≥18 years old; III informed consent. Exclusion criteria: ① age <18 years old; ② death on admission and transfer of patients with imperfect clinical data; ③ patients with organic heart disease: ischaemic cardiomyopathy, congenital heart disease, myocarditis, and other diseases. ④Patients with end-stage chronic and malignant diseases; ⑤Patients who refused to participate in the study or refused to sign the informed consent form; ⑥Patients with a previous history of mental illness or cognitive dysfunction. The blood samples were centrifuged at 4°C and a speed of 2000 rpm for 10 minutes, and the supernatant was collected and immediately frozen at −80°C for storage.
Validation of the Hub Gene Expression
The total RNA was extracted from the serum of the subjects. Reverse transcription was performed under the following conditions: 11 μL of RNA (1 μg) was mixed with 1 μL of random primer (0.2 μg/ mL) and incubated at 65°C for 5 min. Subsequently, a mixture containing 4 μL of 5 × Buffer, 3 μL of dNTP (10 mmol/L), 1 μL of RNase inhibitor (20 U/uL), and 1 μL of reverse transcriptase (20 U/ uL) was added to the reaction. The reaction proceeded at 25°C for 10 min, followed by incubation at 42°C for 1 h and finally at 72°C for 15 min. The real-time PCR reaction system consisted of 10 μL FastStart Universal SYBR Green Master (ROX), 0.5 μL upstream primer (15 μM), 0.5 μL downstream primer (15 μM), 2 μL cDNA, and 7 μL DNAse and RNase-free water, making a total volume of 20 μL. The primer sequence for GAPDH was as follows: upstream primer -CAGGAGGCATTGCTGATGAT-3′ and downstream primer -GAAGGCTGGGGCTCATTT-3′. Additionally, the NETs hub gene primer sequence was included. PCR reaction conditions: The PCR reaction was initiated by pre-denaturation at 94°C for 10 min to activate the Tag enzyme. Subsequently, the amplification process consisted of denaturation at 94°C for 15s, annealing at 60°C for 60s, and a total of 40 cycles. The cycle threshold (CT) value was determined using fluorescence quantitative PCR. Firstly, the difference between the CT value of the target gene and that of the internal control gene GAPDH in each sample was calculated as ΔCT = CT(hub gene)-CT(GAPDH). Then, ΔΔCT was obtained by subtracting the ΔCT value of samples from the experimental group with that from the normal control group. Finally, calculation using 2(−ΔΔCT) indicated the fold change in expression of hub gene in relation to its expression in samples from normal control group. This analysis was designed to assess the expression levels of NETs hub genes in sepsis-associated ARDS. The primer sequences are exhibited in Supplementary Table 2. The catalog numbers for all reagents are exhibited in Supplementary Table 3.
Analysis of the Diagnostic Value of Key Genes for NETs
We first verified the expression of the hub genes of NETs using sepsis-associated ARDS and healthy controls in the GSE32707 dataset. The pROC package in R was further used to evaluate the diagnostic efficacy of the above key genes of NETs in sepsis-associated ARDS. ROC curves were plotted, and AUC values > 0.7 indicated superior diagnostic potential.13
Building of Nomogram and The Assessment of The Prediction Model
The clinically applicable nomogram was developed with the two selected hub genes (LTF and PRTN3) by employing the “rms” package. A calibration curve was established to assess the diagnostic predictive ability of the nomogram for sepsis-associated ARDS.
Single-Sample Gene Set Enrichment Analysis (ssGSEA)
To make a thorough exploration of immune cell infiltration states in sepsis-associated ARDS patients, the “GSVA” function in R software was employed to calculate the immune cell infiltration score of each sample using the gene set composed of immune cell markers.14 Plus, a Spearman correlation method was executed to identify the correlation between the key genes and the immune cells.
