")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 12
Integrated Multi-Omics Profiling Identifies PDZ-Binding Kinase (PBK) as a Novel Prognostic Biomarker in Hepatocellular Carcinoma
Authors Zhang J , Xu Y, Ni X , Mao Z, Xiao H, Chen M, Lin Y, Pan J, Zhang B, Zhang L, Zheng X, Song G, Ge N
Received 30 August 2024
Accepted for publication 26 June 2025
Published 17 July 2025 Volume 2025:12 Pages 1453—1469
DOI https://doi.org/10.2147/JHC.S493907
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Jörg Trojan
Juan Zhang,1,* Yingyu Xu,2,* Xiaojian Ni,2,* Zhiyi Mao,3,* Haitao Xiao,4 Maopei Chen,1 Youpei Lin,5 Jiaomeng Pan,5 Boheng Zhang,6 Lan Zhang,1 Xueying Zheng,3 Guohe Song,5 Ningling Ge1
1Department of Hepatobiliary Oncology, Liver Cancer Institute, Zhongshan Hospital; Key Laboratory of Carcinogenesis and Cancer Invasion (Ministry of Education), Fudan University, Shanghai, People’s Republic of China; 2Department of Biliary Surgery, Zhongshan Hospital, Fudan University; Biliary Tract Disease Center of Zhongshan Hospital, Fudan University; Biliary Tract Disease Institute, Fudan University; Cancer Center, Zhongshan Hospital, Fudan University; Shanghai Biliary Tract Minimal Invasive Surgery and Materials Engineering Research Center, Shanghai, People’s Republic of China; 3Department of Biostatistics, Key Laboratory of Public Health Safety of Ministry of Education, School of Public Health, Fudan University, Shanghai, People’s Republic of China; 4Department of Traumatic Orthopedics, Hainan General Hospital, Haikou, People’s Republic of China; 5Department of Hepatobiliary Surgery and Liver Transplantation, Key Laboratory of Carcinogenesis and Cancer Invasion, Ministry of Education, Liver Cancer Institute, Zhongshan Hospital, Fudan University, Shanghai, People’s Republic of China; 6Department of Hepatobiliary Oncology, Zhongshan Hospital, Fudan University (Xiamen Branch), Xiamen Clinical Research Center for Cancer Therapy, Clinical Research Center for Precision medicine of abdominal tumor of Fujian Province, Xiamen City, Fujian Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guohe Song, Email [email protected] Ningling Ge, Email [email protected]
Background: Hepatocellular carcinoma (HCC) necessitates novel immunotherapeutic targets. PBK, a cancer/testis antigen (CTA), was identified as a pivotal hub gene influencing prognosis, tumor mutation burden (TMB), and immune microenvironment remodeling.
Methods: PBK was prioritized using weighted gene co-expression network analysis (WGCNA) and differential expression screening in the TCGA-LIHC cohort, intersected with curated CTAs. Analyses assessed correlations with clinicopathological features (TNM stage, survival), genomic characterization (mutation frequencies), and functional validation via siRNA-mediated PBK knockdown in Huh7 cells (migration assay). Single-cell RNA sequencing (scRNA-seq) profiled of the tumor immune microenvironment.
Results: PBK overexpression was significantly correlated with advanced TNM stage (P < 0.05) and poor survival (log-rank P = 0.003). Genomic analysis revealed distinct mutation profiles: high-PBK tumors exhibited increased TP53 mutation frequency (39% vs 17%) but decreased CTNNB1 mutations (20% vs 31%). Patients exhibiting with combined PBK overexpression and high TMB demonstrated the poorest prognosis. Functional validation confirmed that PBK knockdown significantly inhibited Huh7 cell migration capacity (P < 0.05). scRNA-seq analysis showed PBK-enriched tumors contained elevated proportions of immunosuppressive SPP1(+) macrophages (22.33% vs 6.6%, FDR corrected P < 0.001) and CD8(+) SLC4A10(+) MAIT cells (9.82% vs 4.7%, FDR corrected P < 0.001).
Conclusion: PBK synergistically drives HCC progression through three synergistic mechanisms: (1) promoting oncogenic mutation accumulation (eg, TP53), (2) increasing metastatic potential, and (3) reprogramming an immune-suppressive microenvironment enriched for SPP1(+) macrophages and CD8(+)SLC4A10(+) MAIT cells. This establishes PBK as a dual-purpose biomarker for prognostic stratification and immunotherapy resistance prediction, providing a mechanistic rationale for developing PBK-targeted therapies in HCC.
