")
Back to Journals » Journal of Inflammation Research » Volume 17
Inhibition of EGFR Pathway Suppresses M1 Macrophage Polarization and Osteoclastogenesis, Mitigating Titanium Particle-Induced Bone Resorption
Authors Jia Q , Liu L, Yu Y , Wulamu W, Jia L, Liu B, Zheng H, Peng Z , Zhang X, Zhu R
Received 22 August 2024
Accepted for publication 19 November 2024
Published 26 November 2024 Volume 2024:17 Pages 9725—9742
DOI https://doi.org/10.2147/JIR.S484529
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Tara Strutt
Qiyu Jia,1,* Lu Liu,1,* Yunyuan Yu,2,* Wuhuzi Wulamu,1 Lin Jia,1 Bo Liu,1 Hao Zheng,3 Zhenlei Peng,1 Xiaogang Zhang,1 Ruixia Zhu2
1The First Affiliated Hospital of Xinjiang Medical University, Urumqi, Xinjiang, People’s Republic of China; 2Department of orthopedics, the Affiliated Changzhou Second People’s Hospital of Nanjing Medical University, Changzhou, People’s Republic of China; 3The First Affiliated Hospital of Zhejiang Chinese Medical University (Zhejiang Provincial Hospital of Chinese Medicine), Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ruixia Zhu, Department of Orthopaedics, the Affiliated Changzhou No.2 People’s Hospital of Nanjing Medical University, 29 Xinglong Alley, Changzhou, 213003, People’s Republic of China, Email [email protected] Xiaogang Zhang, The First Affiliated Hospital of Xinjiang Medical University, 137 Liyue Shan South Road, Xinshi District, Urumqi, 830054, People’s Republic of China, Email [email protected]
Purpose: The polarization of macrophages towards the pro-inflammatory M1 phenotype and osteoclast overactivation play a significant role in the pathogenesis of aseptic loosening of orthopedic implants. This study sought to examine the expression and activation of macrophages and osteoclasts in implant biopsies with respect to epidermal growth factor receptor (EGFR) signaling and to assess the potential of EGFR inhibition in mitigating titanium particle-induced bone resorption in a cranial resorption murine model.
Methods: Bone marrow-derived macrophages (BMDMs) were stimulated with Tumor Necrosis Factor-alpha (TNF-α) and Interferon-gamma (IFN-γ) initially. Subsequently, Osteoclast differentiation was initiated after Gefitinib was added to the treatment groups. Male C57BL/6 mice were treated with Gefitinib or 0.5% Carboxymethyl Cellulose-Sodium (CMC-Na) by oral gavage daily for two weeks. A sham group received no further intervention, while the other groups had titanium particles implanted. Tissues were collected and analyzed by measurements such as micro-computed tomography (micro-CT) analyses, histology, immunofluorescence stainings, cell viability assays, assays for resorption pit formation, Reverse Transcription-Polymerase Chain Reactions (RT-PCRs), and Western blots were conducted.
Results: The study demonstrated a significant upregulation of EGFR in response to titanium particle exposure. Inhibition of EGFR phosphorylation with gefitinib effectively reduced bone degradation at osteolytic sites in a murine model. Gefitinib treatment led to a notable reduction in M1 macrophage polarization, as indicated by immunofluorescence staining and Western blot analysis of macrophage markers. Mechanistically, selective EGFR inhibitors mitigate osteoclastogenesis and osteoclast resorption by inhibiting the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) and nuclear factor-κB (NF-κB) signaling pathways.
Conclusion: Our findings provide compelling evidence of the essential role of EGFR-related pathways in M1 polarization, osteoclast activation, and ensuing periprosthetic osteolysis. Overall, EGFR presents a novel target for addressing bone resorption-associated conditions triggered by particles or modulated by macrophages and osteoclasts.
Keywords: EGFR, Wear particles, osteoclast, bone resorption, macrophage polarization
Introduction
Over the past few years, titanium (Ti) implants have experienced a global increase in usage within orthopedics and dentistry. Despite their high success rate,1,2 issues such as orthopedic implant loosening and oral peri-implantitis continue to pose significant challenges for healthcare providers. Current evidence suggests that implant failure is linked to aseptic inflammation surrounding the implant due to the release of debris, Ti ions, and particles.2 Likewise, prior findings have unveiled that the tissue-prosthesis interface could experience aseptic loosening resulting from interface stress and wear particles caused by friction following the insertion of artificial hip and knee prostheses in orthopedics, a key factor in surgical implant failure.3 Pathological examinations have disclosed pronounced inflammatory reactions around failed orthopedic4,5 and oral6–8 implants associated with Ti particles.
As mentioned above, implantation inherently leads to long-term tissue damage, initiating a sustained inflammatory response known as chronic inflammation. Chronic inflammation, triggered by the generation of inflammatory cytokines and tissue damage, is a pathological phenomenon observed in various chronic conditions like malignancy, metabolic syndrome, and vascular disorders.9–11 The hallmark of chronic inflammation is the consistent recruitment of leukocytes to the injury site.12 Unlike acute inflammation, chronic inflammation typically entails a persistent shift of leukocytes, culminating in inflammation primarily driven by macrophages rather than neutrophils. In this respect, Michal Eger et al13 demonstrated that Ti particles evoke a pronounced inflammatory response in macrophages and stimulate osteoclastogenesis.
Macrophage activation constitutes a critical stage in macrophage functioning, with M1 macrophages being activated in response to pathogens and pro-inflammatory cytokines.14–17 M1 macrophages express an array of inflammatory mediators, including interleukin (IL)-1β, IL-6, and tumor necrosis factor-alpha (TNF-α), which can directly or indirectly activate periprosthetic osteolysis, ultimately leading to prosthetic loosening.18,19 It has been established that TNF’s mechanism of action involves expanding the pool of osteoclast precursors, promoting osteoclast formation by differentiating macrophage colony-stimulating factor (M-CSF)-induced M2 into M1 inflammatory macrophages, suggesting that M1 macrophages can foster osteoclast formation.20
Osteoclastogenesis is a complex process further regulated by epidermal growth factor receptor (EGFR) signaling. EGFR, a widely expressed member of the ErbB receptor family of tyrosine kinase receptors, has been documented in numerous malignancies.21 Furthermore, EGFR is critical in regulating cell growth, proliferation, and differentiation.22 EGFR signaling is widely believed to facilitate the recruitment of osteoclast precursors and osteoclastogenesis during bone development, involving the downregulation of osteoprotegerin (OPG),23–25 acting as a decoy receptor for RANKL and the upregulation of monocyte chemoattractant protein 1 (MCP1) in stromal osteoblasts.26
Beyond osteoclast regulation, recent findings indicate that tyrosine kinase signaling also influences macrophage activation.27 In this respect, studies have linked tyrosine kinase signaling with macrophage function in mouse models of glaucoma, colitis, and cancer.28–32 However, the potential impact of macrophage EGFR signaling on periprosthetic osteolysis remains unexplored. Hardbower et al32 explored this aspect in a mouse model of inflammatory bowel disease and demonstrated EGFR signaling’s crucial involvement in macrophage activation and function. In mouse models, inhibiting EGFR signaling or genetic knockout reduced M1 and Mreg activation. The above studies overlap in their assertion that macrophage EGFR targeting holds promise as a therapeutic approach for diseases and cancers linked to chronic inflammation. Another study proposed that bone marrow-specific EGFR knockout mice experienced altered tumorigenesis due to reduced macrophage, neutrophil, and T cell infiltration, highlighting EGFR’s significance to macrophage function.33
Based on these findings, EGFR signaling emerges as a potential biomarker of chronic inflammation and a target for chemoprevention in patients with periprosthetic osteolysis. This investigation aimed to examine the expression and activation of macrophages and osteoclasts in implant biopsies with respect to EGFR signaling and to assess the potential of EGFR inhibition in mitigating titanium particle-induced bone resorption in a cranial resorption murine model. Additionally, we examined the role of EGFR in macrophage polarization, osteoclastogenesis, and osteoclast bone resorption using bone marrow-derived macrophage cultures.
