")
Back to Journals » Journal of Inflammation Research » Volume 17
Identifying and Validating Extracellular Matrix-Related Gene CTSH in Diabetic Foot Ulcer Using Bioinformatics and Machine Learning
Authors Wu PY , Yu YL, Zhao WR, Zhou B
Received 9 March 2024
Accepted for publication 24 August 2024
Published 30 August 2024 Volume 2024:17 Pages 5871—5887
DOI https://doi.org/10.2147/JIR.S467507
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Pei-Yu Wu,1,2 Yan-Lin Yu,1 Wen-Rui Zhao,1 Bo Zhou1
1Department of Endocrinology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China; 2Department of VIP, Chongqing General Hospital, Chongqing University, Chongqing, People’s Republic of China
Correspondence: Bo Zhou, Department of Endocrinology, The First Affiliated Hospital of Chongqing Medical University, No. 1 Friendship Road, Yuzhong District, Chongqing, 400042, People’s Republic of China, Email [email protected]
Background: Diabetic foot ulcer (DFU) is a serious clinical problem with high amputation and mortality rates, yet there is a lack of desirable therapy. While the extracellular matrix (ECM) contributes significantly to wound healing, ECM-related biomarker for DFU is still unknown. The study was designed to identify ECM-related biomarker in DFU using bioinformatics and machine learning and validate it in STZ-induced mice models.
Methods: GSE80178 and GSE134431 microarray datasets were fetched from the GEO database, and differentially expressed genes (DEGs) analysis was performed, respectively. By analyzing DEGs and ECM genes, we identified ECM-related DEGs, and functional enrichment analysis was conducted. Subsequently, three machine learning algorithms (LASSO, RF and SVM-RFE) were applied to filter ECM-related DEGs to identify key ECM-related biomarkers. Next, we conducted immune infiltration analysis, GSEA, and correlation analysis to explore the hub gene underlying mechanism. A lncRNA-miRNA-mRNA and drug regulatory network were constructed. Finally, we validated the key ECM-related biomarker in STZ-induced mice models.
Results: One hundred and forty-five common DEGs in adult DFU between the two datasets were identified. Taking the intersection of 145 common DEGs and 964 ECM genes, we identified 13 ECM-related DEGs. Thirteen ECM-related DEGs were mainly enriched in pathways associated with tissue remodeling, inflammation and defense against infectious agents. Ultimately, CTSH was identified as the key ECM-related biomarker. CTSH was associated with difference immune cells during the occurrence and development of DFU, and it influenced hedgehog, IL-17 and TNF signaling pathway. Additionally, CTSH expression is correlated with many ECM- and immune-related genes. A lncRNA-miRNA-mRNA and drug regulatory network were constructed with 10 lncRNAs, 2 miRNAs, CTSH and 1 drug. Finally, CTSH was validated as a key biomarker for DFU in animal models.
Conclusion: Our study found that CTSH can be used for both diagnostic and prognostic purposes and might be a potential therapeutic target.
Keywords: diabetic wound healing, extracellular matrix, bioinformatics, machine learning, CTSH
Graphical Abstract:

Introduction
Diabetic foot ulcer (DFU) is defined as a break of the foot skin in diabetic patients that affects at least the epidermis and part of dermis and is commonly accompanied by peripheral neuropathy and/or peripheral artery disease in the lower extremity.1 DFU, a severe complication of diabetes, is linked to higher rates of amputation and mortality. DFUs are a significant health concern, currently affecting 6.3% of the global population with diabetes, with annual recurrence rates of 40%, and a 5-year mortality rate of 49.1%.2,3 The standard care/conventional treatment for patients with DFU includes good glycemic control, wound debridement, offloading, infection control and the use of medical dressing. Furthermore, several other approaches like intralesional epidermal growth factor therapy4 platelet-rich gel,5 negative pressure wound therapy,6 exosome7 and stem cell therapy8 also provide a viable alternative in the management of DFU. However, current treatments cannot achieve satisfactory results. DFU has become an urgent problem to be solved. Thus, it is essential to investigate the molecular changes and mechanism of DFU to improve the efficiency of DFU treatment.
Wound healing is a dynamic and complex process that includes four overlapping stages: hemostasis or coagulation, inflammation, proliferation, and remodeling. The coordinated action of the extracellular matrix (ECM), numerous cell types and soluble mediators drive this process.9 Given the importance of ECM in the process, investigating the ECM-related genes and mechanisms that are involved in diabetic wound healing is essential to find potential therapeutic targets. ECM is a complicated three-dimensional network structure, which not only provides essential physical scaffolding for cells but also provides a proper environment to induce cell adhesion, growth, migration, and differentiation.10 In the process of wound repairing, ECM play a significant role. Delayed healing of wounds in diabetes partly results from the dysfunction and dysregulation of ECM.11 However, the ECM-related biomarker of DFU and its regulation mechanism in DFU remain unknown. Consequently, further research is required to uncover the ECM-related biomarker and its regulation mechanism in DFU. Gene expression profiling is a pivotal technology in the field of molecular medicine. By comparing gene expression patterns between disease status and normal controls, medical researchers can identify differentially expressed genes (DEGs). These DEGs may serve as potential biomarkers for disease diagnosis and prognosis. Furthermore, they can provide valuable insights into the molecular mechanisms of various diseases. Some researchers extract gene expression data from the Gene Expression Omnibus (GEO) database to explore molecular biomarkers associated with diseases. In addition, the combination of gene expression profiling technology and machine learning algorithms has made significant progress in the medical field, changing our ability to diagnose and predict disease outcomes. This combination provides a novel method for identifying molecular targets.
The objective of this study was to explore potential biomarkers and mechanisms of ECM-related genes involved in the development and progression of DFU. A single-gene CTSH was identified based on three machine learning methods (least absolute shrinkage selection operator (LASSO), random forest (RF) and support vector machine recursive feature elimination (SVM-RFE)). Then, CTSH was analyzed by multidimensional bioinformatics. At first, we sought to investigate the immune cells differences, and the pathways of DFU were identified using gene set enrichment analysis (GSEA). Additionally, we assessed the correlation between CTSH expression and ECM-related genes as well as immune-related genes. Subsequently, a lncRNA-miRNA-mRNA and drug regulatory network were revealed. Finally, CTSH has not been investigated in the field of wound healing, and the expression of CTSH was first validated in animal models. In conclusion, this research hoped to reveal ECM-related biomarker for early identification of DFU and can serve as a new treatment strategy for DFU. These findings may provide new insights into the role of ECM in the development and progression of DFU. They highlight the potential of CTSH as a diagnostic and prognostic biomarker, thus laying the groundwork for further research.
Materials and Methods
Raw Data Collection
Two microarray data were acquired from the GEO database (www.ncbi.nlm.nih.gov/geo/) by the following criteria: a) datasets included should be from patients with diabetic foot ulcer; b) samples were wound tissue biopsies. The mRNA expression profiles were obtained from GSE80178 (GPL16686 Affymetrix Human Gene 2.0 ST Array, 6 adult DFU and 3 adult non-Diabetic Foot Skin samples) and GSE134431 (GPL18573 Illumina NextSeq 500, 6 adult unhealed DFU and 7 adult healed DFU samples).
ECM-Related DEGs Identification
GEO2R tool on the GEO website (www.ncbi.nlm.nih.gov/geo/) was performed to screen DEGs for the two datasets. Samples from GSE80178 with |log2(fold change, FC)|>1.0 and an adj. P value <0.05 were defined as the thresholds of DEGs, while samples from GSE134431 with |log2(fold change, FC)|>1.0 and a P value <0.05 were defined as the thresholds of DEGs. Additionally, the difference analysis results were presented by volcano plots and VennDiagram using an online platform (https://hiplot.com.cn/cloud-tool/drawing-tool/list). A total of 964 extracellular matrix and remodeling genes were downloaded.12 The ECM genes and common genes were visualized by VennDiagram using an online platform (https://hiplot.com.cn/cloud-tool/drawing-tool/list).