Keywords: PDZ-binding kinase, bioinformatics, prognosis, cancer immunotherapy, hepatocellular carcinoma
Introduction
Heatocellular carcinoma (HCC), accounting for 75–85% of primary liver cancer cases,1 poses a formidable clinical challenge, with 5-year survival rates for advanced-stage disease below 20%.2 This dismal prognosis underscores the critical need to develop innovative therapeutic strategies targeting molecular vulnerabilities.3 Cancer-testis antigens (CTAs), initially characterized as tumor-specific immunogens through serological analysis of recombinant cDNA expression libraries (SEREX),4 have emerged as dual-functional biomarkers with both prognostic and immunotherapeutic significance.5 The CTA family comprises X chromosome-encoded (XCTA) and autosomal members, with approximately 50% mapping to the X chromosome.6 These evolutionarily conserved proteins exhibit restricted physiological expression in germline cells within immune-privileged sites like the testes,7 while demonstrating aberrant reactivation in diverse malignancies including breast cancer, non-small cell lung cancer (NSCLC), and melanoma.8 In HCC, systematic analyses reveal that overexpression of melanoma-associated antigen A (MAGEA)1/3/4, melanoma-associated antigen C2(MAGEC2), and the New York esophageal squamous cell carcinoma (NY-ESO-1) correlates with aggressive clinicopathological features, including metastatic progression, early recurrence, and reduced 5-year survival.9–15 This tumor-selective expression pattern, coupled with inherent immunogenicity, positions CTAs as promising targets for immunotherapy development.16
Despite these advances, critical knowledge gaps persist regarding the hierarchical regulatory networks governing CTA-mediated oncogenesis. Recent single-cell transcriptomic studies have unveiled the dynamic heterogeneity of tumor-infiltrating immune cells,17 particularly highlighting CD8(+) T cell exhaustion as a key mediator of immune evasion.18 However, the precise mechanisms by which CTAs modulate the immune microenvironment remodeling in HCC remain incompletely understood. Furthermore, a lack of systematic multi-omics analyses has hindered the identification of master regulatory CTAs that concurrently drive tumor progression and immune suppression.
To address these limitations, we performed an integrative multi-omics investigation of CTA networks in HCC. Using weighted gene co-expression network analysis (WGCNA) of TCGA-LIHC data combined with functional enrichment studies, we identified PDZ-binding kinase (PBK) as a pivotal regulatory hub. Building on PBK’s established role in maintaining genomic stability during spermatogenesis, we hypothesized that its oncogenic reactivation in HCC may: (1) potentiate mutational accumulation through DNA repair dysregulation, (2) enhance metastatic capacity via cytoskeletal remodeling, and (3) promote an immunosuppressive microenvironment through cytokine signaling. Leveraging the unique immunobiological properties of CTAs, we hypothesized that PBK is involved in modulating immune cell infiltration patterns and the tumor mutational burden (TMB) in HCC. To test these hypotheses, we employed a multi-faceted approach: (1) systematic analysis of associations between PBK expression, clinicopathological features, TMB, and immune cell abundance; (2) in vitro functional validation of PBK’s role in HCC progression; and (3) single-cell RNA sequencing (scRNA-seq) to dissect PBK mediated immune evasion mechanisms. This systematic approach establishes PBK as a novel prognostic biomarker, but also elucidates its dual role in promoting oncogenesis and immune evasion, providing a mechanistic rationale for developing PBK-targeted strategies to overcome immunotherapy resistance in HCC.
Materials and Methods
Data Collection and Preprocessing
RNA-seq gene expression data, clinical information and immune subtypes from 33 tumor types were obtained from The Cancer Genome Atlas (TCGA) and UCSC Xena databases (https://xenabrowser.net/). The TCGA-LIHC dataset was analyzed using weighted gene co-expression network analysis (WGCNA) to construct co-expression networks. The IMvigor210 immunotherapy cohort was utilized to assess response to immunotherapy across various cancer types.19 Two external scRNA-seq datasets, GSE14961420 and GSE15153021 were retrieved from NCBI Gene Expression Omnibus (GEO) databases and included pre-annotated clinical data.
WGCNA Module Construction, Functional Annotation, and Pathway Enrichment Analysis
The TCGA-LIHC dataset, consisting of 343 HCC samples, was utilized to construct a co-expression network using the WGCNA R package. The key gene modules were identified based on their correlation(Pearson) with clinical variables including survival time, survival status, gender, age, and TNM stage. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were conducted for the functional annotation of module genes. Gene set enrichment analysis (GSEA) and gene set variation analysis (GSVA) were employed to assess pathway enrichment differences between PBK-high and PBK-low expression groups.
Immune Microenvironment Profiling via CIBERSORT and ESTIMATE Algorithms
Using the LM22 gene signature (characterizing 22 immune cell subtypes), the relative abundance of tumor-infiltrating immune cells using the CIBERSORT algorithm. The ESTIMATE algorithm was to calculate stromal score, immune score, and estimate tumor purity. Differences in PBK expression stratified by immune cell infiltration, stromal score, immune score, and immune checkpoint expression status were evaluated in the TCGA-LIHC dataset. Sankey plot visualizing PBK expression levels and immune subtypes using the R packages “ggplot2” and “ggalluvial”.
Genomic Instability and Mutation Burden Analysis
Somatic mutation data were analyzed using the “Maftools” R package to visualize genomic alterations and calculate tumor mutation burden (TMB). Oncoplots depicting mutation landscapes for PBK-high and PBK-low groups were generated using the “oncoplot” function.
Immunohistochemical Staining
Formalin-fixed paraffin-embedded (FFPE) HCC tissue microarray (TMA) sections were deparaffinized, rehydrated, and subjected to antigen retrieval using citrate buffer. Endogenous peroxidase activity was quenched with 0.3% hydrogen peroxide. Non-specific binding was blocked with 5% bovine serum albumin. Anti-PBK antibody (Cell Signaling Technology, 4942T) overnight at 4 °C, washed, and incubated with a species-appropriate secondary antibody. PBK protein expression was visualized using 3,3′-diaminobenzidine (DAB) substrate and counterstained with hematoxylin.