Materials and Methods
Preparation of Titanium Wear Particles
Alfa Aesar (Shanghai, China) provided Ti particles (average diameter 3.0 µm, 90% below 15 µm) that were mixed in a 75% ethanol solution, stirred with a magnetic stirrer for 48 h, and then dried in a 100 °C oven to eliminate endotoxins. The quantitative levels of endotoxins were checked using the Limulus Amebocyte Lysate Assay (BioWhittaker, Walkersville, MD).34,35
Procedures for Operating and Administering Drugs
The protocols implemented in this study adhered to internationally recognized standards and were approved by our institution’s Ethics Committee. Titanium particles were implanted into the calvaria of 48 male C57BL/6 mice,36 ensuring that each group had 6 samples for different analyses. These were divided into four groups: (1) Sham group (mice were administered a 0.5% solution of carboxymethyl cellulose sodium (CMC-Na) via gavage), (2) Vehicle group (mice received a subperiosteal implantation of 20 mg Ti particles and were administered a 0.5% CMC-Na solution via gavage), (3) Low-dose gefitinib group (mice were subjected to 20 mg Ti particles along with a daily dose of 10 mg/kg gefitinib (AstraZeneca, Macclesfield, UK), and (4) High-dose gefitinib group (mice were exposed to 20 mg Ti particles and a daily dose of 50 mg/kg gefitinib, a dose selected based on previous studies demonstrating its efficacy in inhibiting EGFR phosphorylation while maintaining high survival rates37–39). Following the administration of anesthesia, an 8 mm sagittal incision was performed at a mid-calvarial point. A 0.5×0.5 cm area of periosteum was carefully exposed via an incision anterior to the interaural line. While the sham group received no further treatment, the remaining groups underwent titanium particle implantation. Fifteen microliters of titanium particle suspension were uniformly distributed over the intact periosteum using pipettes. Gefitinib was dissolved in a solution of 0.5% CMC-Na and given orally through gavage daily for two weeks. In contrast, the group that did not receive gefitinib was administered a 0.5% CMC-Na solution by oral gavage.
Micro-CT Analysis
Following euthanasia, cranial bones (n=6/group) were collected and fixed in 4% paraformaldehyde. The extent of calvarial damage in the murine specimens was assessed using micro-CT (SkyScan 1176; SkyScan, Aartselaar, Belgium).40 Calvaria were scanned at an isometric resolution of 9 μm, with X-ray energy parameters set to 80 kV and 100 mA. Cone-beam reconstruction software (SkyScan) was employed to reconstruct 3D images, and correlation analyzer software (SkyScan) was utilized to perform quantitative analysis. To minimize potential bias, a volume of interest (VOI) of the same size (3 mm diameter) was established in the center of each calvarium.40 SkyScan software was utilized to analyze the number of VOI pores, bone mineral density (BMD), and bone volume/tissue volume (BV/TV).
Patient-Derived Implant Biopsies
Synovium samples were obtained from patients who underwent primary arthroplasty or periprosthetic osteolysis. The specimens were obtained from areas of bone loss during either revision or primary arthroplasty. The First Affiliated Hospital Ethics Committee of Xinjiang Medical University approved all procedures, and informed consent was obtained from patients or their relatives. The process strictly followed the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Histology
Following 48 h of tissue fixation, cranial bones (n=6/group) were decalcified for one month in 10% ethylenediaminetetraacetic acid (EDTA, Vetec, Germany). The human joint synovium and mouse calvaria specimens were subjected to fixation, decalcification, and paraffin embedding before being sectioned into 5 μm-thick slices. Subsequently, the slices were stained with tartrate-resistant acid phosphatase (TRAP), and hematoxylin, eosin (H&E) and examined under a microscope to obtain images. The bone thickness (mm) and erosion surface (mm2) were measured following the methodology described by Parfitt et al.41 TRAP-positive cells were quantified by measuring the pixel area of their dark purple staining granules.42 Image Pro-Plus 6.0 software (Media Cybernetics, Bethesda, Maryland, USA) was used to determine the ratio of osteoclast surface to bone surface (OcS/BS, %).
The assessment of RANKL, EGFR, and P-EGFR expression was conducted through immunohistochemistry utilizing primary antibodies that were deemed appropriate (Abcam, ab216484, Cambridge, UK; Abcam, ab52894, Cambridge, UK and Abcam, ab40815, Cambridge, UK, respectively). After dewaxing, gradient hydration, and antigen recovery, sections were incubated with primary antibodies for 12 h at 4 °C in the dark. Following washing, the sections were exposed to secondary antibodies for 35 minutes at ambient temperature. Positive staining cells were observed and quantified using a microscope.
Colocalization by Immunofluorescence Staining
Immunofluorescence staining was performed to assess the colocalization of two proteins in a murine skull model. The samples were incubated in a 2% BSA solution for 1 hour, followed by overnight incubation at 4 °C with primary antibodies (IgG, 1:500; CD206, 1:1000; CD86, 1:200; CD68, 1:200; Abcam) to target the proteins of interest. Afterward, the samples were immersed in PBS and treated with antibodies for 60 minutes at ambient temperature. Specifically, goat-derived rabbit and rat IgG H&L antibodies, labeled with Alexa Fluor 647 and 488, respectively, were used for our experiments. Finally, the specimens were examined using laser confocal microscopy (Leica, TCS SP8, Wetzlar, Hessian, Germany) per established academic protocols.
Image analysis was conducted using ImageJ and Wolfram Mathematica software (version 10.0) to determine the pixel ratios of CD86/nuclei and CD206/nuclei as a function of distance from the surface of individual mesh fibers. Positive cells were quantified in at least three distinct regions within each sample.
Cell Culture and Differentiation
Bone marrow-derived macrophages (BMDMs) were isolated from the bone marrow of C57 BL/6 mice and cultured in Dulbecco’s Modified Eagle Medium (HyClone, Logan, Utah, USA) comprising 30 ng/mL M-CSF (R&D Systems, Minnesota, USA), 10% fetal bovine serum (Gibco, California, USA) and 100 U/mL penicillin. After four days, the cells were divided into four groups: control, vehicle (TNF-α, R&D Systems; Interferon-gamma (IFN-γ), Sigma) (RANKL, Abcam), low-dose Gefitinib group (0.1 μmol/L Gefitinib) and high-dose Gefitinib groups (10 μmol/L Gefitinib). The cells were seeded in a 96-well, 24-well, or 6-well plate at a density of 6×103, 9×104, or 5×105 cells per well, respectively. The cells were exposed to stimulation with 10 ng/mL TNF-α (R&D Systems) and 10 ng/mL IFN-γ (Sigma) for 24 hours. After the initial stimulation with TNF-α and IFN-γ, Gefitinib was added to the respective groups, while some groups were left without Gefitinib treatment. The vehicle group received the same treatment without the addition of Gefitinib to serve as a control for any effects of the solvent. The process of osteoclast differentiation was initiated by applying a concentration of 100 ng/mL of RANKL. The determination of the induction time was carried out according to the experimental specifications.
Macrophages were identified using F4/80 as a surface marker. M1 macrophages were characterized by the presence of CD86 (1:500) and iNOS (1:200), while M2 macrophages were characterized by CD206 (1:500) and Arg-1(1:200). Cells were incubated with specific antibodies, washed, and labeled with fluorescent antibodies, such as Alexa Fluor 647 (red) and 488 (green), for visualization using confocal microscopy (Leica).
To stimulate the differentiation of osteoclasts, BMDMs were cultured in an induction medium supplemented with 100 ng/mL RANKL. Following five days of osteoclast induction, TARP-stained microscopy (Zeiss) revealed the presence of multinucleated cells. Immunofluorescence staining was used to detect p65 localization in the cytoplasm and nucleus, with DAPI (blue) as a nuclear counterstain. Positive cells were observed using the Olympus fluorescence microscope and quantified using Image Pro Plus 6.0 software. Experimental protocols followed the manufacturer’s instructions.
Cell Viability Assay
BMDMs were cultivated in an induction medium in 96-well plates for 24 hours. Subsequently, cells were incubated with varying concentrations of gefitinib (0–50 μM) for 48 h to assess its potential cytotoxicity against BMDMs. Cell viability was assessed using the Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan) following the manufacturer’s instructions. The inhibition rate of BMDMs was calculated with GraphPad.