Functional Enrichment Analysis
Metascape was utilized to conduct Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis.13 Visual results were obtained after inputting ECM-related DEGs and selecting H. sapiens species.
Hub Genes Identification via Machine Learning Methods
Hub genes were analyzed using three machine learning methods (LASSO, RF and SVM-RFE). The LASSO algorithm is a regressive analytical arithmetic which makes use of regularisation to improve the accuracy of prediction. First, we implemented LASSO regressive arithmetic through the R package “glmnet” to identify the co-existing genes that are significantly correlated with the development and progression of DFU. RF algorithm was carried out using the R package “random Forest”. The most important variables were identified through RF analysis based on the decision tree algorithm. Following that, the SVM algorithm is a monitored machine learning technology widely used in classification and regression. Using the RFE algorithm, the optimum genes were selected from metadata cohorts in order to avoid overfitting. Subsequently, “e1071” package of R software was used to construct SVM modeling. Through sequential backward feature elimination, SVM-RFE determines optimal hub genes by selecting feature variables. Finally, the intersection of these results was taken as the hub gene of the occurrence and development of DFU.
Assessment of the Immune Landscape
Immune cell infiltration analysis was conducted using the “CIBERSORT” R package to evaluate the expression levels of 22 immune cells for each sample of GSE80178 and GSE134431. Next, our team analyzed the correlation between the expression of hub gene CTSH and those immune cells. ‘Vioplot’, ‘ggplot2’ and ‘ggExtra’ packages of R were used to visualize the results.
Single-Gene GSEA of Hub Gene CTSH
To reveal the pathways involved in the single-gene CTSH, dataset GSE147890 (GPL571 Affymetrix Human Genome U133A 2.0 Array, 12 diabetic skin and 12 non-diabetic skin samples) was collected from GEO database. We grouped the samples based on a single hub gene expression level in the dataset. Subsequently, the “clusterProfiler” R was used to perform GSEA. The first 5 up- and down-regulated pathways were visualized. Furthermore, the top 10 most significant adj.P value pathways were also visualized.
Correlation Analysis of CTSH Expression Level with ECM-Related Genes and Immune-Related Genes
Given the importance of ECM and immunization for DFU, Spearman’s approach was applied to evaluate the association between CTSH expression level and ECM-related genes as well as immune-related genes. The “ggplot2” and “ggExtra” packages of R were applied to visualize the results.
Construction of lncRNA-miRNA-mRNA and Drug Regulatory Network
Comparative Toxicogenomics Database (CTD, https://ctdbase.org/) was used to search molecular compounds or drugs related to the hub gene. To further explore the upstream regulatory molecules of CTSH expression, miRNAs and lncRNAs were predicted. Target miRNAs of CTSH were predicted based on the miRDB (https://mirdb.org/), StarBase (https://starbase.sysu.edu.cn/) and miRWalk (http://mirwalk.umm.uni-heidelberg.de/) databases, and the intersection of the three databases was identified as target miRNAs. The StarBase and miRNet (https://www.mirnet.ca) databases were used to forecast upstream lncRNAs of target miRNAs, and the intersection of the two databases was identified as target lncRNAs. Then, a significant lncRNA-miRNA-mRNA and drug regulatory network were builded and visualized using the Cytoscape software (version 3.9.1).
Animal Models Construction
C57/B6 mice (male, 6–8 weeks old) were procured. All C57BL/6 mice were raised under controlled environmental conditions (22–23°C, light/dark cycle of 12/12h, humidity of 55% ± 5%) with free access to food and water. C57/B6 mice were anaesthetized through intraperitoneal injection with 50 mg/kg sodium pentobarbital, subsequently, a 6 mm skin full-thickness wounds were created on their upper back after shaving. The wound tissues were collected at 1,3,5,7,9 and 11 days post-injury. To further test the accuracy, we searched for mice skin wounds on days 0 to 10 during the skin wound healing process in the GSE23006 dataset for external validation. For diabetic model, mice were induced through intraperitoneal injection with 50 mg/kg streptozotocin (STZ) for 5 consecutive days, and the level of fasting blood glucose ≥300 mg/dL after 1 week was considered to have diabetes. We obtained approval for all animal experiments from the animal ethics committee of the First Affiliated Hospital of Chongqing Medical University, Chongqing, China (Ethics Approval Number: IACUC-CQMU-2023-0444). All animal experiment conformed to the national standard of Laboratory animal-Guideline for ethical review of animal welfare (GB/T 35892–2018).
Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR) Validation
SteadyPure Universal RNA Extraction Kit II (Accurate Biotechnology (Hunan) Co., Ltd., China) was used to extract total RNA from skin wound tissues. Evo M-MLV Mix Kit with gDNA Clean for qPCR (Accurate Biotechnology (Hunan) Co., Ltd., China) was used to synthesize complementary DNA. SYBR Green Premix Pro Taq HS qPCR Kit (Accurate Biotechnology (Hunan) Co., Ltd., China) was used for RT-qPCR analysis. The 2−ΔΔCT method was applied to analyse the relative expression levels of CTSH. The primer sequences were as follows: F:TTGTGAAAAACTCTTGGGGCTC, R:TGAGGAATGGGATAGGAGGCA, and GADPH served as the internal reference gene.
Western Blot Validation
The extraction of total protein from skin wound tissue was determined by bicinchoninic acid method. An equal number of proteins (skin wound tissue 30 µg) were subjected to sodium dodecyl sulfate-poly acrylamide gel electrophoresis gels and later transferred to polyvinylidene fluoride membranes. The membranes were blocked for an hour using 5% non-fat milk at room temperature, and later sequentially incubated with primary antibodies (CTSH, 1:3000, 10,315-1-AP, Proteintech, China; β-actin, 1:10000, M1210-2, Huabio, China) at 4 °C overnight and corresponding secondary antibodies at room temperature for 1 h. Finally, the protein bands were visualized, and the gray values of the protein bands were quantified using ImageJ software.
Statistical Analysis
All gene expression data and statistical analyses were processed using R version 4.3.1. The data processing and analysis were performed using online platform, online databases, ImageJ software and the GraphPad Prism software (Version 9.0). Differences in the CTSH expression were validated in animal models, and all data were present as mean ± SEM. The two groups were compared using a two-sample independent t-test if the data accorded with a normal distribution. Between-group comparisons were performed with Wilcoxon tests when the data did not accord with a normal distribution. A p-value below 0.05 was deemed to be statistically significant.
Results
The Flowchart of the Analysis Process
Figure 1 shows a flow diagram of the analysis process. The differentially expressed hub gene CTSH was identified based on machine learning algorithms. Then, CTSH was analyzed by multidimensional bioinformatics including immune cell infiltration analysis, GSEA analysis, correlation analysis, and lncRNA-miRNA-mRNA and drug regulatory network in our study. Finally, differences in CTSH expression were validated with wound tissues by RT-qPCR and Western blot in normal mice and diabetes mice.
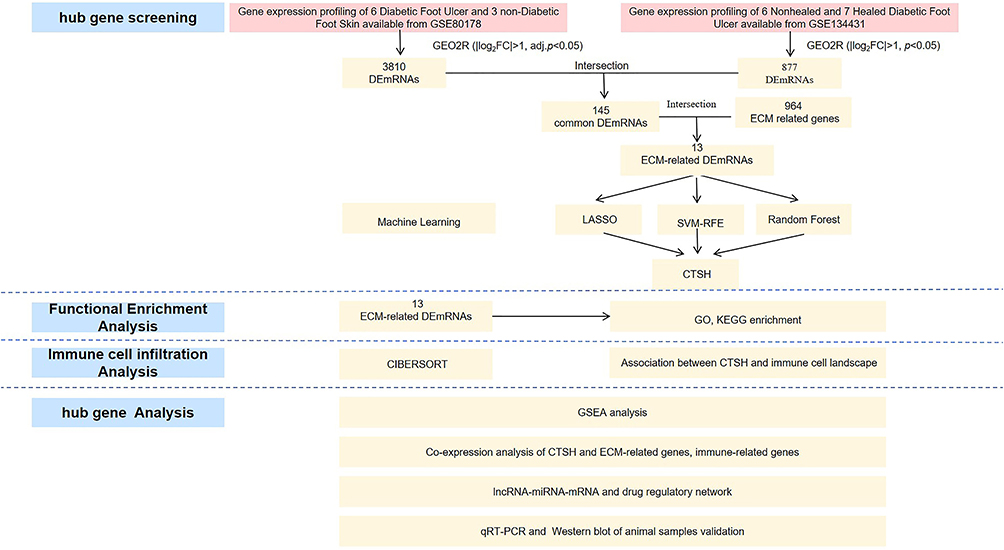 |
Figure 1 The flowchart of the analysis process. |
Identification of ECM-Related DEGs
In total, 3810 DEGs were identified in the GSE80178 dataset (Figure 2A), while 877 were identified in the GSE134431 dataset (Figure 2B). One hundred and forty-five common DEGs were identified between the two datasets, of which 49 were upregulated genes and 96 were downregulated genes (Figure 2C and D).