Prediction of Therapeutic Drug with Hub Genes via Molecular Docking
Molecular docking experiments were further implemented to explore the interaction between potential therapeutic drugs and the screened hub genes. Firstly, the Drug Signatures Database (DSigDB) on the Enrichr website (https://maayanlab.cloud/Enrichr/) was accessed to acquire potential drugs toward core genes.15,16 Afterward, the protein structures corresponding to the hub genes and small molecular drugs were obtained from the UniProt database (https://www.uniprot.org/) and PubChem website (https://pubchem.ncbi.nlm.nih.gov/), respectively. Ultimately, the AutoDock (https://ccsb.scripps.edu/mgltools/downloads/) and PyMOL software were adopted to simulate the binding of targeted proteins and small molecular drugs.
Statistical Analyses
Statistical analysis of raw confidence analysis data was performed using R software (version 4.3.1) and R Studio (version 2023.09.1), while Prism software was used for clinical trial data analysis. Normally distributed measures were described as mean ± standard deviation ( ± s), and comparisons between two groups were made using the independent samples t-test. Non-normally distributed measures were described as median (interquartile range) [M (QL, QU)], and the Mann–Whitney U-test was used for between-group comparisons. Categorical data were presented as frequencies and percentages, and comparisons between groups were performed using the chi-square (χ²) test or Fisher’s exact test, as appropriate. A two-sided P-value of less than 0.05 was considered to indicate statistical significance.
± s), and comparisons between two groups were made using the independent samples t-test. Non-normally distributed measures were described as median (interquartile range) [M (QL, QU)], and the Mann–Whitney U-test was used for between-group comparisons. Categorical data were presented as frequencies and percentages, and comparisons between groups were performed using the chi-square (χ²) test or Fisher’s exact test, as appropriate. A two-sided P-value of less than 0.05 was considered to indicate statistical significance.
Results
Differential Gene Expression Analysis of Sepsis-Associated ARDS
The brief research procedure is illustrated in Figure 1. Analysis of the GSE32707 dataset using R software identified 485 differentially expressed genes (DEGs) associated with sepsis-associated ARDS, including 271 up-regulated and 214 down-regulated genes, based on the criteria adj. P < 0.05 and |log FC| > 1. The volcano and heat maps are shown in Figure 2A and B.
 |
Figure 1 The working flow chart of this study. Abbreviations: GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; ssGSEA, Single-Sample Gene Set Enrichment Analysis; WGCNA, Weighted Gene Co-expression Network AnalysisWeighted Gene Co-expression Network Analysis; ROC, Receiver Operating Characteristic Curve. |
 |
Figure 2 DEGs in sepsis-associated ARDS. (A) Shows the volcano map of differentially expressed genes. (B) Shows the heat map of differentially expressed genes. |
Functional Enrichment Analysis of the Differentially Expressed Genes
We then performed GO and KEGG enrichment analysis of DEGs based on the above results. For biological processes (BP), biological processes (BP) were mainly enriched in leukocyte and neutrophil activation, chemotaxis, migration and immune response; for cellular components (CC), in vesicle lumen, specific granules and secretory granules; and molecular function (MF), mainly in peroxidase activity and oxidoreductase activity (Figure 3A–C). The KEGG analysis highlighted the neutrophil extracellular trap (NETs) signaling pathways (Figure 3D and E).
 |
Figure 3 GO and KEGG. (A) Shows the bar graph of GO enrichment analysis. (B) Shows the bubble graph of GO enrichment analysis. (C) Shows the GO functional enrichment analysis. (D and E) Shows the KEGG enrichment analysis. |
Establishment of Co-Expression Network and Deciphering Hub Gene Modules
Weighted Gene Co-expression Network Analysis (WGCNA) results indicated that a soft threshold of 14 was optimal (Figure 4A and B). This led to the generation of merged cluster tree modules (Figure 4C) and subsequent evaluation of module correlations (Figure 4D), identifying the grey modules with the highest correlation coefficients, encompassing 332 genes.