Assay for Resorption Pit Formation
BMDMs were cultured in osteoclast resorption plates (OAP, Corning, NY, USA) supplemented with 100 ng/mL RANKL and varying concentrations of gefitinib (0.1–10 μM) until the 5th day, at which point osteoclasts were observed. After plate washing and sonication, the resorption pits were examined using a microscope (Zeiss). Consequently, areas of resorption were evaluated by Image J software (National Institutes of Health, Bethesda, MD, USA).
Analysis of Quantitative Real-Time PCR (RT-PCR)
Total RNA extraction from the cells was conducted using TRIzol reagent (Sigma) according to the recommended protocols. To ensure robustness and accuracy, each sample was subjected to triplicate analysis. The reverse transcription process was conducted with Takara reverse transcriptase (Otsu, Japan). Subsequently, the resulting cDNA was amplified using a SYBR Premix Ex Tag Kit (Takara). Table 1 provides the primer sequences.
 |
Table 1 Primers Used for RT-PCR |
Western Blot Analysis
The expression of the target protein was analyzed by Western blotting. The primary antibodies used included iNOS (1:1000 dilution, Abcam), Arg-1 (1:1000 dilution, Sigma-Aldrich), CD86 (1:1000 dilution, Abcam), CD206 (1:1000 dilution, Sigma-Aldrich), EGFR (1:1000 dilution, Abcam), P-EGFR (1:1000 dilution, Abcam), AKT (1:1000 dilution, Abcam), P-AKT (1:1000 dilution, Abcam), p65 (1:1000 dilution, Abcam), P-p65 (1:1000 dilution, Abcam), Iκbα (1:1000 dilution, Abcam), P-Iκbα (1:1000 dilution, Abcam) and β-actin (1:2000 dilution, weiao). After rinsing the membranes with TBS-Tween, they were exposed to the appropriate secondary antibodies for a reaction. Ultimately, the visualization of blots was achieved using a Bio-Rad chemiluminescence detection system.
Statistical Analysis
The data were analyzed using mean and standard deviation, and statistical significance was determined using paired t-tests or one-way ANOVA with Tukey’s post-hoc test for multiple comparisons. A significance level of P < 0.05 was considered statistically significant. In the figures, significance levels were denoted as *indicating P < 0.05, and **indicating P < 0.01.
Results
EGFR Signaling Was Significantly Upregulated in Periprosthetic Membranes of Aseptic Loosening Patients
Immunohistochemical staining was performed on primary and revision periprosthetic osteolysis (PPO) arthroplasty tissues to reveal EGFR and P-EGFR expression. Multinucleated macrophage-like cells were identified as the primary sources of EGFR and P-EGFR expression within human coxa osteoarthritic synovium. Moreover, the soft tissues from the revision arthroplasty joint exhibited higher P-EGFR expression than primary arthroplasty cells (Figure 1A-B). RANKL expression, predominantly observed in osteocytes, osteoblasts, activated T-cells, and stromal fibroblasts within the human coxa synovial membrane,43–46 was consistent with P-EGFR expression in soft tissues obtained from primary and revision PPO arthroplasty (Figure 1C).
 |
Figure 1 P-EGFR was overexpressed in aseptic implant-loosening-associated human periprosthetic membranes. Immunostaining was performed for EGFR (A), P-EGFR (B), and RANKL (C). Scale bars represent 50 and 100 μm. |
EGFR Inhibition Reduced Ti Particle-Induced Bone Destruction in vivo
After establishing the upregulation of P-EGFR in human hip synovium after revision surgery, the feasibility of EGFR inhibition to mitigate particle-induced bone destruction was assessed. The selective EGFR inhibitor gefitinib was utilized within a titanium particle-induced osteolysis model to assess their impact on pathological osteolysis. 3D reconstructions of murine calvaria from distinct groups were generated by micro-CT scans. Calvaria from the vehicle group exhibited multiple bone disruptions, while the sham group showed fewer disruptions.
Administration of gefitinib through daily gavage significantly inhibited bone pitting induced by titanium particles, exhibiting a dose-dependent effect (Figure 2A). Quantitative analysis revealed significant reductions in BMD by 42.3%, bone volume fraction (BV/TV) by 14.1%, and a notable increase of 3.5-fold in porosity (%) and 5.0-fold in pore number in the vehicle group relative to the sham group. Gefitinib-treated mice exhibited improved BMD and BV/TV compared to the vehicle group (Figures 2B-C). Gefitinib administration also reduced pore number and porosity (%) (Figure 2D-E).
 |
Figure 2 Illustrates the effectiveness of inhibiting P-EGFR with gefitinib in reducing titanium particle-induced bone destruction in a murine PPO model. Micro-CT images demonstrated the 3D reconstruction of the calvaria (A) with 1 mm scale bars. Quantitative measurements included BMD (B), BV/TV (C), number of pores (D), and areas of porosity (E, %). Histological analysis was conducted using H&E and TRAP staining, with scale bars indicating a length of 100 μm (F and I). The study measured the eroded surface (G), bone thickness (H), TRAP-positive cell numbers (J), and OCs/BS (K). *P<0.05, **P<0.01. |
Histological analysis indicated that EGFR inhibition mitigated particle-induced osteolysis, with reduced erosion surface and increased bone thickness in the gefitinib-treated group compared to the vehicle group (Figure 2F–H).
TRAP staining showed TRAP-positive cells primarily co-localized with bone pitting (Figure 2I). The histomorphometric analysis validated gefitinib’s significant inhibitory effect on bone pitting and the OCs/BS ratio, alongside enhanced bone density in the presence of titanium particles in a dose-dependent manner (Figure 2J–K).
Reduced P-EGFR Levels in Murine Particle-Induced Osteolysis Model with Gefitinib Treatment
Immunohistochemical staining for EGFR and P-EGFR revealed a dose-dependent reduction of P-EGFR expression by gefitinib (Figure 3C and D). Low and high concentrations of gefitinib lowered the abundance of P-EGFR-positive cells by 21.5% (P < 0.01) and 47.4% (P < 0.01), respectively, compared to the vehicle group. However, EGFR-positive cell abundance showed no significant disparity among low-dose and high-dose gefitinib treatment groups and the vehicle group (Figure 3A and B). Taken together, these findings corroborate that gefitinib could reduce bone resorption via EGFR phosphorylation inhibition.
 |
Figure 3 Gefitinib decreased P-EGFR levels in a mouse model of calvarial osteolysis induced by titanium particle exposure. EGFR immunostained images (A) and P-EGFR immunostained images (C) were shown with a scale bar indicating 100 μm. The numbers of EGFR-positive cells (B) and P-EGFR-positive cells (D) were determined. **P<0.01. |
3.4. Gefitinib induced macrophage phenotype shift from M1 to M2 in the murine model of particle-induced osteolysis.
Macrophage secretion of inflammatory cytokines significantly contributes to peri-implant inflammation and is strongly associated with aseptic loosening.47 Macrophages exist in two phenotypes: pro-inflammatory M1 and pro-healing M2. To investigate the effect of gefitinib on macrophage polarization during peri-implant inflammation, dual fluorescence staining using CD68 (green) and CD86 (or CD206; red) was performed on murine skulls (Figure 4A). Evidence of CD68+ macrophages was found in the tissue surrounding titanium particles in the vehicle group. Most CD68+ macrophages expressed the M1 marker CD86, with a minority expressing the M2 marker CD206, consistent with the literature.48 Low and high-dose gefitinib-treated groups demonstrated reduced infiltration of M1 macrophages (CD68+CD86+) and increased ratios of M2 macrophages (CD68+CD206+) with anti-inflammatory properties (Figure 4B).