Cell Culture and Transient Transfection
Huh7, L02, SMMMC-7721, MHCC97H, PLC-RPF-5, Hep3B, and HCCLM3 cell lines were sourced from the Institute of Liver Cancer at Fudan University (Shanghai, China). These cells were cultured in either Dulbecco’s Modified Eagle Medium (DMEM) or Roswell Park Memorial Institute Medium 1640 (RPMI-1640), supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin, and maintained at 37 °C in a humidified incubator with 5% CO2. Cells were transiently transfected with PBK-targeting or control small interfering RNAs (siRNAs; Genomeditech, Shanghai, China) using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol.
Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) and reverse transcripted into cDNA using a commercial kit (Yeasen Biotech, Shanghai, China). The concentration and purity of RNA was assessed spectrophotometrically. qRT-PCR was performed using SYBR Green Master Mix Kit (Yeasen Biotech, Shanghai, China). Primer sequences used were: GAPDH, 5′-GAGTCAACGGATTTGGTCGT-3′ (forward) and 5′-GACAAGCTTCCCGTTCTCA-G-3′ (reverse); PBK, 5′-CCAAACATTGTTGGTTATCGTGC-3′ (forward) and 5′-GGCTGGCTTTATATCGTTCTTCT-3′ (reverse). Relative gene expression was calculated using the 2−ΔΔCt method with GAPDH as the endogenous control.
Western Blotting
Total cellular protein was extracted using RIPA lysis buffer. The protein concentration was determined using a BCA protein assay kit (Beyotime Biotechnology, China). Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were blocked with 5% non-fat milk and probed with primary antibodies against PBK (Cell Signaling Technology, 4942T; 1:1000), β-actin (Cell Signaling Technology, 4970S; 1:1000), or GAPDH (Cell Signaling Technology, 2118S; 1:1000) overnight at 4 °C. After washing, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies. Protein bands were detected using a Western Bright ECL kit (Beyotime Biotechnology, China) and visualized using a chemiluminescence imaging system.
Transwell Migration and Invasion Assays
Cell migration and invasion were assessed using 24-well Transwell chamber (Corning, USA) with 8.0-μm pore polycarbonate membranes. For invasion assays, membranes were pre-coated with Matrigel (BD Biosciences, USA). Briefly, 2×104 Huh7 cells in 200 μL serum-free medium were seeded to the upper chambers. The lower chamber contained 500 µL medium supplemented with 10% FBS as a chemoattractant. After incubation for 24h (migration) or 48h (invasion), non-migrated/invaded cells on the upper surface were removed. Cells on the lower surface were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet, and counted in five random fields per membrane under a light microscope. Experiments were performed in triplicate.
scRNA-Seq Datasets Analysis
Data Preprocessing, Filtering, and Normalization
Single-cell RNA sequencing (scRNA-seq) data from GSE14961420 and GSE15153021 were processed independently using the Seurat R package. Quality control filtering was applied: For GSE149614, cells with 500–6000 detected genes, 1000–100,000 unique molecular identifiers (UMIs), and <15% mitochondrial gene content were retained. For GSE151530, cells with 300–8000 genes, 1000–80,000 UMIs, and <30% mitochondrial gene content were retained. UMI counts were normalized and variance-stabilized using the SCTransform function. The top 3000 highly variable genes (HVGs) were identified for downstream analysis.
Clustering and Cell Type Annotation
To distinguish malignant hepatocytes, inferCNV22 was applied to predict copy number alterations (CNAs), using non-malignant cell annotations (eg, immune, stromal) from the original GEO metadata as the reference. Batch effects between samples/datasets were corrected using Harmony (v0.1.1),23 and uniform manifold approximation and projection (UMAP) was used for dimensionality reduction and visualization. Graph-based clustering was performed at a resolution of 0.5. Cell clusters were annotated based on the expression of canonical marker genes (Table S1).
Cell Distribution Patterns and Phenotypic Signature Scoring
The spatial distribution of cell types across PBK expression groups was quantified by using the observed-to-expected (R_o/e) cell ratio.24 Expected frequencies were derived from chi-square tests based on overall tissue and cluster distributions. Phenotypic signatures (eg, T/NK cell exhaustion: hepatitis A Virus Cellular Receptor 2(HAVCR2), ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1), layilin(LAYN), LAG3(lymphocyte activation gene 3); co-inhibitory receptor expression: programmed cell death protein 1(PDCD1), cytotoxic T lymphocyte-associated antigen 4(CTLA4), T cell immunoreceptor with immunoglobulin and ITIM domain(TIGIT) were scored at the single-cell level using the AUCell algorithm.25
Differential Expression and Functional Enrichment
Differentially expressed genes (DEGs) between specialized cell clusters or conditions were identified using the “FindMarkers” or “FindAllMarkers” functions in Seurat, with significance defined as an adjusted P-value < 0.05 (Benjamini-Hochberg method). Enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways among DEGs were identified using the cluster Profiler R package.26
Statistical Analysis
Statistical analyses were performed using R software (version 4.1.0). Continuous variables were analyzed using the Mann–Whitney U-test, while categorical variables were assessed using Fisher’s exact test. Bivariate correlations between continuous variables were evaluated using Pearson’s correlation coefficients. Multivariable Cox proportional hazards regression models were employed to estimate hazard ratios (HRs) and corresponding 95% confidence intervals (CIs). Survival curves were generated using the Kaplan-Meier method, and between-group differences were assessed using the Log rank test. For all differentially expressed gene (DEG) analyses, appropriate multiple testing corrections (eg, Benjamini-Hochberg) were applied. Statistical significance was defined as a two-sided P < 0.05 for all analyses.