 |
Figure 4 WGCNA. (A) Shows a clustered dendrogram of sepsis-associated ARDS and healthy controls. (B) Shows a soft threshold analysis diagram. (C) Shows the clustering tree module diagram. (D) Shows the gene module and clinical trait association plot. |
Screening of Hub Genes for NETs in Sepsis-Associated ARDS
Through a literature search, NETs gene sets were obtained, which contained 137 genes. It was overlapped with 332 genes of WGCNA and 485 genes of the above DEGs, and the five key genes of NETs were selected as PRTN 3, LTF, CTSG, CD177 and MPO (Figure 5).
 |
Figure 5 Analysis of key genes for NETs in sepsis-associated ARDS. |
Identification of Hub Genes via Machine Learning Algorithm
Machine learning algorithms were adopted to screen underlying diagnostic biomarkers from 5 shared key genes previously determined. The LASSO algorithm identified 4 hub genes (Figure 6A and B). Meanwhile, the SVM-REF method showed that 2 genes achieved the highest accuracy (Figure 6C). In addition, the RF algorithm identified 5 potential biomarkers and ranked them according to their importance (Figure 6D and E). By intersecting the results of the three algorithms, 2 candidate biomarkers were obtained: PRTN3 and LTF (Figure 6F).
 |
Figure 6 Machine learning in screening candidate diagnostic markers for sepsis-associated ARDS. (A and B) Biomarkers screening in the LASSO model. (C) Two crosstalk genes were selected by using the SVM-RFE algorithm. (D and E) The error rate confidence intervals and the relative importance of genes for random forest algorithm. (F) The Venn diagram of the intersection of LASSO, SVM-REF, and random forest signature genes. |
Hub Genes of NETs Were Verified by Dataset and RT-qPCR
To validate the mRNA abundance of the two diagnostic markers in clinical samples, RT-PCR was then conducted on the blood samples from the healthy controls and sepsis-associated ARDS patient (Figure 7A and B). Compared with the healthy controls, quantification of the mRNA abundance of LTF (p < 0.05) and PRTN3 (p < 0.01) were both markedly up-regulated in the sepsis-associated ARDS patients (Figure 7C and D).
 |
Figure 7 Hub genes of NETs were verified by dataset and RT-qPCR. (A and B) LTF and PRTN3 expression was significantly higher in sepsis-associated ARDS patients than in healthy controls. (C and D) RT-qPCR verified that LTF and PRTN3 expression were significantly higher in sepsis-associated ARDS patients than in healthy controls. Note:***: Indicates a highly significant difference with a p-value less than 0.001. |
Validation and Diagnostic Efficacy Analysis of the Two Core Genes
We detected the expression of PRTN3 and LTF in the sepsis-associated ARDS datasets. Figure shows that the expression of PRTN3 and LTF are significantly increased in the sepsis-associated ARDS. Subsequently, ROC analysis was applied to assess the specificity and sensitivity of these two hub genes in diagnosing sepsis-associated ARDS. The results showed that LTF (AUC = 0.832), PRTN3 (AUC = 0.790) (Figure 8).
 |
Figure 8 Analysis of the efficacy of NETs key genes for disease diagnosis. (A) LTF ROC curve. (B) PRTN3 ROC curve. (C) LTF and PRTN3 ROC curves. |
Construction of the Nomogram and the Prediction of the Risk of Disease Occurrence
A nomogram was constructed based on the two hub genes, and a calibration plot was created to assess the model’s predictive value (Figure 9A and B). The calibration plot illustrated minimal deviation between the actual event risk and the predicted event risk, indicating the acceptable reliability of the model.