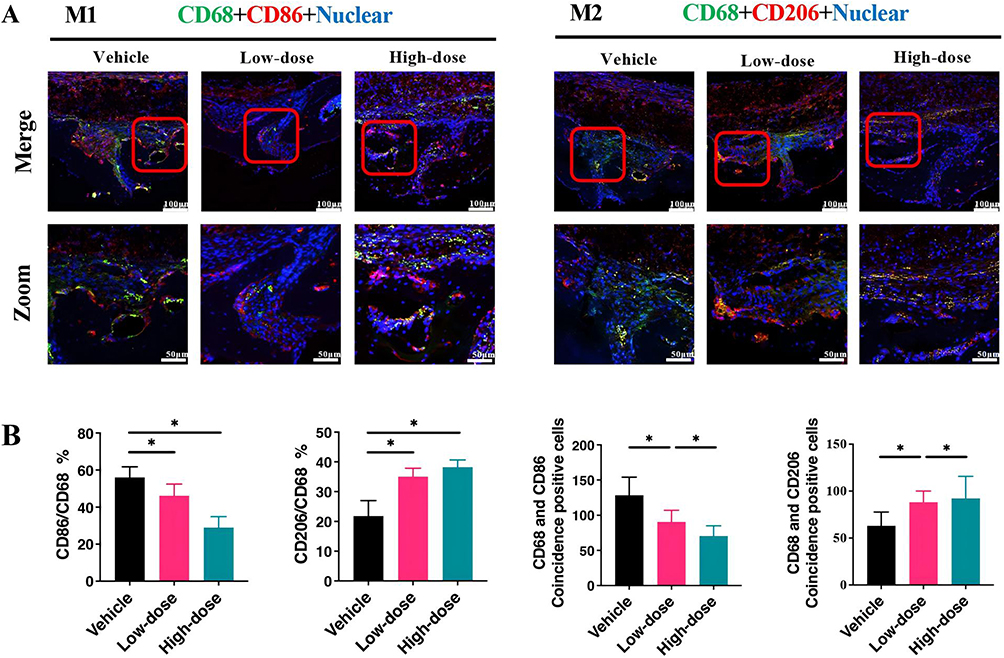 |
Figure 4 The phenotype of macrophages in vivo. Representative immunofluorescence colocalization was performed using staining images for F4/80 (green) and CD86 (or CD206; red) (A) with a scale bar of 100 μm. The percentage of double-positive macrophages was determined and quantified (B) (n=6 per group). *P<0.05. |
Macrophage Polarization in vitro
We next mimicked the inflammatory response induced by wear particles using TNF-α for in vitro M1 polarization. CCK-8 assay confirmed gefitinib’s low cytotoxicity to BMDMs at doses below 20 μM (Figure 5A). Gefitinib at 50 μM significantly inhibited BMDM proliferation (Figure 5A). Gefitinib treatment in TNF-α-induced BMDMs led to a dose-dependent decrease in EGFR phosphorylation levels, verified by Western blotting (Figure 5B and C).
 |
Figure 5 Effect of gefitinib treatment on macrophage polarization. Following treatment of BMDMs with TNF-α (10 ng/mL) + IFN-α (10 ng/mL) for 24 h, and the in vitro determination of macrophage polarization was performed across different treatment groups. The impact of gefitinib on BMDMs at various stimulation concentrations was assessed using the CCK-8 assay (A). Western blot analysis was conducted to examine the expression of EGFR and P-EGFR (B), followed by quantitative analysis of EGFR and P-EGFR (C) (n = 3/group). Immunofluorescence staining was performed to visualize F4/80 (green) and CD86 (or CD206; red), with a scale bar indicating a distance of 25 μm (D). The double-staining positivity was quantified by determining the percentage of macrophages that were positive for both CD86 and CD206 (E) (n = 6/group). Western blot analysis was performed to assess the expression of CD86, iNOS, CD206, and Arg-1 (F). Quantitative analysis of Western blot results (G) (n = 3/group). *P<0.05, **P<0.01. |
Dual fluorescence staining with F4/80 (green) and CD86 (or CD206; red) revealed a decrease in F4/80+CD68+ M1 macrophages and an increase in F4/80+CD206+ M2 macrophages with increased doses of gefitinib (Figure 5D and E). Western blot analysis demonstrated decreased iNOS and CD86 expression and increased Arg-1 and CD206 expression with increasing doses of gefitinib, highlighting its ability to prompt macrophage conversion from M1 to M2 phenotype (Figure 5F and G).
In line with our findings, EGFR inhibition could induce a shift in macrophages from the M1 to the M2 phenotype, facilitating the transition from a pro-inflammatory to an anti-inflammatory state by inhibiting EGFR under inflammatory conditions, consistent with the outcomes observed in our in vivo experiments.
Inhibition of EGFR-Suppressed RANKL-Induced Osteoclastogenesis
After establishing reduced osteoclast and M1 macrophage abundance in the titanium particle-induced osteolysis model following EGFR inhibition, we sought to explore whether inhibiting EGFR could impact osteoclast formation. To validate this hypothesis, we established an in vitro experimental setup, culturing BMDMs in induction media supplemented with 30 ng/mL M-CSF and 100 ng/mL RANKL while introducing different concentrations of gefitinib (0, 0.1, 10 μM). After five days, we observed a reduction in mature osteoclasts with increasing concentrations of gefitinib (Figure 6A and B).
 |
Figure 6 EGFR inhibition with gefitinib suppressed RANKL-induced osteoclastogenesis. BMDMs were cultured in an induction medium supplemented with gefitinib at 0, 0.1, and 10 μM concentrations (A). Following five days of osteoclast induction, TRAP staining was conducted to assess the abundance of multinucleated osteoclasts generated from BMDMs (B). The BMDMs were cultured in 24-well resorption plates at a density of 1×103 cells per well, using an induction medium supplemented with varying concentrations of gefitinib and 100 ng/mL of RANKL (C). The resorption area was quantified by visualizing complete resorption pits under an inverted microscope after a five-day incubation period (D). To differentiate BMDMs into osteoclasts, M-CSF and RANKL were added, and cells were cultured for five days, followed by staining with F-actin (green) and visualization of nuclei using DAPI (blue) (E). RT-PCR analysis was conducted to assess the expression levels of TRAP, NFATc1, CTSK, Oscar, and C-Fos (F). Scale bars represent 50 and 200 μm. *P<0.05, **P<0.01. |
We also examined the effect of EGFR inhibition on osteoclast function by culturing BMDMs on resorption plates with varying gefitinib concentrations (Figure 6C). Our analysis revealed a significant decrease in the resorption area compared to the control group (Figure 6D).
It is widely acknowledged that upon attachment to bone, osteoclasts initiate membrane reorganization, leading to the formation of specific membrane domains crucial for bone resorption. Consequently, we conducted a study examining the impact of EGFR on osteoclast function, specifically focusing on the structures of F-actin (cytoskeleton).49 Immunostaining with Acti stain 488 fluorescent Phalloidin demonstrated that RANKL-treated BMDMs displayed the characteristic structure and spread of the F-actin ring (Figure 6E). In contrast, the gefitinib-treated group exhibited a significant disruption of F-actin ring integrity, accompanied by a decrease in ring size (Figure 6E). These findings suggest that EGFR inhibition hinders the formation of F-actin rings during osteoclastogenesis, disrupting the cytoskeletal organization, structure, and function of mature osteoclasts.
Furthermore, RT-PCR results demonstrated that EGFR inhibition through gefitinib at a concentration of 10 μM downregulated the expression of osteoclast-associated genes, including C-FOS, TRAP, NF-ATC1, CTSK, and OSCAR (Figure 6F). These findings indicate that EGFR suppression induces the inhibition of RANKL-induced osteoclastogenesis.