Results
Identification of PBK as a Prognostic Hub Gene in HCC
HCC is characterized by poor clinical outcomes attributable to its aggressive biological behavior, including high-grade malignancy, propensity for extrahepatic metastasis, and limited therapeutic response. To systematically identify prognostic biomarkers, we integrated weighted gene co-expression network analysis (WGCNA) with clinicopathological parameters. Module-trait association analysis revealed that the turquoise module demonstrated the strongest correlation with survival status (Pearson’s r = 0.16, P < 0.001) and tumor grade (Pearson’s r = 0.37, P < 0.001) in patients diagnosed with HCC (Figure 1A). This module contained 948 candidate genes (Table S2), suggesting their collective involvement in HCC pathogenesis.
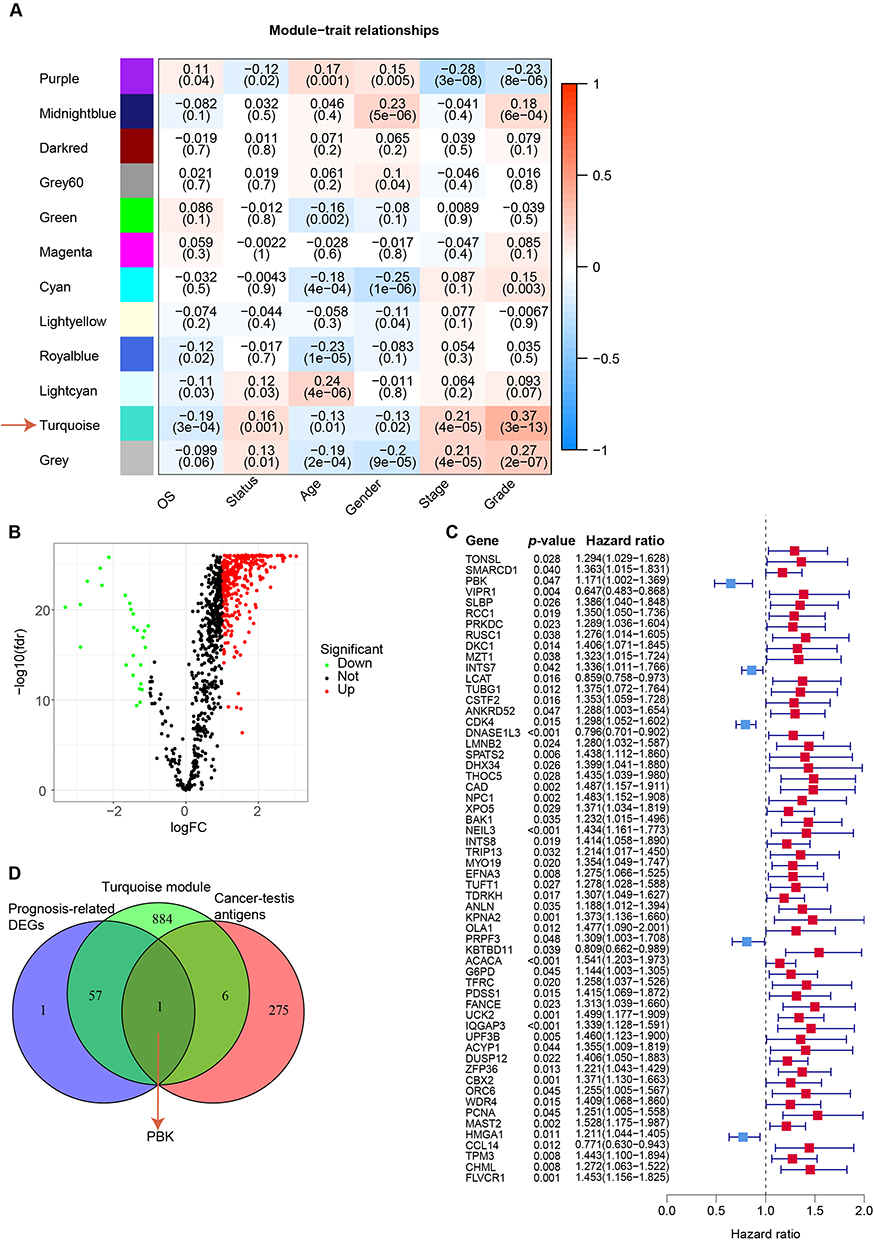 |
Figure 1 Identification of PBK as a hub gene. (A) Correlation of module signature genes with clinical features. The values in the cells are presented as “Pearson r (P-value). (B) Differential gene expression within the turquoise module by tumor grade. Volcano plot showing significantly upregulated (red) and downregulated (green) genes (|log2 (fold change)| > 1, FDR < 0.05). (C) Prognostic impact of PBK expression. Forest plot displaying hazard ratios (HRs) and 95% confidence intervals (CIs) for overall survival. HR > 1 indicates worse prognosis with high PBK expression; dashed line indicates HR = 1. (D) Identification of PBK as the overlapping hub gene. Venn diagram illustrating the intersection of prognosis-associated differentially expressed genes (DEGs) from the turquoise module, cancer-testis antigens (CTAs), and WGCNA-selected genes. |
Subsequent differential expression analysis within the turquoise module identified 340 significantly upregulated and 26 downregulated genes (|log2(fold change)| > 1, false discovery rate [FDR] < 0.05) in HCC tumor tissues compared with paired adjacent non-tumor tissues (Figure 1B and C; complete gene list in Table S3). Given the tumor-specific expression pattern characteristic of CTAs, we integrated 276 experimentally validated CTAs from the Cancer-Testis Antigen Database (CTAdb) into our screening pipeline. Through intersection analysis of prognosis-associated DEGs and CTAs within the turquoise module, PBK emerged as the unique hub gene (Figure 1D). The selection of PBK was further supported by two key considerations: (1) Limited existing data regarding its expression dynamics and functional role in HCC pathogenesis, particularly within tumor microenvironment interactions; (2) Preliminary evidence suggesting its dual regulatory capacity in both cell cycle progression and immune modulation. These knowledge gaps position PBK as a priority candidate for mechanistic exploration in HCC biology.