 |
Figure 9 Nomogram construction and diagnostic performance validation. (A) Nomogram construction and diagnostic performance validation. (B) The calibration plot showing the accuracy of the nomogram. |
Immune Infiltration Analysis of Differential Genes and Hub Genes
To further confirm the role played by immune cells in sepsis-associated ARDS, we evaluated the degree of immune cell infiltration using data from GSE32707 and the single-sample gene set enrichment analysis (ssGSEA) function of the GSVA package in R. Further, we analyzed the correlations between sepsis and infiltration of 23 types of immune cell (Figure 10A and B). According to the immune infiltration levels in the two groups (patients with sepsis-associated ARDS and healthy controls), Central memory CD4 T cell, Effector memory CD4 T cell, Memory B cell, Natural killer cell, CD56bright natural killer cell and CD56dim natural killer cell were down-regulated in the sepsis-associated ARDS group. Further, Activated CD8 T cell, Neutrophil, Activated dendritic cell, Plasmacytoid dendritic cell, Immature dendritic cell, Macrophage, Eosinophil, Mast cell and Monocyte were significantly up-regulated in sepsis (Figure 10C). These results suggest that immune infiltration has an important role in sepsis-associated ARDS. The expression of key NETs genes (PRTN3, LTF) in sepsis-associated ARDS was closely associated with immune cell infiltration, including Neutrophil, Mast cell, Myeloid derived suppressor cell and Activated dendritic cell (Figure 10D).
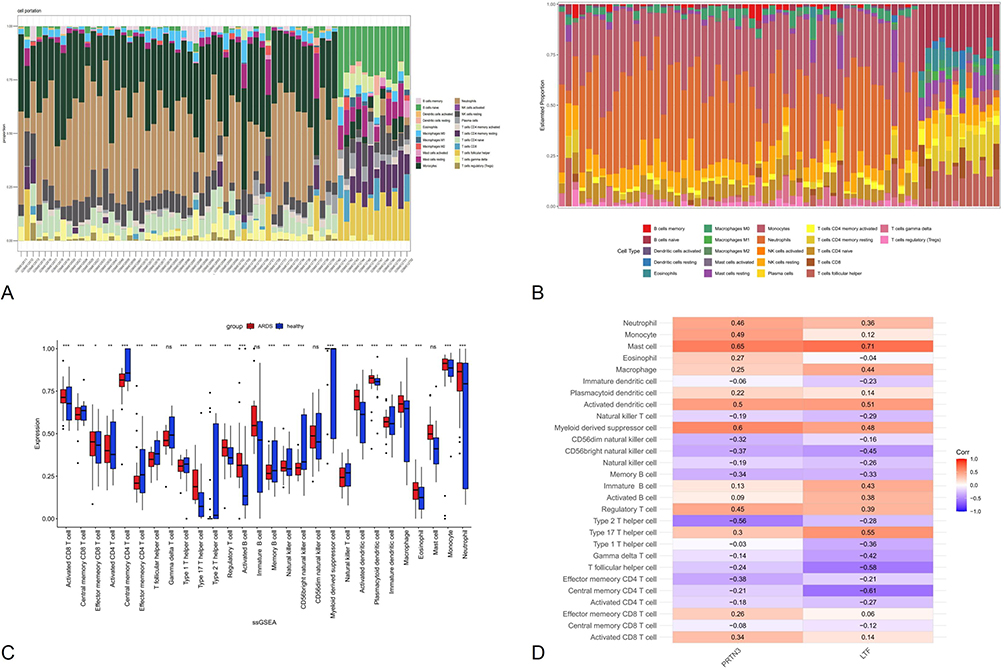 |
Figure 10 Immune cell infiltration analysis with the ssGSEA algorithm. (A and B) The heatmap exhibited the difference between healthy control and sepsis-associated ARDS of 28 types of immune cells. (C) Comparison of 28 types of immune cells between healthy control and sepsis-associated ARDS. (D) Correlation analysis between hub genes and immune cells in sepsis-associated ARDS patients. Notes: *: Indicates a statistically significant difference with a p-value < 0.05. **: Indicates a more significant difference with a p-value < 0.01. ***: Indicates a highly significant difference with a p-value < 0.001. |
Visualization of Molecular Docking Simulation Toward Hub Genes
To explore drugs targeting two key genes, PRTN 3 and LTF, with potential therapeutic effects against sepsis-associated ARDS, we sought to screen small molecule drugs targeting PRTN 3 and LTF by molecular docking. We downloaded the crystal structure of PRTN3 and LTF with high resolution.The binding poses and sites are shown in Figure 11, where yellow dashed lines and black numbers represent hydrogen bonds and their lengths, and green represents the small-molecule drugs.The results showed that a potential small molecule drug, nimesulide, had a LTF binding energy value of −5.81kcal/mol (Figure 11A). The binding energies of the two potential small-molecule drugs, minocycline and pyrantel with PRTN3 were −5.1kcal/mol and −4.3kcal/mol, respectively (Figure 11B and C).