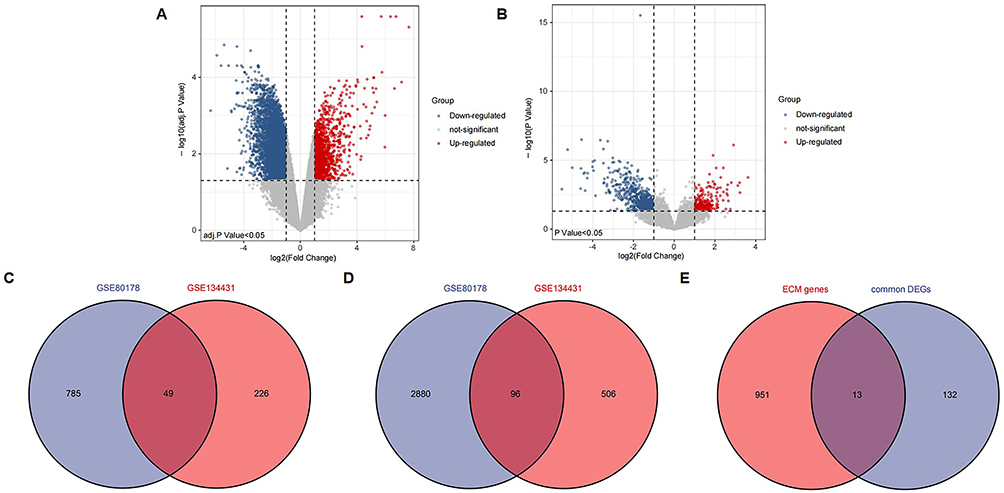 |
Figure 2 Identifying ECM-related DEGs. (A) Volcano plot of DEGs between DFU and non-diabetic foot skin in GSE80178. (B) Volcano plot of DEGs between unhealed DFU and healed DFU in GSE134431. (C) Venn diagram of up-regulated DEGs in GSE80178 and GSE134431. (D) Venn diagram of down-regulated DEGs in GSE80178 and GSE134431. (E) The Venn diagram of 13 ECM-related DEGs was obtained by 145 common DEGs and 964 ECM-related genes. Abbreviations: ECM, extracellular matrix; DEGs, differentially expressed genes; DFU, Diabetic Foot Ulcer. |
To explore the role of ECM in the occurrence and development of DFU, we overlapped 145 common DEGs with 964 ECM-related genes, and 13 overlapping genes were obtained, namely ECM-related DEGs, as shown in the Venn diagram (Figure 2E).
Functional Enrichment Analysis
We investigated the potential biological changes in the occurrence and development of DFU by analyzing 13 ECM-related DEGs for functional annotation and enrichment. GO analysis results demonstrated that the ECM-related DEGs were mainly enriched in the collagen-containing extracellular matrix, lysosomal lumen, tertiary granule lumen, immune response-regulating signaling pathway, response to wounding, immune effector process and tube morphogenesis (Figure 3A-B). In terms of KEGG pathway, the significant enrichment pathways were lysosome and phagosome (Figure 3C). Interestingly, the ECM-related genes were mainly involved in pathways related to tissue remodeling, inflammation, and defense against infectious agents.
 |
Figure 3 Functional enrichment analysis of 13 ECM-related DEGs. (A) Heatmap of GO enriched terms, colored by p value. (B) Network of GO enriched terms, colored by Cluster ID. (C) Heatmap of KEGG pathways, colored by p value. Produced using Metascape (http://metascape.org/gp/index.html#/main/step1) with parameters set as follows. Abbreviations: ECM, extracellular matrix; DEGs, differentially expressed genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes. Notes: minimum overlap = 3, P value cutoff = 0.01 and minimum enrichment = 1. |
Identification of Hub Genes Based on Machine Learning Algorithms
To further select the most promising diagnostic and prognostic genes related to the occurrence and development of DFU, we applied three different methods (LASSO, SVM-RFE and RF) based on the above 13 ECM-related genes, yielding 2, 6, 3 genes, respectively (Figure 4A-E). Then, the gene CTSH was selected by overlapping above three algorithms for further analysis in this study (Figure 4F). These results suggested that CTSH may play a pivotal role in the occurrence and development of DFU.
 |
Figure 4 Identification of hub genes via machine learning. (A) Hub genes screening in the Lasso model. LASSO algorithm was used to screen out 2 mRNAs. (B and C) Hub genes screening in the SVM-RFE model. SVM-RFE algorithm was used to screen out 6 mRNAs. (D and E) Hub genes screening in the RF model. RF algorithm was used to screen out 3 mRNAs. (F) Venn diagram was used to screen for overlapping genes identified by the three algorithms. Abbreviations: LASSO, Least Absolute Shrinkage and Selection Operator; SVM-RFE, Support Vector Machine-Recursive Feature Elimination; RF, Random Forest. |
Association Between CTSH and Immune Cell Landscape
Considering that immune cell takes part in wound healing, CIBERSORT was used to analyze immune cell abundances in different groups. Compared with the healed DFU samples, the T cells follicular helper (Tfh) were decreased in the unhealed DFU samples (Figure 5B). Unfortunately, the immune landscape was no statistically significant difference between DFU and non-diabetic foot skin (Figure 5A). Furthermore, the relationship between the expression of CTSH and immune cell contents was investigated. Correlation analysis showed a positive correlation between CTSH expression and infiltration levels of mast cells resting and NK cells activated in foot ulcer patients with and without diabetes (Figure 5C). Instead, CTSH was significantly negatively correlated with dendritic cells activated, NK cells resting and monocytes (Figure 5C). There was a significant positive correlation between CTSH and neutrophils in DFU patients with and without healed ulcers (Figure 5D). In contrast, Tfh correlated negatively with CTSH (Figure 5D).