Elevated PBK Expression Associates with Aggressive HCC Phenotypes and Poor Prognosis
Elevated PBK expression demonstrated significant clinicopathological correlations in HCC. Comparative analysis revealed age-dependent PBK upregulation, with patients < 65 years exhibiting higher mRNA levels than those ≥ 65 years (P = 0.007; Figure 2A). High-grade tumors (G3) showed elevated PBK expression compared to low-grade (G1) lesions (P < 0.001; Figure 2B), while stage III tumors displayed higher PBK levels than stage I tumors (P < 0.001; Figure 2C). Survival analysis further confirmed PBK’s prognostic impact: high expression correlated with significantly reduced median overall survival (P = 0.003), accelerated disease progression (P < 0.001), and early recurrence (P = 0.005; Figure 3D–F). These multi-dimensional associations support PBK as a key driver of HCC aggressiveness and a candidate therapeutic target.
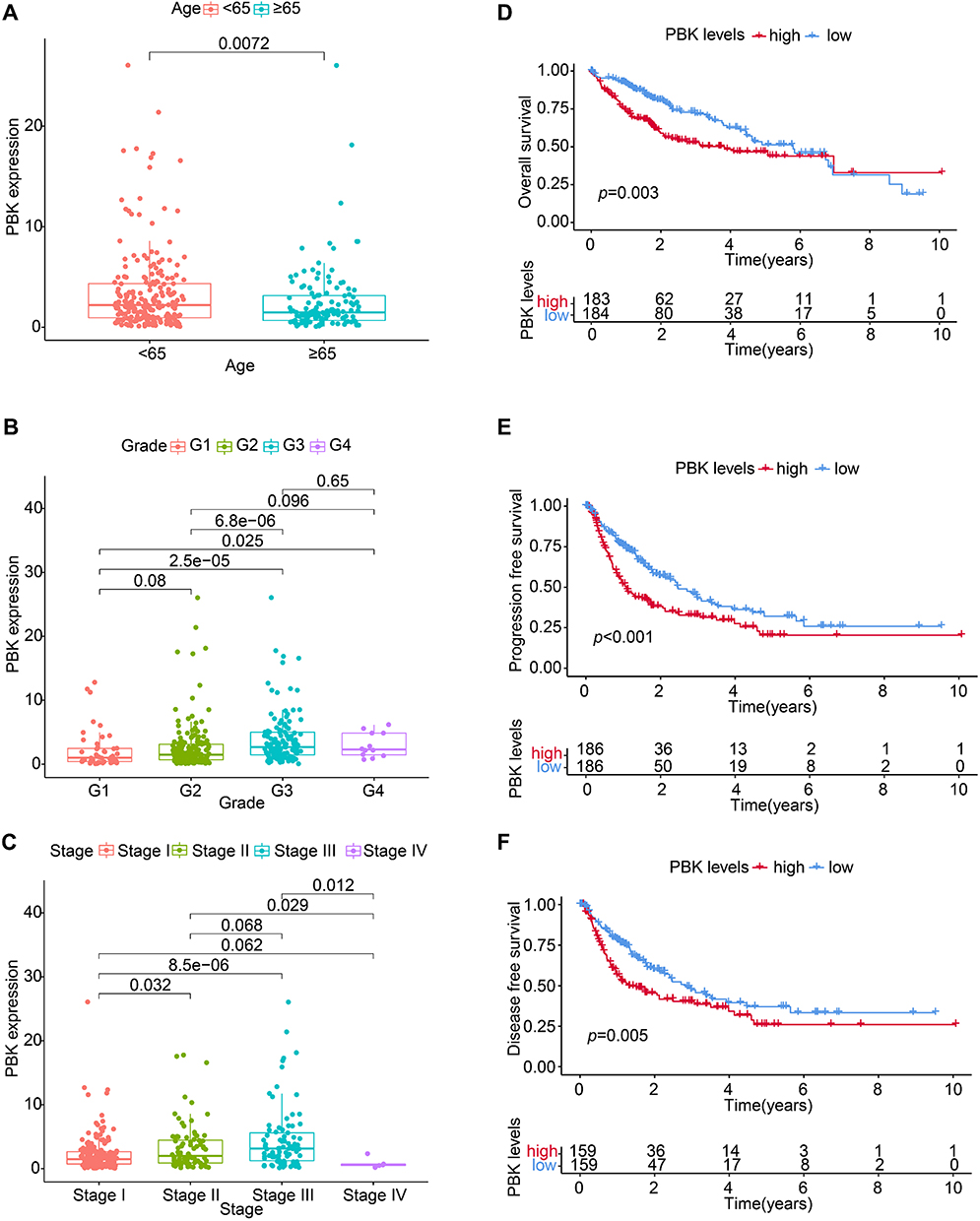 |
Figure 2 Association of PBK expression with clinicopathological features and prognosis in HCC. (A–C) PBK mRNA expression stratified by (A) patient age (< 65 vs ≥ 65 years), (B) tumor grade (G1 vs G3), and (C) tumor stage (I vs III). Boxplots display median, quartiles, and range; P-values indicate significance (Mann–Whitney U-test). (D–F) Kaplan-Meier survival curves comparing patients with high vs low PBK expression (stratified by median): (D) Overall survival (OS), (E) Progression-free survival (PFS), and (F) Disease-free survival (DFS). Log-rank P-values are shown. |
 |
Figure 3 Genomic mutation profiles and tumor mutational burden (TMB) in PBK-high and PBK-low HCC cohorts. (A and B) Mutation spectra of the top 20 most frequently altered genes in the (A) PBK-low and (B) PBK-high groups. Waterfall plots detail mutation types per sample; adjacent bar plots show mutation frequencies and type distributions. Mutation key is provided.(C) Kaplan-Meier overall survival analysis stratified by TMB level (high vs low, based on median).(D) Kaplan-Meier overall survival analysis stratified by combined PBK expression and TMB status. |
Genome Mutation Spectrum and TMB in PBK-High and PBK-Low Groups
Comparative genomic analysis revealed distinct mutation profiles between PBK-high and PBK-low HCC groups. Analysis of the top 20 most frequently mutated genes identified differential mutation frequencies: TP53(39%), CTNNB1(20%), TTN (24%), and MUC16(17%) showed elevated rates in PBK-high tumors, whereas CTNNB1(31%), TTN (23%), TP53(17%), and ALB (15%) were more frequently mutated in PBK-low tumors (Figure 