 |
Figure 11 Molecular docking patterns. (A) LTF complexed with Nimesulide (B) PRTN3 complexed with Minocycline, and (C) PRTN3 complexed with Fucose. |
Discussion
Sepsis-associated ARDS is a severe condition with high mortality rates, making early diagnosis and timely intervention critical for reducing case fatality. In this study, we identified 485 differentially expressed genes (DEGs) associated with sepsis-associated ARDS through analysis of the GSE32707 dataset using the limma package in R. These DEGs were significantly enriched in biological processes such as neutrophil activation and chemotaxis, cellular components like vesicle lumen and secretory granules, and molecular functions including peroxidase and oxidoreductase activity. KEGG analysis further revealed enrichment in pathways associated with neutrophil extracellular traps (NETs). These findings suggest that the DEGs in the ARDS associated with sepsis are involved in the production and signaling of NETs. Previous studies found that a large number of NETs were detected in mice with sepsis-associated acute lung injury and sepsis-associated ARDS patients, which is consistent with the results of this study.17
Weighted gene co-expression network analysis (WGCNA) identified gene modules that were significantly associated with ARDS. By intersecting key module genes identified by DEG, WGCNA and NETs, we screened and identified five hub genes of NETs associated with ARDS associated with sepsis: PRTN 3, LTF, CTSG, CD177 and MPO. Subsequently, we used three machine learning algorithms to further identify two NETs hub genes, LTF and PRTN 3. Validation of its RT-qPCR in dataset 32707 and clinical samples showed that both LTF and PRTN 3 expression were significantly upregulated in sepsis-associated ARDS patients compared with controls. Previous studies showed that the protein product encoded by the LTF gene is significantly present in the secondary granules of neutrophils and is considered to be the innate immunity protein against infection.18 The LTF protein, through its ability to reduce the levels of c-reactive protein, is a potent regulator of inflammatory homeostasis.19 PRTN3 was associated with endothelial dysfunction in sepsis,20 along with a positive correlation with increased leukocyte and neutrophil counts in hematological parameters.21 It suggests that the two hub NETs genes, LTF and PRTN 3, are involved in the pathophysiology of sepsis-associated ARDS. Previous studies have proposed various biomarkers for sepsis, including procalcitonin, which is widely used in clinical settings due to its association with bacterial infections.22 But procalcitonin may not specifically distinguish sepsis from other common inflammatory conditions. In contrast, our study identifies LTF and PRTN3, which are specifically associated with NETs and show strong upregulation in sepsis-associated ARDS patients. This specificity may provide a more accurate diagnostic tool. Further ROC curve analysis and Nomogram analysis of two hub NETs genes, LTF and PRTN3, showed that LTF and PRTN 3 may serve as effective diagnostic biomarkers for sepsis-associated ARDS. However, the exact mechanism of its action in sepsis-associated ARDS should be further explored.