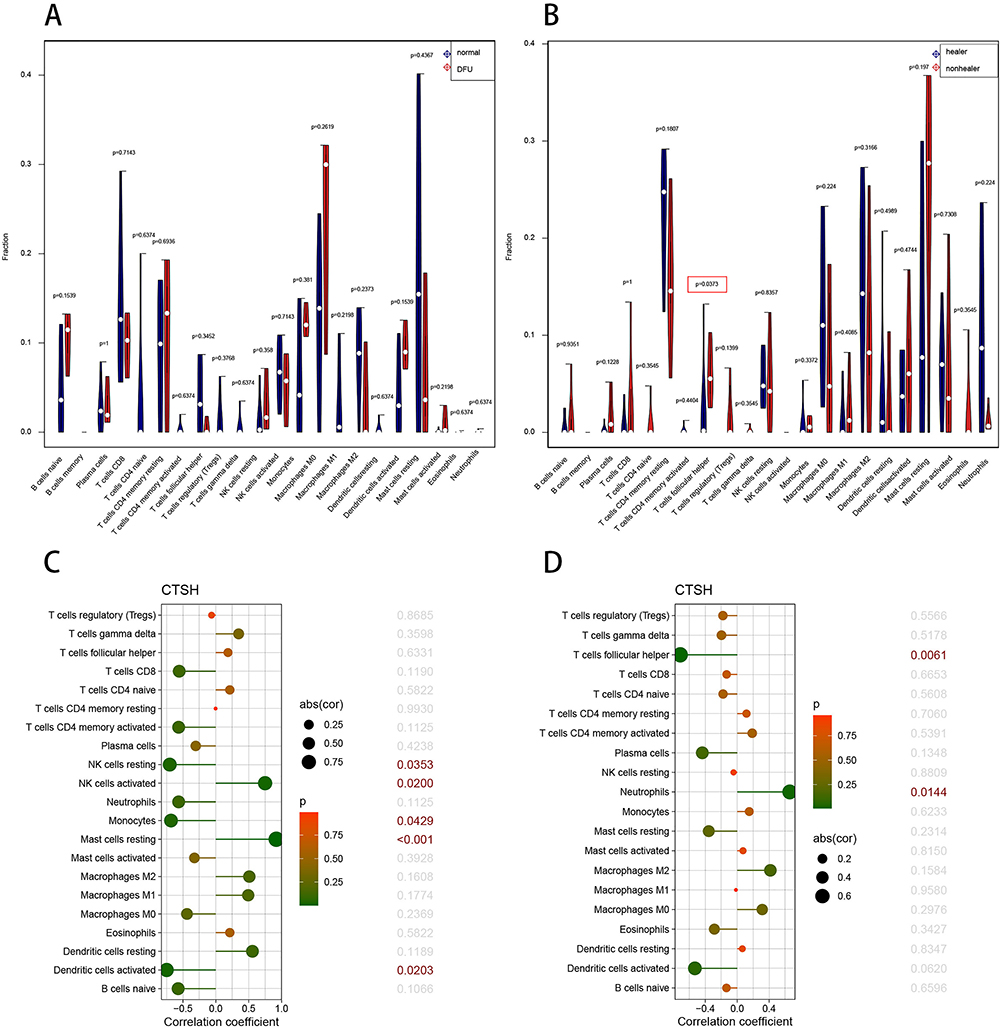 |
Figure 5 Landscape of immune cell infiltration. (A) Violin graph shows the difference of infiltrated immune cells between DFU and non-diabetic foot skin in GSE80178 (x-axis: each immune cell). The normal group is shown in blue and DFU group is shown in red. (B) Violin graph shows the difference of infiltrated immune cells between unhealed DFU and healed DFU in GSE134431 (x-axis: each immune cell). The healer DFU group is shown in blue and nonhealer DFU group is shown in red. (C) Relationship between the expression of CTSH and immune infiltrating cells in foot ulcer patients with and without diabetes. (D) Relationship between the expression of CTSH and immune infiltrating cells in DFU patients with and without healed ulcers. Abbreviation: DFU, Diabetic Foot Ulcer. Note: P < 0.05 was highlighted. |
The GSEA Analysis of CTSH
Subsequently, the single-gene GSEA analysis was performed via KEGG pathways in GSE147890. The single-gene (CTSH) GSEA analysis results revealed that drug metabolism−cytochrome P450, glycosylphosphatidylinositol (GPI)−anchor biosynthes, hedgehog signaling pathway, melanogenesis and metabolism of xenobiotics by cytochrome P450 were the top 5 upregulated pathways (Figure 6A). Bladder cancer, DNA replication, IL−17 signaling pathway, proteasome and rheumatoid arthritis were the top 5 downregulated pathways (Figure 6B). In terms of the most significant adj.P value pathways, the results showed that several pathways were markedly enriched, including TNF signaling pathway and IL-17 signaling pathway (Figure 6C).
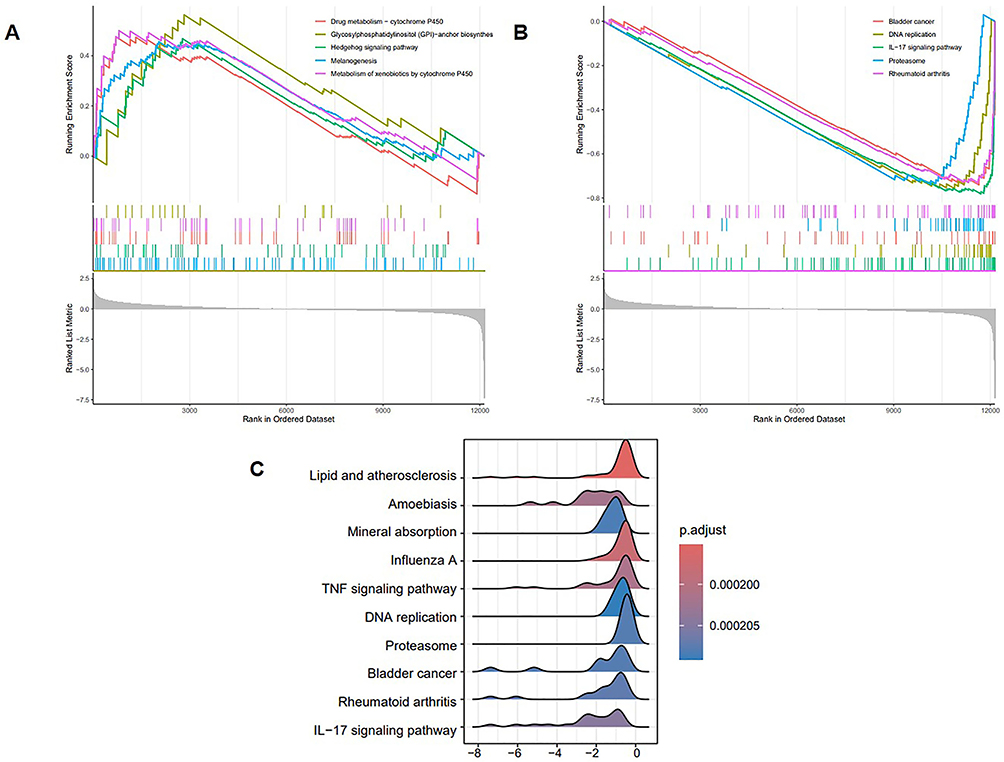 |
Figure 6 Single-gene GSEA for CTSH. (A) Top 5 upregulated KEGG pathways ranked by the enrichment score from single-gene GSEA. Red line represents the pathway of drug metabolism-cytochrome P450; Yellow line represents the pathway of glycosylphosphatidylinositol (GPI)-anchor biosynthes; Green line represents the pathway of hedgehog signaling pathway; Blue line represents the pathway of melanogenesis; Purple line represents the pathway of metabolism of xenobiotics by cytochrome P450. (B) Top 5 downregulated KEGG pathways ranked by the enrichment score from single-gene GSEA. Red line represents the pathway of bladder cancer; Yellow line represents the pathway of DNA replication; Green line represents the pathway of IL-17 signaling pathway; Blue line represents the pathway of proteasome; Purple line represents the pathway of rheumatoid arthritis. (C) Top 10 KEGG pathways ranked by adj.P value from single-gene GSEA. Abbreviations: GSEA, Gene Set Enrichment Analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes. |
Correlation Analysis of CTSH Expression Level with ECM-Related Genes and Immune-Related Genes
The correlation between the CTSH gene and ECM-related genes was analyzed. The correlation analysis showed that CTSH expression correlated with 362 ECM-related genes in GSE80178, while CTSH expression correlated with 309 ECM-related genes in GSE134431 (p < 0.05). CTSH expression both correlated with 102 ECM-related genes in the two datasets, including genes related to wound healing such as MMPs, TLRs, ADAMTSs, TGFBs, proteoglycan (DCN, VCAN and BGN), collagen (COL21A1, COL3A1, COL5A2 and COL6A3) (Table 1).
 |
Table 1 The Correlation Analysis Between CTSH and ECM-Related Genes Expression |
Given the importance of immunization for DFU, we studied the relationship between CTSH expression and immune-related genes. In total, 1793 immune-related genes were fetched from the Immunology Database and Analysis Portal (http://www.immport.org/).14 The correlation analysis showed that CTSH expression both correlated with 93 immune-related genes in the two datasets (p < 0.05). Of the 93 genes, IL7, IL33, TLR1, TLR3 and TLR4 were highly correlated with CTSH (Figure 7A-J).