3A and B). Integrated analysis of tumor mutational burden (TMB) demonstrated synergistic prognostic effects: PBK-high/TMB-high patients exhibited the worst median overall survival (P < 0.001), contrasting with PBK-low/TMB-low subgroup outcomes (Figure 3C and D). These findings establish PBK expression as a molecular modulator of oncogenic mutagenesis and TMB-mediated immune evasion in HCC progression.
PBK Regulates Signaling Pathways Involved in the Cell Cycle
Functional enrichment analysis of TCGA-LIHC data revealed PBK’s central role in cell cycle regulation. Gene Set Enrichment Analysis (GSEA) demonstrated significant enrichment pathways related to cell cycle checkpoints, G2/M transition, and DNA replication pathways in PBK-high tumors (Figure 4A). Conversely, Gene Set Variation Analysis (GSVA) identified suppression of metabolic processes in PBK-low group, particularly in branched-chain amino acid catabolism and fatty acid metabolism (Figure 4B and C). The reciprocal regulation between proliferative signaling and metabolic reprogramming suggests that PBK orchestrates HCC progression through dual mechanisms: driving mitotic acceleration while constraining energy metabolism - a hallmark of aggressive tumor phenotypes.
 |
Figure 4 Pathway enrichment analysis associated with PBK expression in HCC. (A) GSVA enrichment scores for hallmark and KEGG pathways in PBK-high vs PBK-low groups.(B) Gene Ontology (GO) enrichment analysis results for the PBK-high group.(C) KEGG pathway enrichment analysis results for the PBK-high group (GSEA). |
PBK Promotes HCC Tumorigenesis via Epithelial-Mesenchymal Transition
Both mRNA and protein levels of PBK were significantly higher in HCC tissues compared to adjacent non-cancerous tissues (Figure 5A, D and E). Immunohistochemical analysis revealed detectable PBK expression in 73.14% of HCC tissues (Figure 5E–F). qRT-PCR analysis showed that among eight tested HCC cell lines, Huh7 cells exhibited the highest expression level of PBK (Figure 5B). Consistent with clinical observations, statistical analysis confirmed that high PBK expression was significantly associated with a poor prognosis in patients diagnosed with hepatocellular carcinoma (P = 0.031, Figure 5G). To further investigate the functional role of PBK, we employed small interfering RNA (siRNA) to knock down PBK expression in Huh7 cells and subsequently evaluated cell migration and invasion capabilities. Results demonstrated that PBK knockdown significantly reduced both the migration and invasion rates of Huh7 cells (Figure 5C and H–J).