Suppression of PI3K/AKT and NF-κB Signaling Pathways During Osteoclastogenesis After EGFR Inhibition
To elucidate the precise mechanisms underlying the downregulation of osteoclastogenesis resulting from EGFR inhibition, we employed Western blotting analysis to examine the key signaling pathways implicated in osteoclast formation and differentiation. Specifically, BMDMs were stimulated with 100 ng/mL RANKL for various durations (0, 15, 30, 45, or 60 min), with or without gefitinib at a concentration of 10 μM. Our results indicated a significant increase in AKT, p65, and IκBα phosphorylation upon RANKL stimulation (Figure 7A–D). This effect was subsequently countered by the administration of gefitinib (Figure 7A–D). Moreover, RANKL stimulation led to a notable translocation of p65 from the cytoplasm to the nucleus, which was dose-dependently inhibited by gefitinib treatment (Figure 7E). These results suggest that EGFR inhibition interferes with RANKL-induced osteoclastogenesis by suppressing the PI3K/AKT and NF-κB signaling pathways.
 |
Figure 7 EGFR inhibition suppressed the activation of PI3K/AKT and NF-κB pathways. BMDMs were subjected to stimulation with 100 ng/mL RANKL for durations (0, 15, 30, 45, or 60 min) with or without gefitinib. Western blotting against (A, B) AKT and P-AKT, (A, C) IκB-α and P-IκB-α, (A, D) p65 and P-p65. (E) Following stimulation with 100 ng/mL RANKL for 60 min, representative images of p65 translocation into the nucleus are displayed. The scale bar represents 50 μm. *P<0.05, **P<0.01. |
Discussion
EGFR, known for its influence on epithelial cell function and its oncogenic role in various cancers.50–54 has been established in the context of inflammation and cancer for its impact on macrophages,28,29 However, our study substantiated the crucial role of EGFR in regulating macrophage polarization and osteoclastogenesis, which holds huge potential for mitigating implant-induced osteolysis. Our study provided hitherto undocumented evidence of the predominance of EGFR and P-EGFR in multinucleated macrophage-like cells within human coxarthrosis synovial samples. Additionally, significant activation of P-EGFR was noted in coxa synovial membrane samples from patients undergoing revision arthroplasty with periprosthetic osteolysis. The recruitment of macrophages by wear particles and their transition to M1 macrophages with pro-inflammatory attributes are key contributors to chronic inflammation and osteolysis.55,56 Given that macrophage polarization significantly influences inflammatory osteolysis,57–59 it is highly conceivable that EGFR activation could trigger M1 polarization, contributing to particle-induced osteolysis.
Earlier studies have emphasized the dependence of macrophage activation and function on EGFR signaling, a key factor in generating pro-inflammatory chemokines in vivo.28,32 Prior research has also indicated that EGFR can reprogram macrophages into the M1 phenotype to regulate inflammatory macrophage responses.60 Building on this understanding, we postulated that inhibiting EGFR could mitigate the production of M1-type macrophages and mitigate particle-induced osteolysis. To test this hypothesis, we administered the EGFR inhibitor gefitinib in a murine model of titanium particle-induced osteolysis, and the observed outcomes were in line with our expectations. Our findings indicated that gefitinib treatment increased BMD and BV/TV while reducing porosity. Additional histological bone staining substantiated that inhibiting EGFR effectively countered titanium particle-induced bone degradation.
The intricate link between chronic inflammation and various diseases underscores the pivotal role of macrophages. These adaptable cells exhibit distinct polarized phenotypes, encompassing the pro-inflammatory M1 phenotype and the anti-inflammatory M2 phenotype, enabling them to fulfill diverse functions.15 The emergence of excessive M1 macrophage polarization is attributed to peri-implant inflammation, contributing to the aseptic loosening of implants. This polarization potentiates the expression of inflammatory cytokines, subsequently activating osteoclasts.61 To further elucidate the mechanism by which gefitinib mitigates titanium particle-induced osteolysis, we explored the hypothesis that EGFR inhibition would reduce M1 macrophage formation. This hypothesis was investigated both in vivo and in vitro. Herein, gefitinib effectively inhibited M1 polarization and facilitated a shift towards an M2-like phenotype within an inflammatory environment by blocking EGFR activity. In conclusion, our study underscores the role of EGFR inhibition in curtailing the generation of macrophages implicated in particle-induced osteolysis, suggesting its potential as a therapeutic avenue for treating osteolytic disorders.
Osteoclasts, pivotal for bone homeostasis and density maintenance, are also key players in bone resorption.62,63 Research has emphasized the critical role of activated osteoclasts in the pathophysiology of particle-induced periprosthetic osteolysis, leading to escalated bone resorption.34,64 RANKL, a central regulator of osteoclastogenesis, is integral to bone resorption by promoting osteoclast differentiation and formation while regulating wear debris-induced osteolysis.64,65 Our study revealed higher levels of RANKL expression in tissues from revision arthroplasty patients with PPO, consistent with elevated P-EGFR expression. This correlation between P-EGFR and RANKL levels in human coxa osteoarthritic synovium samples highlights P-EGFR’s role in regulating particle-induced osteolysis akin to RANKL. As RANKL downregulation has been shown to mitigate osteoclast formation and differentiation, suppressing bone destruction.66,67 We hypothesized that EGFR inhibition could similarly hinder osteoclastogenesis and mitigate particle-induced osteolysis. Hence, we employed the EGFR inhibitor gefitinib, which demonstrated efficacy in a murine model of titanium particle-induced osteolysis, in alignment with our hypothesis. While increasing bone mass and decreasing osteoclast numbers, gefitinib diminished EGFR expression in a murine particle-induced osteolysis model. These findings imply that gefitinib’s inhibitory impact could stem from EGFR expression suppression, reducing osteolytic bone resorption and osteoclastogenesis. Accordingly, we investigated the effect of EGFR inhibitors on RANKL-induced osteoclastogenesis in vitro. As expected, the EGFR inhibitor gefitinib hindered osteoclast formation and associated resorption, concurrently suppressing osteoclast-associated gene expression by downregulating EGFR.
Prior research has demonstrated EGFR’s influence over diverse cellular processes such as proliferation, growth, survival, metabolism, and migration through the activation of the PI3K-AKT pathway, subject to temporal and spatial control.68 Furthermore, the PI3K/AKT pathway regulates osteoclast activation and survival.69–71 Studies have indicated that inhibiting AKT, like with LY2940, considerably diminishes mature osteoclast numbers stimulated by RANKL.72,73 In this context, we investigated whether PI3K/AKT pathway inhibition contributed to the reduced osteoclast activation due to gefitinib. Western blotting confirmed that RANKL-stimulated cells displayed heightened AKT phosphorylation, markedly reversed with gefitinib treatment. The decreased AKT phosphorylation after gefitinib administration suggests its potential to inhibit osteoclast activation through direct or indirect PI3K/AKT pathway modulation.
Previous research has shown that RANKL-induced osteoclastogenesis is mediated by NF-κB signaling,72,74–76 which can be modulated by PI3K/AKT activation.77 Furthermore, NF-κB activation and PI3K/AKT signaling involve the translocation of p65, an NF-κB subunit, from the cytoplasm to the nucleus.78 These pathways collectively drive osteoclast activation and gene expression.79 In light of PI3K/AKT’s impact on NF-κB, we assessed the effects of gefitinib on IκB-α and p65 phosphorylation. As expected, gefitinib effectively reversed the RANKL-induced elevation of P-p65 and P-IκB-α in BMDMs. Additionally, gefitinib indirectly validated IκB-α phosphorylation inhibition by countering RANKL-induced IκB-α degradation. Moreover, gefitinib mitigated RANKL-induced p65 translocation from the cytoplasm to the nucleus. These results are consistent with the established role of EGFR inhibitors in synergistically inhibiting cell proliferation through the targeting of the NF-κB signaling pathway.80 In summary, EGFR inhibition suppressed RANKL-induced osteoclastogenesis by blocking the PI3K/AKT and NF-κB pathways.
Furthermore, our study revealed that multinucleated macrophage-like cells in human hip synovium express EGFR and P-EGFR. Notably, P-EGFR expression was elevated in multinucleated macrophage-like cells within human coxa synovial membrane samples from patients undergoing revision arthroplasty. This parallel with osteoclasts found in revised tissue implies that the multinucleated macrophage-like cells could potentially serve as precursor cells for osteoclasts.45 These findings emphasize EGFR’s role in forming macrophages and osteoclasts, where inhibiting EGFR activity impedes osteoclast precursor cells and osteoclastogenesis.