Results of immune cell infiltration analysis showed that neutrophils were significantly more active in sepsis-associated ARDS patients compared with healthy controls. This finding underscores the importance of hub NETs genes in disease progression. And perform the molecular docking drug prediction for the hub NETs genes. Two meaningful small molecule compounds were obtained (nimesulide and minocycline). Strikingly, nimesulide had the lowest binding energy of - 5.8 kcal/mol, implying that it may most strongly minimize the pathogenic gene expression in sepsis-associated ARDS patients. Nimesulide (NIM) is a non-steroidal anti-inflammatory drug and a specific inhibitor of cyclooxygenase-2 (COX 2) for the treatment of various inflammation-associated diseases.23 NIM has been reported to inhibit the lPS-induced iNOS expression in the alveolar macrophages.24 Inhibition of COX-2 provides anti-inflammatory therapy in acute respiratory distress syndrome.25 Minocycline is a semisynthetic tetracycline analogue with broad-spectrum anti-pathogenic microbial activity. It has also been reported that the anti-inflammatory and immunomodulatory properties of minocycline may be beneficial for the treatment of COVID-19 patients, especially in the case of combined ARDS and multiple organ function impairment.26
It is essential to acknowledge the limitations of our study. First, It is the small sample size dataset and small clinical cohort, which may limit the generalizability of the findings. Therefore, experimental validation in larger sample size dataset and larger cohorts is needed. Second, the mechanistic studies of key NETs genes are scarce, which is needed to further clarify the regulatory mechanism. Third, the molecular docking simulations predict potential therapeutic compounds, but in vivo validation studies are lack, which are required to design so as to confirm their efficacy and safety. These limitations are precisely the direction we will work towards in the future.
Conclusions
In conclusion, our study uncovers LTF and PRTN3 as novel hub genes in NETs pathways associated with sepsis-associated ARDS (not previously reported for ARDS), positioning them as potential diagnostic biomarkers and therapeutic targets for this life-threatening condition. By employing molecular docking, we identified nimesulide and minocycline as promising small-molecule candidates, offering a new therapeutic strategy. These findings represent a critical advancement in understanding NETs-driven pathology in sepsis-associated ARDS, with implications for early diagnostic screening and drug repurposing. Future validation in larger cohorts and mechanistic studies will clarify the roles of LTF and PRTN3, potentially enabling targeted interventions to improve patient outcomes in this high-mortality disease.
Abbreviations
NETs, Neutrophil Extracellular Traps; ARDS, Acute Respiratory Distress Syndrome; DEGs, Differentially expressed genes; WGCNA, Weighted Gene Co-expression Network Analysis; TLRs, Toll-like receptor; GEO, Gene Expression Omnibus; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; BP, biological processes; CC, cellular components; MF, molecular functions; ssGSEA, Single-Sample Immune Cell Infiltration Analysis.
Data Sharing Statement
The data supporting the findings of this study are available in the NCBI Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/).
Ethics Approval and Consent to Participate
The study was approved by the Ethics and Research Committee of Shanxi Bethune Hospital. All the patients who took part in this study were provided informed consent prior to participation in the study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
2023 Talent Introduction Support Program of Shanghai Pudong New Area Zhoupu Hospital (ZP-XK-2023B-4). 2023 Public Institution People’s Livelihood Research Special Project, Pudong New Area Science and Technology Development Fund (PKJ2023-Y12). 2024 Teaching Research Project for Faculty, Shanghai University of Medicine & Health Sciences (CFDZ20240025P).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interests.
References
1. Gong H, Chen Y, Chen M, et al. Advanced development and mechanism of sepsis-related acute respiratory distress syndrome. Front Med. 2022;9:1043859. doi:10.3389/fmed.2022.1043859
2. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
3. Sartini C, Landoni G, Belletti A, et al. Beyond the Surviving Sepsis Campaign Guidelines: a systematic review of interventions affecting mortality in sepsis. Panminerva Med. 2024;66(1):55–62. doi:10.23736/S0031-0808.23.04986-8
4. Ghanem-Zoubi N, Bitterman H, Laor A, Yurin V, Vardi M. The accuracy of clinical prediction of prognosis for patients admitted with sepsis to internal medicine departments. Ann Med. 2015;47(7):555–560. doi:10.3109/07853890.2015.1089361
5. Chang Y, Yoo HJ, Kim SJ, et al. A targeted metabolomics approach for sepsis-induced ARDS and its subphenotypes. Crit Care. 2023;27(1):263. doi:10.1186/s13054-023-04552-0
6. Wang H, Shi Y, Xu X, et al. A novel neutrophil extracellular traps-related lncRNA signature predicts prognosis in patients with early-stage lung adenocarcinoma. Ann Med. 2023;55(2):2279754. doi:10.1080/07853890.2023.2279754
7. Demkow U. Molecular Mechanisms of Neutrophil Extracellular Trap (NETs) Degradation. Int J Mol Sci. 2023;24(5):4896. doi:10.3390/ijms24054896
8. Arneth B, Arneth R. Neutrophil Extracellular Traps (NETs) and Vasculitis. Int J Med Sci. 2021;18(7):1532–1540. doi:10.7150/ijms.53728
9. Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007;23(14):1846–1847. doi:10.1093/bioinformatics/btm254
10. Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013;29(14):1830–1831. doi:10.1093/bioinformatics/btt285
11. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9(1):559. doi:10.1186/1471-2105-9-559
12. Motamedi F, Pérez-Sánchez H, Mehridehnavi A, Fassihi A, Ghasemi F. Accelerating Big Data Analysis through LASSO-Random Forest Algorithm in QSAR Studies. Bioinformatics. 2022;38(2):469–475. doi:10.1093/bioinformatics/btab659
13. Obuchowski NA, Bullen JA. Receiver operating characteristic (ROC) curves: review of methods with applications in diagnostic medicine. Phys Med Biol. 2018;63(7):07TR01. doi:10.1088/1361-6560/aab4b1
14. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14(1):7. doi:10.1186/1471-2105-14-7
15. Yoo M, Shin J, Kim J, et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics. 2015;31(18):3069–3071. doi:10.1093/bioinformatics/btv313
16. Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–W97. doi:10.1093/nar/gkw377
17. Ronchetti L, Boubaker NS, Barba M, Vici P, Gurtner A, Piaggio G. Neutrophil extracellular traps in cancer: not only catching microbes. J Exp Clin Cancer Res. 2021;40(1):231. doi:10.1186/s13046-021-02036-z
18. Lin G, Li N, Liu J, et al. Identification of key genes as potential diagnostic biomarkers in sepsis by bioinformatics analysis. PeerJ. 2024;12:e17542. doi:10.7717/peerj.17542
19. Pammi M, Suresh G. Enteral lactoferrin supplementation for prevention of sepsis and necrotizing enterocolitis in preterm infants. Cochrane Database Syst Rev. 2017;6(6):CD007137. doi:10.1002/14651858.CD007137.pub5
20. Liu H, Sun L, Zhao H, et al. Proteinase 3 depletion attenuates leukemia by promoting myeloid differentiation. Cell Death Differ. 2024;31(6):697–710. doi:10.1038/s41418-024-01288-4
21. Van Nynatten LR, Slessarev M, Martin CM, et al. Novel plasma protein biomarkers from critically ill sepsis patients. Clin Proteomics. 2022;19(1):50. doi:10.1186/s12014-022-09389-3
22. Tan M, Lu Y, Jiang H, Zhang L. The diagnostic accuracy of procalcitonin and C-reactive protein for sepsis: a systematic review and meta-analysis. J Cell Biochem. 2019;120(4):5852–5859. doi:10.1002/jcb.27870
23. Yang Z, Ji W, Li M, et al. Protective effect of nimesulide on acute lung injury in mice with severe acute pancreatitis. Am J Transl Res. 2019;11(9):6024–6031.
24. Khanduja KL, Sohi KK, Pathak CM, Kaushik G. Nimesulide inhibits lipopolysaccharide-induced production of superoxide anions and nitric oxide and iNOS expression in alveolar macrophages. Life Sci. 2006;78(15):1662–1669. doi:10.1016/j.lfs.2005.07.033
25. Caiazzo E, Maione F, Morello S, et al. Adenosine signalling mediates the anti-inflammatory effects of the COX-2 inhibitor nimesulide. Biochem Pharmacol. 2016;112:72–81. doi:10.1016/j.bcp.2016.05.006
26. Singh H, Kakkar AK, Chauhan P. Repurposing minocycline for COVID-19 management: mechanisms, opportunities, and challenges. Expert Rev Anti Infect Ther. 2020;18(10):997–1003. doi:10.1080/14787210.2020.1782190
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.