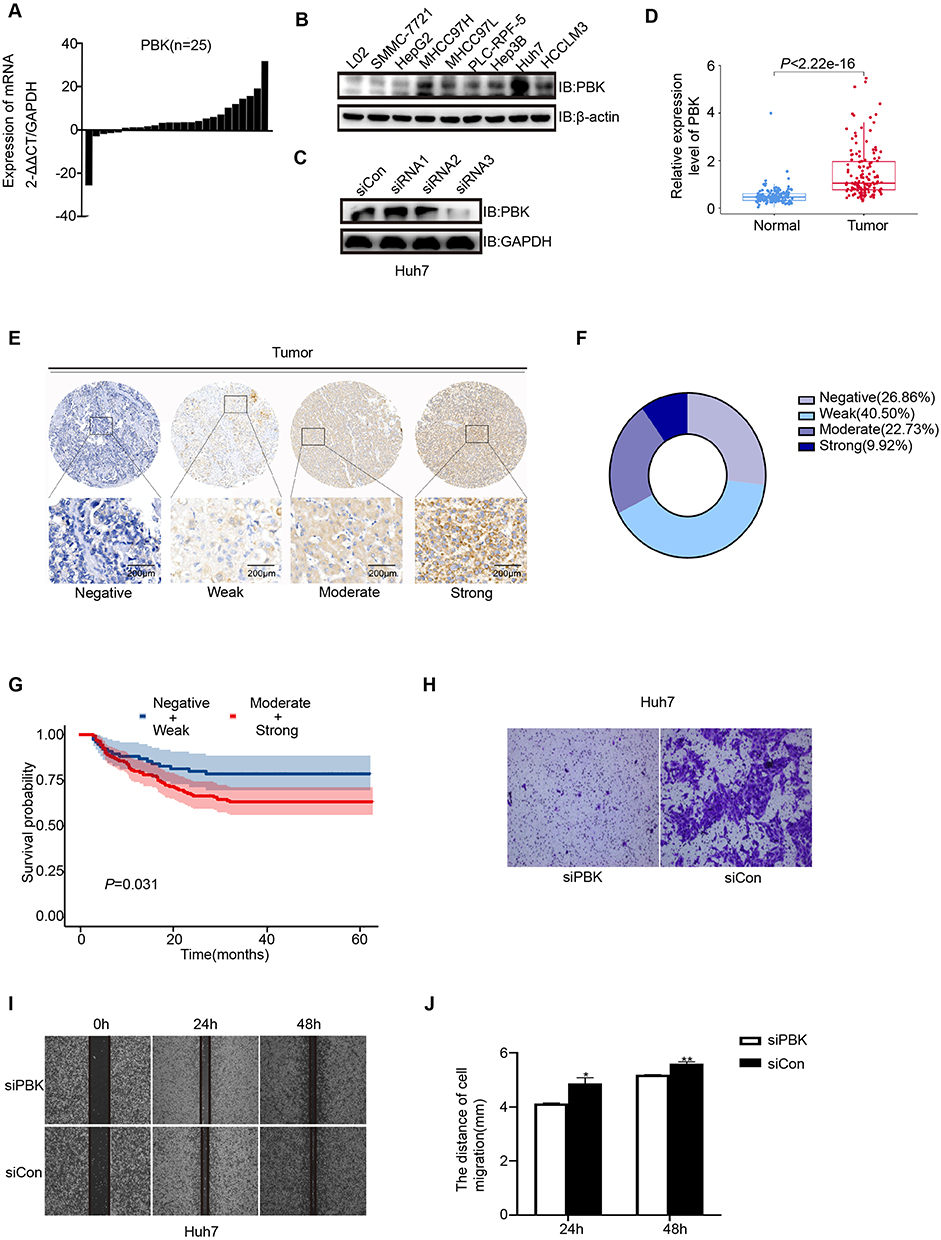 |
Figure 5 Validation of PBK expression and functional role in HCC. (A) PBK mRNA levels across eight HCC cell lines (qRT-PCR).(B) PBK protein levels across nine HCC cell lines (Western blot).(C) PBK protein knockdown validation in Huh7 cells transfected with siRNA (Western blot).(D) PBK protein expression in 159 HCC tumor tissues from the CHCC-HBV cohort (proteomics).(E) Representative immunohistochemical (IHC) staining of PBK in HCC tissue. Scale bar = 200 µm.(F) Distribution of PBK IHC staining intensity in HCC tissues.(G) Kaplan-Meier overall survival analysis based on PBK IHC staining intensity (Negative/Weak vs Medium/Strong).(H) Transwell migration assay comparing control (siCon) and PBK-knockdown (siPBK) Huh7 cells.(I) Representative images from wound healing assays (siCon vs siPBK).(J) Quantitative analysis of wound closure percentage. *P < 0.05, **P < 0.01. |
Interplay Between PDZ-Binding Kinase (PBK) and the Tumor Immune Microenvironment in HCC
To investigate the association between PBK expression and immune heterogeneity in HCC, we performed comparative analyses of immune cell infiltration between PBK-high and PBK-low subgroups. The PBK-high group exhibited elevated infiltration of naïve B cells, monocytes, and M2 macrophages, whereas the PBK-low group showed increased proportions of plasma cells, resting memory CD4(+) T cells, follicular helper T cells, NK cells, M0 macrophages, resting dendritic cells, and stromal cells (Figure 6A). Immune subtype stratification revealed significant enrichment of PBK-high tumors in the C1 and C2 subtypes, while PBK-low tumors predominated in the C3 and C4 subtypes (Figure 6B). Consistent with overall prognostic trends, PBK-high patients exhibited poorer survival within C1/C2 subtypes. Notably, PBK-high tumors displayed significant enrichment of T-cell exhaustion markers, including programmed cell death protein 1(PD-1), programmed death-ligand 1(PD-L1), cytotoxic T lymphocyte-associated antigen 4(CTLA-4), and T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT) (Figure 6C–F). Paradoxically, in the IMvigor210 immunotherapy cohort, elevated PBK expression correlated with improved clinical response to immunotherapy (P = 0.0027; Figure 6G), despite its association with adverse overall survival. Considering PBK’s classification as a cancer-testis antigen and its potential role in MHC-I-mediated antigen presentation, these findings suggest PBK may serve as a dual biomarker, indicative of both an immunosuppressive microenvironments and potential sensitivity to immunotherapy in HCC.