Maintaining the delicate balance between osteoclastic bone resorption and osteoblastic bone formation, known as bone remodeling, is crucial for various physiological functions like bone development, fracture repair, and mineral metabolism.81 Disruption of this equilibrium, often due to aberrant osteoclast activity, can lead to metabolic bone disorders such as PPO. Additionally, macrophages play a significant role in prosthesis-related inflammation, exacerbating PPO progression.82 In osteolytic diseases, wear particle stimulation triggers osteoclast generation and activation,83 heightened M1 macrophage expression, and aggravated local inflammation,84 ultimately resulting in PPO. In this context, inhibiting M1 macrophage polarization and osteoclast generation and activation can mitigate particle-induced periprosthetic osteolysis.82,85 Our study offers a unique perspective on the role of EGFR in macrophage polarization and osteoclastogenesis within the context of periprosthetic osteolysis, providing a detailed mechanistic understanding. Our research demonstrates that inhibiting EGFR with gefitinib can reduce the number of M1 macrophages and osteoclasts, thereby alleviating bone destruction in a murine model of titanium particle-induced osteolysis. Furthermore, we highlight that gefitinib’s inhibitory effects on osteoclastogenesis and activation involve the EGFR/PI3K/Akt and NF-κB signaling pathways. Given the importance of regulating bone regeneration in wear particle-induced osteolytic diseases, these findings emphasize the potential therapeutic benefits of targeting EGFR to mitigate such conditions.
While our study contributes valuable insights, there are certain limitations to consider. Titanium particles were utilized to induce osteolysis, although ultrahigh molecular weight polyethylene debris is the primary material used in medical practice.86 The rationale behind choosing titanium particles was their stability and adhesive properties. It should be borne in mind that prior studies have reported similar osteolytic effects between the two materials.36,87 However, due to the differing conditions in our in vitro and in vivo models (TNF-α/IFN-γ vs titanium particles), there is room for further optimization and alignment. We plan to address this in future iterations to enhance experimental rigor. Additionally, we recognize the importance of distinguishing the role of EGFR in macrophage polarization versus osteoclast differentiation, which was not specifically isolated in our study. We have now added this aspect to our considerations. Furthermore, our experiment focused on the relevance of the EGFR pathway to osteoblasts and macrophages without extensively exploring and targeting downstream genes, which we intend to pursue in future research.
Conclusions
Overall, our study uncovers the pivotal role of activated EGFR-related pathways in M1 polarization, osteoclast activation, and subsequent periprosthetic osteolysis. Importantly, we demonstrate that inhibiting EGFR phosphorylation effectively mitigates titanium-induced osteolysis, reduces bone destruction at affected sites, and suppresses osteoclastogenesis and related gene expression by robustly targeting the RANKL-induced PI3K/AKT and NF-κB pathways. Consequently, EGFR emerges as a promising therapeutic target for addressing periprosthetic osteolysis and other bone resorption and destruction processes orchestrated by macrophages and osteoclasts.
Abbreviations
EGFR, epidermal growth factor receptor; Ti, Titanium; IL, interleukin; OPG, osteoprotegerin; TNF-α, tumor necrosis factor-alpha; CCK-8, Cell Counting Kit 8; MCP1, monocyte chemoattractant protein 1; EDTA, ethylenediaminetetraacetic acid; OcS/BS, the ratio of osteoclast surface to bone surface; BMDMs, The bone marrow-derived macrophages; RT-PCR, Real-time PCR; PPO, periprosthetic osteolysis; BV/TV, Bone Volume Fraction/Total Volume; BMD, Bone Mineral Density; M-CSF, macrophage colony-stimulating factor; IFN-γ, Interferon-gamma.
Data Sharing Statement
Data will be made available on request.
Ethics Statement
The animal study was reviewed and approved by Ethics Committee of the First Affiliated Hospital of Xinjiang Medical University. The process was strictly followed the National Research Council’s Guide for the Care and Use of Laboratory Animals.
Consent for Publication
Written informed consent for publication were obtained from patients.
Acknowledgments
The authors would like to express their sincere appreciation to Dr. Gongyin Zhao for his invaluable and outstanding contribution to the review process.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. All authors have read and approved the final submitted manuscript.
Funding
This research was funded by the Natural Science Foundation of Xinjiang Uygur Autonomous Region (2022D01D56, 2022D01C756); The “Tianshan Talents” High-Level Medical and Health Personnel Training Program (TSYC202301A008); the National Natural Science Foundation of China (82160421) and The First Affiliated Hospital of Xinjiang Medical University “Youth Research Launch” Special Fund (2023YFY-QKMS-05).
Disclosure
The authors declare that they have no conflict of interest.
References
1. Moraschini V, Poubel LA, Ferreira VF, Barboza Edos S. Evaluation of survival and success rates of dental implants reported in longitudinal studies with a follow-up period of at least 10 years: a systematic review. Int J Oral Maxillofac Surg. 2015;44(3):377–388. doi:10.1016/j.ijom.2014.10.023
2. Revell PA. The combined role of wear particles, macrophages and lymphocytes in the loosening of total joint prostheses. J R Soc Interface. 2008;5(28):1263–1278. doi:10.1098/rsif.2008.0142
3. Fretwurst T, Nelson K, Tarnow DP, Wang HL, Giannobile WV. Is metal particle release associated with peri-implant bone destruction? An emerging concept J Dent Res. 2018;97(3):259–265.
4. Witt JD, Swann M. Metal wear and tissue response in failed titanium alloy total Hip replacements. J Bone Joint Surg Br. 1991;73(4):559–563. doi:10.1302/0301-620X.73B4.2071635
5. Scales JT. Black staining around titanium alloy prostheses--an orthopaedic enigma. J Bone Joint Surg Br. 1991;73(4):534–536. doi:10.1302/0301-620X.73B4.2071632
6. Wilson TG Jr, Valderrama P, Burbano M, et al. Foreign bodies associated with peri-implantitis human biopsies. J Periodontol. 2015;86(1):9–15. doi:10.1902/jop.2014.140363
7. Olmedo DG, Paparella ML, Spielberg M, Brandizzi D, Guglielmotti MB, Cabrini RL. Oral mucosa tissue response to titanium cover screws. J Periodontol. 2012;83(8):973–980. doi:10.1902/jop.2011.110392
8. Schlegel KA, Eppeneder S, Wiltfang J. Soft tissue findings above submerged titanium implants--a histological and spectroscopic study. Biomaterials. 2002;23(14):2939–2944. doi:10.1016/S0142-9612(01)00423-9
9. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11(2):98–107. doi:10.1038/nri2925
10. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011;12(8):715–723. doi:10.1038/ni.2060
11. Rader DJ, Daugherty A. Translating molecular discoveries into new therapies for atherosclerosis. Nature. 2008;451(7181):904–913. doi:10.1038/nature06796
12. Potteaux S, Gautier EL, Hutchison SB, et al. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe-/- mice during disease regression. J Clin Invest. 2011;121(5):2025–2036. doi:10.1172/JCI43802
13. Eger M, Hiram-Bab S, Liron T, et al. Mechanism and prevention of titanium particle-induced inflammation and osteolysis. Front Immunol. 2018;9:2963. doi:10.3389/fimmu.2018.02963
14. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi:10.12703/P6-13
15. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi:10.1038/nri2448
16. Benoit M, Desnues B, Mege JL. Macrophage polarization in bacterial infections. J Immunol. 2008;181(6):3733–3739. doi:10.4049/jimmunol.181.6.3733
17. Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73(2):209–212. doi:10.1189/jlb.0602325