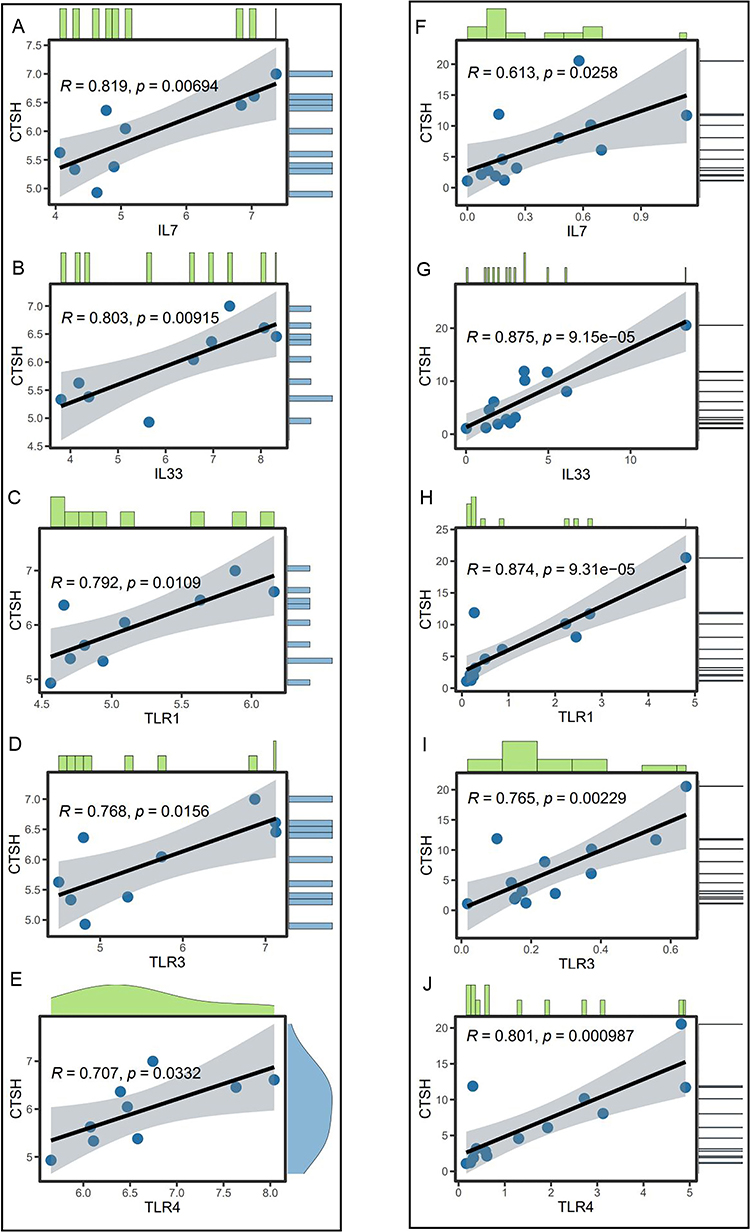 |
Figure 7 Co-expression analysis of CTSH and immune-related genes in GSE80178 dataset. (A) IL-7; (B) IL-33; (C) TLR1; (D) TLR3; (E) TLR4. Co-expression analysis of CTSH and immune-related genes in GSE134431 dataset. (F) IL-7; (G) IL-33; (H) TLR1; (I) TLR3; (J) TLR4. |
LncRNA-miRNA-mRNA and Drug Regulatory Network
Only one compound of relevance based on the CTSH gene was screened in the CTD. Benzo(a)pyrene may result in increased methylation of CTSH promoter, thereby regulating the expression level of CTSH (Table 2).
 |
Table 2 Interactions Between Compounds and Gene Related to DFU in the CTD Database |
The miRNAs corresponding to the CTSH gene were predicted, and a total of two miRNAs (miR-124-3p and miR-506-3p) were revealed based on the miRDB, miRWalk and StarBase databases (Figure 8A). Then, the miRNet and StarBase databases predicted 10 lncRNAs (NEAT1, TMEM147-AS1, MALAT1, XIST, MAPKAPK5-AS1, FGD5-AS1, LINC01089, SNHG14, LINC00963 and SNHG16) targeting the two miRNAs (Figure 8B-C).
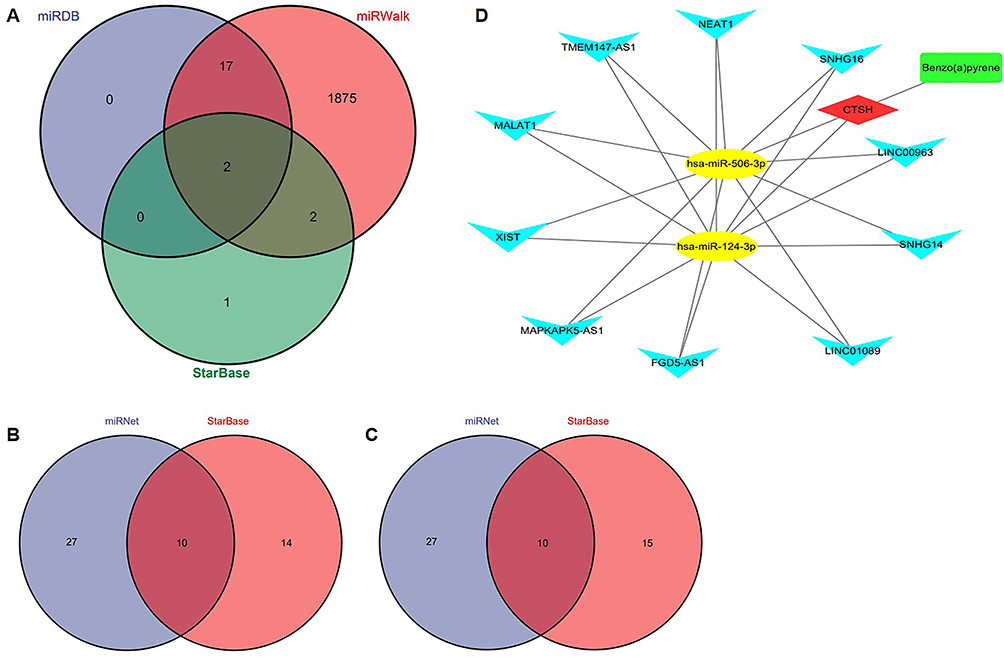 |
Figure 8 Construction of a lncRNA-miRNA-mRNA and drug regulatory network. (A) Venn diagram presenting the intersection of predicted miRNAs based on the miRDB, miRWalk and StarBase databases. (B) Venn diagram presenting the intersection of predicted lncRNAs of hsa-miR-124-3p based on the miRNet and StarBase databases. (C) Venn diagram presenting the intersection of predicted lncRNAs of hsa-miR-506-3p based on the miRNet and StarBase databases. (D) The lncRNA-miRNA-mRNA and drug regulatory network. Red diamond represents a hub gene, yellow nodes represent miRNAs, blue inverted triangles represent lncRNAs and green round rectangle represents a drug. |
The lncRNA-miRNA-mRNA interaction network and drug network are shown in Figure 8D. The result showed that a total of 1 drug, CTSH, 2 miRNAs and 10 lncRNAs were included in the current network.