18. Lin TH, Tamaki Y, Pajarinen J, et al. Chronic inflammation in biomaterial-induced periprosthetic osteolysis: NF-kappaB as a therapeutic target. Acta Biomater. 2014;10(1):1–10. doi:10.1016/j.actbio.2013.09.034
19. Green TR, Fisher J, Stone M, Wroblewski BM, Ingham E. Polyethylene particles of a ‘critical size’ are necessary for the induction of cytokines by macrophages in vitro. Biomaterials. 1998;19(24):2297–2302. doi:10.1016/S0142-9612(98)00140-9
20. Zhao Z, Hou X, Yin X, et al. TNF Induction of NF-kappaB RelB Enhances RANKL-induced osteoclastogenesis by promoting inflammatory macrophage differentiation but also limits it through suppression of NFATc1 expression. PLoS One. 2015;10(8):e0135728. doi:10.1371/journal.pone.0135728
21. Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59(2 Suppl):21–26. doi:10.1016/j.ijrobp.2003.11.041
22. Hubbard SR, Mohammadi M, Schlessinger J. Autoregulatory mechanisms in protein-tyrosine kinases. J Biol Chem. 1998;273(20):11987–11990. doi:10.1074/jbc.273.20.11987
23. Wang K, Yamamoto H, Chin JR, Werb Z, Vu TH. Epidermal growth factor receptor-deficient mice have delayed primary endochondral ossification because of defective osteoclast recruitment. J Biol Chem. 2004;279(51):53848–53856. doi:10.1074/jbc.M403114200
24. Sibilia M, Wagner B, Hoebertz A, et al. Mice humanised for the EGF receptor display hypomorphic phenotypes in skin, bone and heart. Development. 2003;130(19):4515–4525. doi:10.1242/dev.00664
25. Chien HH, Lin WL, Cho MI. Down-regulation of osteoblastic cell differentiation by epidermal growth factor receptor. Calcif Tissue Int. 2000;67(2):141–150. doi:10.1007/s00223001128
26. Zhu J, Jia X, Xiao G, Kang Y, Partridge NC, Qin L. EGF-like ligands stimulate osteoclastogenesis by regulating expression of osteoclast regulatory factors by osteoblasts: implications for osteolytic bone metastases. J Biol Chem. 2007;282(37):26656–26665. doi:10.1074/jbc.M705064200
27. Tietzel I, Mosser DM. The modulation of macrophage activation by tyrosine phosphorylation. Front Biosci. 2002;7(1–3):d1494–1502. doi:10.2741/tietzel
28. Lu N, Wang L, Cao H, et al. Activation of the epidermal growth factor receptor in macrophages regulates cytokine production and experimental colitis. J Immunol. 2014;192(3):1013–1023. doi:10.4049/jimmunol.1300133
29. Lanaya H, Natarajan A, Komposch K, et al. EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat Cell Biol. 2014;16(10):972–977. doi:10.1038/ncb3031
30. Liu B, Neufeld AH. Activation of epidermal growth factor receptor signals induction of nitric oxide synthase-2 in human optic nerve head astrocytes in glaucomatous optic neuropathy. Neurobiol Dis. 2003;13(2):109–123. doi:10.1016/S0969-9961(03)00010-X
31. Scholes AG, Hagan S, Hiscott P, Damato BE, Grierson I. Overexpression of epidermal growth factor receptor restricted to macrophages in uveal melanoma. Arch Ophthalmol. 2001;119(3):373–377. doi:10.1001/archopht.119.3.373
32. Hardbower DM, Singh K, Asim M, et al. EGFR regulates macrophage activation and function in bacterial infection. J Clin Invest. 2016;126(9):3296–3312. doi:10.1172/JCI83585
33. Hardbower DM, Coburn LA, Asim M, et al. EGFR-mediated macrophage activation promotes colitis-associated tumorigenesis. Oncogene. 2017;36(27):3807–3819. doi:10.1038/onc.2017.23
34. Geng D, Xu Y, Yang H, et al. Protection against titanium particle induced osteolysis by cannabinoid receptor 2 selective antagonist. Biomaterials. 2010;31(8):1996–2000. doi:10.1016/j.biomaterials.2009.11.069
35. Ragab AA, Van De Motter R, Lavish SA, et al. Measurement and removal of adherent endotoxin from titanium particles and implant surfaces. J Orthop Res. 1999;17(6):803–809. doi:10.1002/jor.1100170603
36. von Knoch M, Jewison DE, Sibonga JD, et al. The effectiveness of polyethylene versus titanium particles in inducing osteolysis in vivo. J Orthop Res. 2004;22(2):237–243. doi:10.1016/j.orthres.2003.08.013
37. Godugu C, Doddapaneni R, Patel AR, Singh R, Mercer R, Singh M. Novel gefitinib formulation with improved oral bioavailability in treatment of A431 skin carcinoma. Pharm Res. 2016;33(1):137–154. doi:10.1007/s11095-015-1771-6
38. Chen Y, Wang M, Zhong W, Zhao J. Pharmacokinetic and pharmacodynamic study of Gefitinib in a mouse model of non-small-cell lung carcinoma with brain metastasis. Lung Cancer. 2013;82(2):313–318. doi:10.1016/j.lungcan.2013.08.013
39. Ma Y, Tang N, Thompson RC, et al. InsR/IGF1R pathway mediates resistance to EGFR inhibitors in glioblastoma. Clin Cancer Res. 2016;22(7):1767–1776. doi:10.1158/1078-0432.CCR-15-1677
40. Yang H, Xu Y, Zhu M, et al. Inhibition of titanium-particle-induced inflammatory osteolysis after local administration of dopamine and suppression of osteoclastogenesis via D2-like receptor signaling pathway. Biomaterials. 2016;80:1–10. doi:10.1016/j.biomaterials.2015.11.046
41. Parfitt AM, Drezner MK, Glorieux FH, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. report of the ASBMR histomorphometry nomenclature committee. J Bone Miner Res. 1987;2(6):595–610. doi:10.1002/jbmr.5650020617
42. Sawyer A, Lott P, Titrud J, McDonald J. Quantification of tartrate resistant acid phosphatase distribution in mouse tibiae using image analysis. Biotech Histochem. 2003;78(5):271–278. doi:10.1080/10520290310001646668
43. Ledesma-Colunga MG, Adan N, Ortiz G, et al. Prolactin blocks the expression of receptor activator of nuclear factor kappaB ligand and reduces osteoclastogenesis and bone loss in murine inflammatory arthritis. Arthritis Res Ther. 2017;19(1):93. doi:10.1186/s13075-017-1290-4
44. Darowish M, Rahman R, Li P, et al. Reduction of particle-induced osteolysis by interleukin-6 involves anti-inflammatory effect and inhibition of early osteoclast precursor differentiation. Bone. 2009;45(4):661–668. doi:10.1016/j.bone.2009.06.004
45. Holding CA, Findlay DM, Stamenkov R, et al. The correlation of RANK, RANKL and TNFalpha expression with bone loss volume and polyethylene wear debris around Hip implants. Biomaterials. 2006;27(30):5212–5219. doi:10.1016/j.biomaterials.2006.05.054
46. Haynes DR, Crotti TN, Potter AE, et al. The osteoclastogenic molecules RANKL and RANK are associated with periprosthetic osteolysis. J Bone Joint Surg Br. 2001;83(6):902–911. doi:10.1302/0301-620X.83B6.0830902
47. Zaveri TD, Dolgova NV, Lewis JS, Hamaker K, Clare-Salzler MJ, Keselowsky BG. Macrophage integrins modulate response to ultra-high molecular weight polyethylene particles and direct particle-induced osteolysis. Biomaterials. 2017;115:128–140. doi:10.1016/j.biomaterials.2016.10.038
48. Antonios JK, Yao Z, Li C, Rao AJ, Goodman SB. Macrophage polarization in response to wear particles in vitro. Cell Mol Immunol. 2013;10(6):471–482. doi:10.1038/cmi.2013.39
49. Pennanen P, Alanne MH, Fazeli E, et al. Diversity of actin architecture in human osteoclasts: network of curved and branched actin supporting cell shape and intercellular micrometer-level tubes. Mol Cell Biochem. 2017;432(1–2):131–139. doi:10.1007/s11010-017-3004-2
50. Chaturvedi R, Asim M, Piazuelo MB, et al. Activation of EGFR and ERBB2 by Helicobacter pylori results in survival of gastric epithelial cells with DNA damage. Gastroenterology. 2014;146(7):1739–1751e1714. doi:10.1053/j.gastro.2014.02.005
51. Yan F, Cao H, Chaturvedi R, et al. Epidermal growth factor receptor activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Gastroenterology. 2009;136(4):
52. Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res. 2006;12(18):5268–5272. doi:10.1158/1078-0432.CCR-05-1554
53. Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37(Suppl 4):S9–15. doi:10.1016/S0959-8049(01)00231-3
54. Tokunaga A, Onda M, Okuda T, et al. Clinical significance of epidermal growth factor (EGF), EGF receptor, and c-erbB-2 in human gastric cancer. Cancer. 1995;75(6 Suppl):1418–1425.