Validation of CTSH by RT-qPCR and Western Blot in Animal Models
The expression of CTSH was initially investigated spanning all stages of the healing process of wounds in wild-type mice. RT-qPCR results showed dynamic changes in CTSH expression levels, and the level reached the highest point on day 5 after wound (Figure 9A). Then, to verify this result, microarray data (GSE23006) was discovered which analyzed gene expression from day 0 to day 10 during skin wound healing process in mice skin wounds (Figure 9B). Our results and external dataset GSE23006 have the similar patterns of CTSH expression during skin wound repair process, indicating that expression of CTSH in a dynamic manner may improve skin wound healing. Subsequently, STZ (50 mg/kg) was injected intraperitoneally to induce diabetic mice model. Mice with the fasting glucose level >16.7mmol/L over one weeks after the first injection of STZ was considered to have diabetes. Next, wound model was made. The expression of CTSH in skin wound tissues of wild-type or diabetic mice on day 5 was analyzed by RT-qPCR and Western blot. Our results showed that CTSH expression was decreased significantly in diabetic mice (Figure 9C-D).
 |
Figure 9 Validation of CTSH by RT-qPCR and Western blot in animal models. (A) The expression of CTSH in wild-type mouse skin wound tissues was analyzed at the time appointed by RT-qPCR (n=3). (B) The relative expression values of CTSH mRNA during skin wound healing process (1d, 3d, 5d, 7d and 10d) were extracted from microarray dataset (GSE23006). n=3 at every different time point. (C and D) The expression of CTSH in skin wound tissues of wild-type or diabetic mice on day 5 was analyzed by RT-qPCR (n=5) and Western blot (n=4). All results are expressed as means ± SEMs. Notes: *P < 0.05, **P < 0.01. |
Discussion
DFUs are associated with high rates of mortality as well as high costs.15 Therefore, new treatment strategies targeting specific molecules are urgently needed to enhance the efficiency of DFU treatment. Dysregulation of ECM structure, formation and deposition is one of the major reasons for impaired wound healing in diabetes.8,16 In the present study, bioinformatics approach and three machine learning algorithms were used to identify 13 ECM-related DEGs and CTSH as potentially diagnostic and prognostic biomarker for DFU. Additionally, we investigated the molecular mechanisms of DFU. New biomarker and potential molecular mechanism may provide new insights into the role of ECM involved in the development and progression of DFU. Considering the significant impact that DFU has on healthcare systems, early diagnosis and prognostic evaluation of DFU can offer reassurance to healthcare professionals. The study serves as a valuable strategy for healthcare professionals in the future.
CTSH, a lysosomal cysteine protease, plays a crucial role in the development and progression of various disease states. Several studies have suggested that CTSH may promote carcinogenesis. CTSH levels have been found to be elevated, such as in breast carcinoma,17 prostate cancer18 and glioma.19 By contrast, melanoma20 and head and neck carcinoma21 have lower CTSH levers than normal tissue. Compared to other cathepsins, CTSH has fewer studied, and the results are controversial in cancer. After brain injury or infection, CTSH may regulate microglia cell-mediated inflammatory response and even cell death.22 CTSH is an important regulator of β-cell function during progression of Type 1 Diabetes, in which overexpression prevents β-cells apoptosis and downregulation promotes β-cells apoptosis.23,24 Filaggrin is a vital portion of the skin barrier to the outside environment. The skin of CTSH-deficient mice showed reduced filaggrin processing. Furthermore, the mice exhibited a mild proinflammatory phenotype and impaired skin barrier function.24 CTSH raises more and more attention and interest due to its various roles in pathophysiologic processes. However, CTSH has not been investigated in the field of wound healing. Our results demonstrated that CTSH was lowly expressed in the occurrence and development of DFU, and we hypothesized that the high expression of CTSH had a protective factor in DFU. In animal wound models, our results showed that CTSH expression was decreased in diabetic mice compared with normal mice. The RT-qPCR and Western blot verification results of CTSH were consistent with our bioinformatics results.
Wound healing is a complex process involving many cells. Among these cells, immune cells are recruited to the wound site to clean pathogens and debris.25 Downregulation of FOXM1 contributes to decreased immune cell infiltration and results in delayed wound healing.26 Mast cells, an immune cell, take part in all stages of wound healing and contribute to the pathogenesis of unhealed DFU.27 Considering that immune cells participant in wound healing, we analyzed immune cell abundance differences. Compared with healed DFU patients, we found that Tfh was lowly expressed in unhealed DFU patients. Tfh is a subset of CD4+T cells and has been found to be involved in a range of diseases.28,29 Tfh cells are responsible for regulating the activation and differentiation of B cells to produce protective humoral immunity.30 Research suggests that wound healing can be manipulated by directly delivering mature, naive B cells. These cells help regulate the balance of mature immune cells in the wound microenvironment, thereby speeding up the healing process.31 Regrettably, the immune landscape was no statistically difference between DFU and non-diabetic foot skin. These results demonstrated that immune cells may be taking part in DFU prognosis but not in the occurrence of DFU.
To further investigate the pathogenesis in occurrence and progression of DFU, we performed GSEA. The GSEA analysis suggested that CTSH was enriched in hedgehog, IL-17 and TNF signaling pathway. A previous study found that inhibition of casein kinase 2α accelerates skin wound healing in type 1 diabetes mice. This effect is achieved by promoting endothelial cell proliferation through activating hedgehog signaling pathway.32 Research suggests SEMA3C/NRP2/Hedgehog signaling is involved in FOXM1-mediated wound healing in DFU.33 IL-17 is an important inflammatory signaling pathway, and excessive chronic inflammatory is one of the main characteristics of diabetic wounds.34,35 A study has shown that Huangbai liniment, a traditional Chinese medicine, and berberine, a major component of huangbai liniment, inhibit IL-17 signaling pathway and contribute to improve wound healing in diabetes mellitus. This research emphasizes targeting IL-17 signaling pathway as a strategy for diabetic wounds.34 Additionally, several studies have demonstrated the importance role of TNF in inflammatory diseases. As per available information, local oxygen therapy has been shown to promote wound healing in diabetic foot ulcers mainly through the TNF signaling pathway and the apoptosis pathway.36
Due to the importance of the role of ECM and immunization in DFU, we explored the relationship between CTSH expression and ECM-related genes as well as immune-related genes. The results showed that CTSH correlated with many ECM-related genes and immune-related genes, such as MMP2, MMP7, MMP16, MMP24, TIMP2, COL21A1, COL3A1, COL5A2, COL6A3, TLR1, TLR3, TLR4, IL-7 and IL-33. Matrix metalloproteinases (MMPs) play important parts in the wound healing process by degrading ECM proteins.37 Satisfactory wound healing requires strict spatial and temporal expression of MMPs; however, tissue inhibitor of metalloproteinases (TIMPs) specifically regulate the proteolytic activity of MMPs.38 The balance between MMPs and TIMPs, when disrupted, results in delayed wound healing.39 Collagen, a major component of ECM, can be broadly divided into fibrillar and nonfibrillar families and is vital for tissue formation and cell alignment during the wound healing.40 Abnormal collagen metabolism hinders the progress of healing in diabetic wounds.11 Toll-like receptors play a crucial part not only in early host defense against pathogen invasion but also in tissue repair, regeneration, and non-infectious inflammation.41 Previous research has shown that TLR4 modulates wound healing through the expression of TGF-β and CCL5.41 It is reported that skin wound healing is delayed in TLR3-deficient mice.42 Conversely, polyriboinosinic-polyribocytidylic acid, a TLR3 agonist, promotes wound healing.43 Previous studies have shown that the overexpression of IL-7 in Mesenchymal Stem Cells can strengthen the healing potential and help wound healing in diabetic animals through inducing angiogenic genes,44 highlighting its role in wound closure. Recent studies have found that IL-33 promotes skin wound healing, whereas its inhibition delays wound healing.45
In this study, the CTSH gene was found to be lowly expressed in the occurrence and progression of DFU. Subsequently, a lncRNA-miRNA-mRNA and drug regulatory network were constructed including 1 drug, CTSH, 2 miRNAs and 10 lncRNAs. The role of miRNAs in wound healing is attracting more attention in recent years. Our finding demonstrated that miR-124-3p and miR-506-3p may be involved in the targeted modulation of CTSH. DFU and peripheral arterial disease (PAD) have some congenerous risk factors. PAD could increase the risk of infection and non-healing ulcers, thereby aggravating the progression of DFU.46,47 Previous study has shown that miR-124-3p was a key regulator of angiogenesis in PAD and a potential diagnostic, therapeutic and prognostic target for PAD.47 Dermal fibroblasts are a promising candidate therapy for wound healing due to their ability of generating ECM, secreting growth factors and promoting wound contraction.48 The miR-506-3p and its downstream target TGF-β1 regulate the cell migration, viability and synthesis of ECM in human dermal fibroblasts after burns.48 However, miR-124-3p and miR-506-3p both have not been explored in the field of diabetic wound healing. Our findings may provide effective treatment strategies for DFU. By competing with miRNAs, competing endogenous RNAs (ceRNAs) regulate gene expression.49 Furthermore, online databases predicted 10 lncRNAs (NEAT1, TMEM147-AS1, MALAT1, XIST, MAPKAPK5-AS1, FGD5-AS1, LINC01089, SNHG14, LINC00963 and SNHG16) targeting the two miRNAs. Research suggests that MALAT1-miR-124 promotes skin wound healing through activating the Wnt/β-catenin pathway.50 Experimental evidence supports ceRNA networks affecting complex cellular processes.50,51 Research on wound healing in the emerging field of ceRNA networks may provide direction for further scientific exploration.
Inevitably, there are some limitations in this study. First, a relatively small samples of DFU datasets may affect the analytical results. Second, in finite time, we were unable to collect enough human foot skin samples, especially non-diabetic foot skin. The results of bioinformatics analysis need to be validated in more clinical samples. Third, the further urgent action is to validate the function and molecular mechanism of the hub gene CTSH so as to gain an insight into diabetic wound healing. To investigate the potential effects of CTSH on diabetic wound healing, we will use recombination mouse CTSH (rmCTSH) protein in STZ-induced mice wound models in the future. Additionally, future research will focus on the cellular localization of CTSH, and its impact on, as well as regulatory mechanism within, the physiological functions of localized cells. By addressing these limitations, a more comprehensive understanding of DFU will be provided. CTSH would have a potential clinical value in the diagnosis and prognosis of DFU and may be a potential therapeutic target.
Conclusion
In conclusion, CTSH was identified as an ECM-related diagnostic and prognostic biomarkers for DFU using bioinformatics methods and machine learning and may be a potential therapeutic target. Multidimensional bioinformatics analysis and animal experiment validation may provide a basis for future study. However, future research is warranted to validate the results from data analysis in more clinical samples and clarify the detailed function and molecular regulatory mechanism of CTSH in vitro.
Abbreviations
DFU, Diabetic foot ulcer; ECM, Extracellular matrix; DEGs, Differentially expressed genes; GEO, Gene expression omnibus; LASSO, Least absolute shrinkage selection operator; RF, Random forest; SVM-RFE, Support vector machine recursive feature elimination; GSEA, Gene set enrichment analysis; GO, Gene ontology; KEGG, Kyoto encyclopedia of genes and genomes; CTD, Comparative Toxicogenomics Database; STZ, Streptozotocin; RT-qPCR, Real-time quantitative polymerase chain reaction; Tfh, T cells follicular helper; MMPs, Matrix metalloproteinases; TIMPs, Tissue inhibitor of metalloproteinases; PAD, Peripheral arterial disease; ceRNA, Competing endogenous RNA.
Data Sharing Statement
The datasets analyzed in this article are accessible to the public. The datasets were acquired from the Gene Expression Omnibus (GEO, www.ncbi.nlm.nih.gov/geo/) database, and the accession numbers were GSE80178, GSE134431, GSE147890 and GSE23006.
Ethics Approval
Gene Expression Omnibus (GEO) is a public online database. Users can download relevant data for free for the purpose of researching and publishing relevant articles. Our study involving humans was based on data from the publicly available GEO database and was approved by the Medical Ethics Committee of Chongqing General Hospital (approval number: KY S2023-096-01).
Disclosure
All authors declare no conflicts of interest in this work.
References
1. Van Netten JJ, Bus SA, Apelqvist J, et al. Definitions and criteria for diabetes-related foot disease (IWGDF 2023 update). Diabetes Metab Res Rev. 2024;40(3):e3654. doi:10.1002/dmrr.3654
2. Chen L, Sun S, Gao Y, et al. Global mortality of diabetic foot ulcer: a systematic review and meta-analysis of observational studies. Diabetes Obes Metab. 2023;25(1):36–45. doi:10.1111/dom.14840
3. Armstrong DG, Boulton AJM, Bus SA. Diabetic foot ulcers and their recurrence. N Engl J Med. 2017;376(24):2367–2375. doi:10.1056/NEJMra1615439
4. Özker E. Intralesional epidermal growth factor therapy in recalcitrant diabetic foot ulcers. J Wound Care. 2023;32(Sup4):S14–S21. doi:10.12968/jowc.2023.32.Sup4.S14
5. Li Y, Gao Y, Gao Y, et al. Autologous platelet-rich gel treatment for diabetic chronic cutaneous ulcers: a meta-analysis of randomized controlled trials. J Diabetes. 2019;11(5):359–369. doi:10.1111/1753-0407.12850
6. Tang Y, Liu L, Jie R, et al. Negative pressure wound therapy promotes wound healing of diabetic foot ulcers by up-regulating PRDX2 in wound margin tissue. Sci Rep. 2023;13(1):16192. doi:10.1038/s41598-023-42634-9
7. Yu L, Qin J, Xing J, et al. The mechanisms of exosomes in diabetic foot ulcers healing: a detailed review. J Mol Med. 2023;101(10):1209–1228. doi:10.1007/s00109-023-02357-w
8. Panda D, Nayak S. Stem cell-based tissue engineering approaches for diabetic foot ulcer: a review from mechanism to clinical trial. Stem Cell Rev Rep. 2024;20(1):88–123. doi:10.1007/s12015-023-10640-z
9. Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res. 2012;49(1):35–43. doi:10.1159/000339613
10. Shen J, Zhao X, Zhong Y, et al. Exosomal ncRNAs: the pivotal players in diabetic wound healing. Front Immunol. 2022;13:1005307. doi:10.3389/fimmu.2022.1005307
11. Huang Y, Kyriakides TR. The role of extracellular matrix in the pathophysiology of diabetic wounds. Matrix Biol Plus. 2020;6-7:100037. doi:10.1016/j.mbplus.2020.100037
12. Ahluwalia P, Ahluwalia M, Mondal AK, et al. Prognostic and therapeutic implications of extracellular matrix associated gene signature in renal clear cell carcinoma. Sci Rep. 2021;11(1):7561. doi:10.1038/s41598-021-86888-7
13. Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. doi:10.1038/s41467-019-09234-6
14. Bhattacharya S, Andorf S, Gomes L, et al. ImmPort: disseminating data to the public for the future of immunology. Immunol Res. 2014;58(2–3):234–239. doi:10.1007/s12026-014-8516-1
15. Waibel FWA, Uçkay I, Soldevila-Boixader L, et al. Current knowledge of morbidities and direct costs related to diabetic foot disorders: a literature review. Front Endocrinol. 2024;14:1323315. doi:10.3389/fendo.2023.1323315
16. Short WD, Olutoye OO, Padon BW, et al. Advances in non-invasive biosensing measures to monitor wound healing progression. Front Bioeng Biotechnol. 2022;10:952198. doi:10.3389/fbioe.2022.952198
17. Gabrijelcic D, Svetic B, Spaić D, et al. Cathepsins B, H and L in human breast carcinoma. Eur J Clin Chem Clin Biochem. 1992;30(2):69–74.