55. Nich C, Takakubo Y, Pajarinen J, et al. Macrophages-Key cells in the response to wear debris from joint replacements. J Biomed Mater Res A. 2013;101(10):3033–3045. doi:10.1002/jbm.a.34599
56. Ingham E, Fisher J. The role of macrophages in osteolysis of total joint replacement. Biomaterials. 2005;26(11):1271–1286. doi:10.1016/j.biomaterials.2004.04.035
57. Bai J, Wang H, Chen H, et al. Biomimetic osteogenic peptide with mussel adhesion and osteoimmunomodulatory functions to ameliorate interfacial osseointegration under chronic inflammation. Biomaterials. 2020;255:120197. doi:10.1016/j.biomaterials.2020.120197
58. Li B, Hu Y, Zhao Y, et al. Curcumin attenuates titanium particle-induced inflammation by regulating macrophage polarization in vitro and in vivo. Front Immunol. 2017;8:55. doi:10.3389/fimmu.2017.00055
59. Pajarinen J, Kouri VP, Jamsen E, Li TF, Mandelin J, Konttinen YT. The response of macrophages to titanium particles is determined by macrophage polarization. Acta Biomater. 2013;9(11):9229–9240. doi:10.1016/j.actbio.2013.06.027
60. Luo S, Xu R, Xie P, et al. EGFR of platelet regulates macrophage activation and bacterial phagocytosis function. J Inflamm. 2024;21(1):10. doi:10.1186/s12950-024-00382-1
61. Hu X, Xu L, Fu X, et al. The TiO(2)-mu implant residual is more toxic than the Al(2)O(3)-n implant residual via blocking LAP and inducing macrophage polarization. Nanoscale. 2021;13(19):8976–8990. doi:10.1039/D1NR00696G
62. Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4(8):638–649. doi:10.1038/nrg1122
63. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337–342.
64. Mediero A, Ramkhelawon B, Wilder T, et al. Netrin-1 is highly expressed and required in inflammatory infiltrates in wear particle-induced osteolysis. Ann Rheum Dis. 2016;75(9):1706–1713. doi:10.1136/annrheumdis-2015-207593
65. Hofbauer LC, Schoppet M. Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. JAMA. 2004;292(4):490–495. doi:10.1001/jama.292.4.490
66. Hu X, Ping Z, Gan M, et al. Theaflavin-3,3’-digallate represses osteoclastogenesis and prevents wear debris-induced osteolysis via suppression of ERK pathway. Acta Biomater. 2017;48:479–488. doi:10.1016/j.actbio.2016.11.022
67. Bartell SM, Kim HN, Ambrogini E, et al. FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H2O2 accumulation. Nat Commun. 2014;5(1):3773. doi:10.1038/ncomms4773
68. Er EE, Mendoza MC, Mackey AM, Rameh LE, Blenis J. AKT facilitates EGFR trafficking and degradation by phosphorylating and activating PIKfyve. Sci Signal. 2013;6(279):ra45. doi:10.1126/scisignal.2004015
69. Qu Z, Guo S, Fang G, Cui Z, Liu Y. AKT pathway affects bone regeneration in nonunion treated with umbilical cord-derived mesenchymal stem cells. Cell Biochem Biophys. 2015;71(3):1543–1551. doi:10.1007/s12013-014-0378-6
70. Moon JB, Kim JH, Kim K, et al. Akt induces osteoclast differentiation through regulating the GSK3beta/NFATc1 signaling cascade. J Immunol. 2012;188(1):163–169. doi:10.4049/jimmunol.1101254
71. Lee SE, Woo KM, Kim SY, et al. The phosphatidylinositol 3-kinase, p38, and extracellular signal-regulated kinase pathways are involved in osteoclast differentiation. Bone. 2002;30(1):71–77. doi:10.1016/S8756-3282(01)00657-3
72. Abdelmagid SM, Sondag GR, Moussa FM, et al. Mutation in osteoactivin promotes receptor activator of NFkappaB Ligand (RANKL)-mediated osteoclast differentiation and survival but inhibits osteoclast function. J Biol Chem. 2015;290(33):20128–20146. doi:10.1074/jbc.M114.624270
73. Abdelmagid SM, Belcher JY, Moussa FM, et al. Mutation in osteoactivin decreases bone formation in vivo and osteoblast differentiation in vitro. Am J Pathol. 2014;184(3):697–713. doi:10.1016/j.ajpath.2013.11.031
74. Kubota T, Hoshino M, Aoki K, et al. NF-kappaB inhibitor dehydroxymethylepoxyquinomicin suppresses osteoclastogenesis and expression of NFATc1 in mouse arthritis without affecting expression of RANKL, osteoprotegerin or macrophage colony-stimulating factor. Arthritis Res Ther. 2007;9(5):R97. doi:10.1186/ar2298
75. Tsurukai T, Udagawa N, Matsuzaki K, Takahashi N, Suda T. Roles of macrophage-colony stimulating factor and osteoclast differentiation factor in osteoclastogenesis. J Bone Miner Metab. 2000;18(4):177–184. doi:10.1007/s007740070018
76. Quinn JM, Elliott J, Gillespie MT, Martin TJ. A combination of osteoclast differentiation factor and macrophage-colony stimulating factor is sufficient for both human and mouse osteoclast formation in vitro. Endocrinology. 1998;139(10):4424–4427. doi:10.1210/endo.139.10.6331
77. Ardeshna KM, Pizzey AR, Devereux S, Khwaja A. The PI3 kinase, p38 SAP kinase, and NF-kappaB signal transduction pathways are involved in the survival and maturation of lipopolysaccharide-stimulated human monocyte-derived dendritic cells. Blood. 2000;96(3):1039–1046. doi:10.1182/blood.V96.3.1039
78. Baldwin AS Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14(1):649–683. doi:10.1146/annurev.immunol.14.1.649
79. Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40(2):251–264. doi:10.1016/j.bone.2006.09.023
80. Hu Y, Zang J, Cao H, et al. Liver X receptors agonist GW3965 re-sensitizes gefitinib-resistant human non-small cell lung cancer cell to gefitinib treatment by inhibiting NF-kappaB in vitro. Oncotarget. 2017;8(9):15802–15814. doi:10.18632/oncotarget.15007
81. Kodama J, Kaito T. Osteoclast multinucleation: review of current literature. Int J Mol Sci. 2020;21(16):5685. doi:10.3390/ijms21165685
82. Wang L, Wang Q, Wang W, et al. Harmine alleviates titanium particle-induced inflammatory bone destruction by immunomodulatory effect on the macrophage polarization and subsequent osteogenic differentiation. Front Immunol. 2021;12:657687. doi:10.3389/fimmu.2021.657687
83. Guo X, Liu Y, Bai J, et al. Efficient inhibition of wear-debris-induced osteolysis by surface biomimetic engineering of titanium implant with a mussel-derived integrin-targeting peptide. Adv. Biosyst. 2019;3(2). doi:10.1002/adbi.201800253.
84. Rao AJ, Gibon E, Ma T, Yao Z, Smith RL, Goodman SB. Revision joint replacement, wear particles, and macrophage polarization. Acta Biomater. 2012;8(7):2815–2823. doi:10.1016/j.actbio.2012.03.042
85. Guo X, Bai J, Ge G, et al. Bioinspired peptide adhesion on Ti implants alleviates wear particle-induced inflammation and improves interfacial osteogenesis. J Colloid Interface Sci. 2022;605:410–424. doi:10.1016/j.jcis.2021.07.079
86. Qu S, Bai Y, Liu X, Fu R, Duan K, Weng J. Study on in vitro release and cell response to alendronate sodium-loaded ultrahigh molecular weight polyethylene loaded with alendronate sodium wear particles to treat the particles-induced osteolysis. J Biomed Mater Res A. 2013;101(2):394–403. doi:10.1002/jbm.a.34327
87. Wooley PH, Morren R, Andary J, et al. Inflammatory responses to orthopaedic biomaterials in the murine air pouch. Biomaterials. 2002;23(2):517–526. doi:10.1016/S0142-9612(01)00134-X
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.