18. Jevnikar Z, Rojnik M, Jamnik P, et al. Cathepsin H mediates the processing of talin and regulates migration of prostate cancer cells. J Biol Chem. 2013;288(4):2201–2209. doi:10.1074/jbc.M112.436394
19. Sivaparvathi M, Sawaya R, Gokaslan ZL, et al. Expression and the role of cathepsin H in human glioma progression and invasion. Cancer Lett. 1996;104(1):121–126. doi:10.1016/0304-3835(96)04242-5
20. Fröhlich E, Schlagenhauff B, Möhrle M, et al. Activity, expression, and transcription rate of the cathepsins B, D, H, and L in cutaneous malignant melanoma. Cancer. 2001;91(5):972–982. doi:10.1002/1097-0142(20010301)91:5<972::AID-CNCR1087>3.0.CO;2-Q
21. Kos J, Smid A, Krasovec M, et al. Lysosomal proteases cathepsins D, B, H, L and their inhibitors stefins A and B in head and neck cancer. Biol Chem Hoppe-Seyler. 1995;376(7):401–405. doi:10.1515/bchm3.1995.376.7.401
22. Fan K, Li D, Zhang Y, et al. The induction of neuronal death by up-regulated microglial cathepsin H in LPS-induced neuroinflammation. J Neuroinflammation. 2015;12(1):54. doi:10.1186/s12974-015-0268-x
23. Fløyel T, Brorsson C, Nielsen LB, et al. CTSH regulates β-cell function and disease progression in newly diagnosed type 1 diabetes patients. Proc Natl Acad Sci. 2014;111(28):10305–10310. doi:10.1073/pnas.1402571111
24. Wang Y, Zhao J, Gu Y, et al. Cathepsin H: molecular characteristics and clues to function and mechanism. Biochem Pharmacol. 2023;212:115585. doi:10.1016/j.bcp.2023.115585
25. Vasalou V, Kotidis E, Tatsis D, et al. The effects of tissue healing factors in wound repair involving absorbable meshes: a narrative review. J Clin Med. 2023;12(17):5683. doi:10.3390/jcm12175683
26. Sawaya AP, Stone RC, Brooks SR, et al. Deregulated immune cell recruitment orchestrated by FOXM1 impairs human diabetic wound healing. Nat Commun. 2020;11(1):4678. doi:10.1038/s41467-020-18276-0
27. Dong J, Chen L, Zhang Y, et al. Mast cells in diabetes and diabetic wound healing. Adv Ther. 2020;37(11):4519–4537. doi:10.1007/s12325-020-01499-4
28. Yang Q, Zhang F, Chen H, et al. The differentiation courses of the Tfh cells: a new perspective on autoimmune disease pathogenesis and treatment. Biosci Rep. 2024;44(1):BSR20231723. doi:10.1042/BSR20231723
29. Crotty S. T Follicular Helper cell biology: a decade of discovery and diseases. Immunity. 2019;50(5):1132–1148. doi:10.1016/j.immuni.2019.04.011
30. Tangye SG, Ma CS, Brink R, et al. The good, the bad and the ugly - TFH cells in human health and disease. Nat Rev Immunol. 2013;13(6):412–426. doi:10.1038/nri3447
31. Sîrbulescu RF, Boehm CK, Soon E, et al. Mature B cells accelerate wound healing after acute and chronic diabetic skin lesions. Wound Repair Regen. 2017;25(5):774–791. doi:10.1111/wrr.12584
32. Zhu J, Chen P, Liang J, et al. Inhibition of CK2α accelerates skin wound healing by promoting endothelial cell proliferation through the Hedgehog signaling pathway. FASEB J. 2023;37(9):e23135. doi:10.1096/fj.202300478RR
33. Yang Y, Zhang B, Yang Y, et al. FOXM1 accelerates wound healing in diabetic foot ulcer by inducing M2 macrophage polarization through a mechanism involving SEMA3C/NRP2/Hedgehog signaling. Diabet Res Clin Pract. 2022;184:109121. doi:10.1016/j.diabres.2021.109121
34. Zhang JJ, Zhou R, Deng LJ, et al. Huangbai liniment and berberine promoted wound healing in high-fat diet/Streptozotocin-induced diabetic rats. Biomed Pharmacother. 2022;150:112948. doi:10.1016/j.biopha.2022.112948
35. Li S, Ding X, Zhang H, et al. IL-25 improves diabetic wound healing through stimulating M2 macrophage polarization and fibroblast activation. Int Immunopharmacol. 2022;106:108605. doi:10.1016/j.intimp.2022.108605
36. Wang S, Pan LF, Gao L, et al. Randomized research on the mechanism of local oxygen therapy promoting wound healing of diabetic foot based on RNA-seq technology. Ann Palliat Med. 2021;10(2):973–983. doi:10.21037/apm-20-295
37. Nguyen TT, Wolter WR, Anderson B, et al. Limitations of knockout mice and other tools in assessment of the involvement of matrix metalloproteinases in wound healing and the means to overcome them. ACS Pharmacol Transl Sci. 2020;3(3):489–495. doi:10.1021/acsptsci.9b00109
38. Tardáguila-García A, García-Morales E, García-Alamino JM, et al. Metalloproteinases in chronic and acute wounds: a systematic review and meta-analysis. Wound Repair Regen. 2019;27(4):415–420. doi:10.1111/wrr.12717
39. Li G, Zou X, Zhu Y, et al. Expression and influence of matrix metalloproteinase-9/tissue inhibitor of metalloproteinase-1 and vascular endothelial growth factor in diabetic foot ulcers. Int J Low Extrem Wounds. 2017;16(1):6–13. doi:10.1177/1534734617696728
40. Li X, Mao X, Cong J, et al. Recombinantly expressed rhFEB remodeled the skin defect of db/db mice. Appl Microbiol Biotechnol. 2024;108(1):183. doi:10.1007/s00253-024-13021-9
41. Suga H, Sugaya M, Fujita H, et al. TLR4, rather than TLR2, regulates wound healing through TGF-β and CCL5 expression. J Dermatol Sci. 2014;73(2):117–124. doi:10.1016/j.jdermsci.2013.10.009
42. Lin Q, Fang D, Fang J, et al. Impaired wound healing with defective expression of chemokines and recruitment of myeloid cells in TLR3-deficient mice. J Immunol. 2011;186(6):3710–3717. doi:10.4049/jimmunol.1003007
43. Lin Q, Wang L, Lin Y, et al. Toll-like receptor 3 ligand polyinosinic:polycytidylic acid promotes wound healing in human and murine skin. J Invest Dermatol. 2012;132(8):2085–2092. doi:10.1038/jid.2012.120
44. Khalid RS, Khan I, Zaidi MB, et al. IL-7 overexpression enhances therapeutic potential of rat bone marrow mesenchymal stem cells for diabetic wounds. Wound Repair Regen. 2019;27(3):235–248. doi:10.1111/wrr.12706
45. Gao Y, Cai L, Li D, et al. Extended characterization of IL-33/ST2 as a predictor for wound age determination in skin wound tissue samples of humans and mice. Int J Legal Med. 2023;137(4):1287–1299. doi:10.1007/s00414-023-03025-x
46. Ward R, Dunn J, Clavijo L, et al. Outcomes of critical limb ischemia in an urban, safety net hospital population with high wifi amputation scores. Ann Vasc Surg. 2017;38:84–89. doi:10.1016/j.avsg.2016.08.005
47. Zou J, Zhang W, Chen X, et al. Data mining reveal the association between diabetic foot ulcer and peripheral artery disease. Front Public Health. 2022;10:963426. doi:10.3389/fpubh.2022.963426
48. Yang M, Yang Z, Pan X, et al. miR-506-3p regulates TGF- 1 and affects dermal fibroblast proliferation, migration and collagen formation after thermal injury. Tissue Cell. 2021;72:101548. doi:10.1016/j.tice.2021.101548
49. Kim S. LncRNA-miRNA-mRNA regulatory networks in skin aging and therapeutic potentials. Front Physiol. 2023;14:1303151. doi:10.3389/fphys.2023.1303151
50. He L, Zhu C, Jia J, et al. ADSC-Exos containing MALAT1 promotes wound healing by targeting miR-124 through activating Wnt/β-catenin pathway. Biosci Rep. 2020;40(5):BSR20192549. doi:10.1042/BSR20192549
51. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–352. doi:10.1038/nature12986
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.