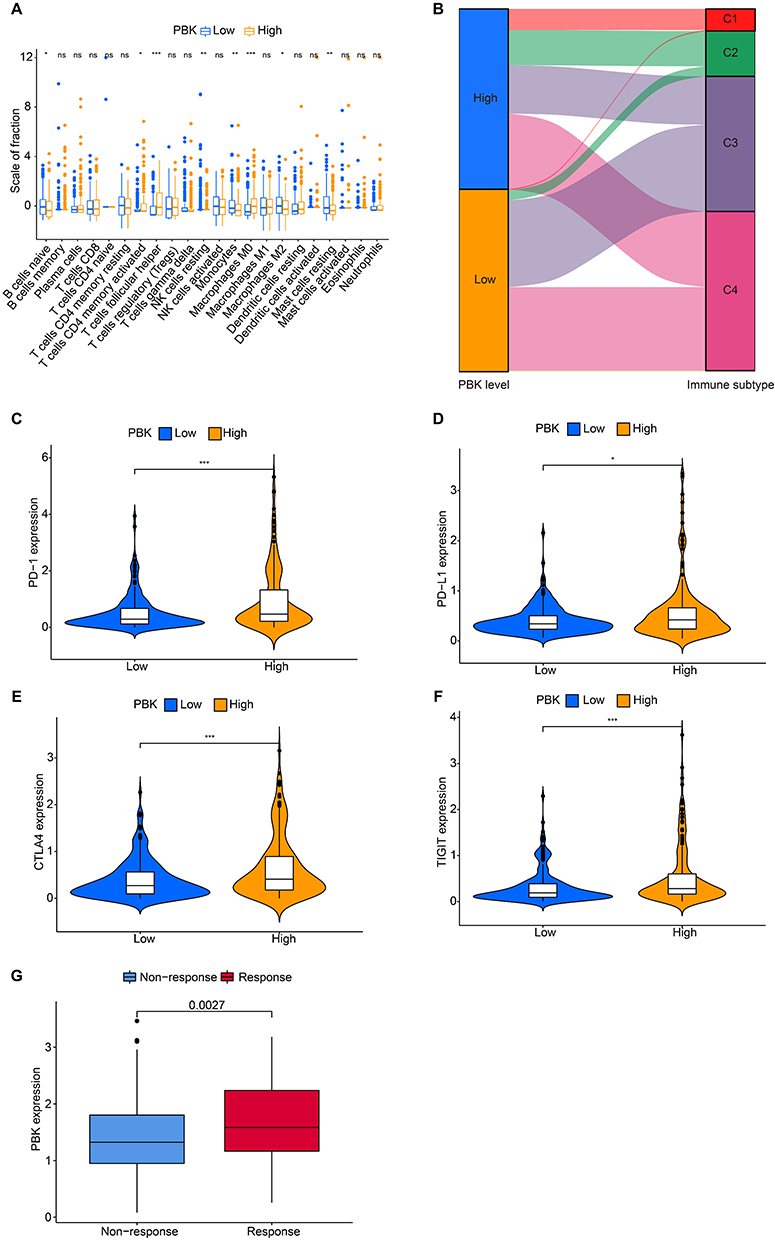 |
Figure 6 Association between PBK expression and the tumor immune microenvironment in HCC.(A) Differential infiltration levels of immune cell subsets in PBK-high vs PBK-low HCC tumors.(B) Sankey diagram illustrating the association between PBK expression groups (high/low) and immune subtypes (C1-C4).(C–F) Correlation between PBK expression and immune checkpoint molecule expression: (C) PD-1 (PDCD1), (D) PD-L1 (CD274), (E) CTLA-4, (F) TIGIT.(G) PBK expression levels in responders vs non-responders to immunotherapy within the IMvigor210 cohort. *P < 0.05, **P < 0.01. ***P < 0.001. |
Single-Cell Profiling Reveals PBK Expression-Driven Heterogeneity in the HCC Tumor Microenvironment
To delineate tumor microenvironment heterogeneity between PBK-high and PBK-low HCC, we analyzed scRNA-seq data from 24 treatment-naïve HCC samples (GSE149614/GSE151530). After rigorous quality control, 119,095 cells were retained (23,687 non-tumor cells, 84,437 tumor cells, 5971 tumor thrombus cells). Utilizing the original dataset annotations, we categorized tumor and tumor thrombus-derived cells into five major lineages, visualized via uniform manifold approximation and projection (UMAP; Figure 7A). PBK-high tumors demonstrated significantly higher infiltration of T/NK and B cells, while PBK-low tumors showed increased proportions of endothelial cells and fibroblasts (Figure 7B). Differentially expressed gene (DEG) analysis identified upregulation of SPP1 in myeloid cells of PBK-high tumors (Figure 7C). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis revealed distinct pathway activation in macrophages and CD8(+)T cells (Figure 7D). Unsupervised clustering of myeloid and T/NK cells resolved eight functional subsets. Notably, PBK-high tumors exhibited higher frequencies of CD8_Trm_ZNF683, CD8_Tem_GZMK, CD4_Treg_FOXP3, and CD4_Tfh_CXCR5 cells, alongside elevated T-cell exhaustion and co-inhibitory receptor scores (P < 0.05; Figure 7E). Conversely, CD8(+) SLC4A10(+) MAIT cells were reduced in PBK-high tumors (4.7% vs 9.82%, FDR-adjusted P < 0.001), while immunosuppressive SPP1(+) macrophages predominated (22.33% vs 6.6%, FDR-adjusted P < 0.001; Figure 7F). These findings suggest that PBK expression drives the remodeling of an immunosuppressive niche in HCC, warranting mechanistic validation.
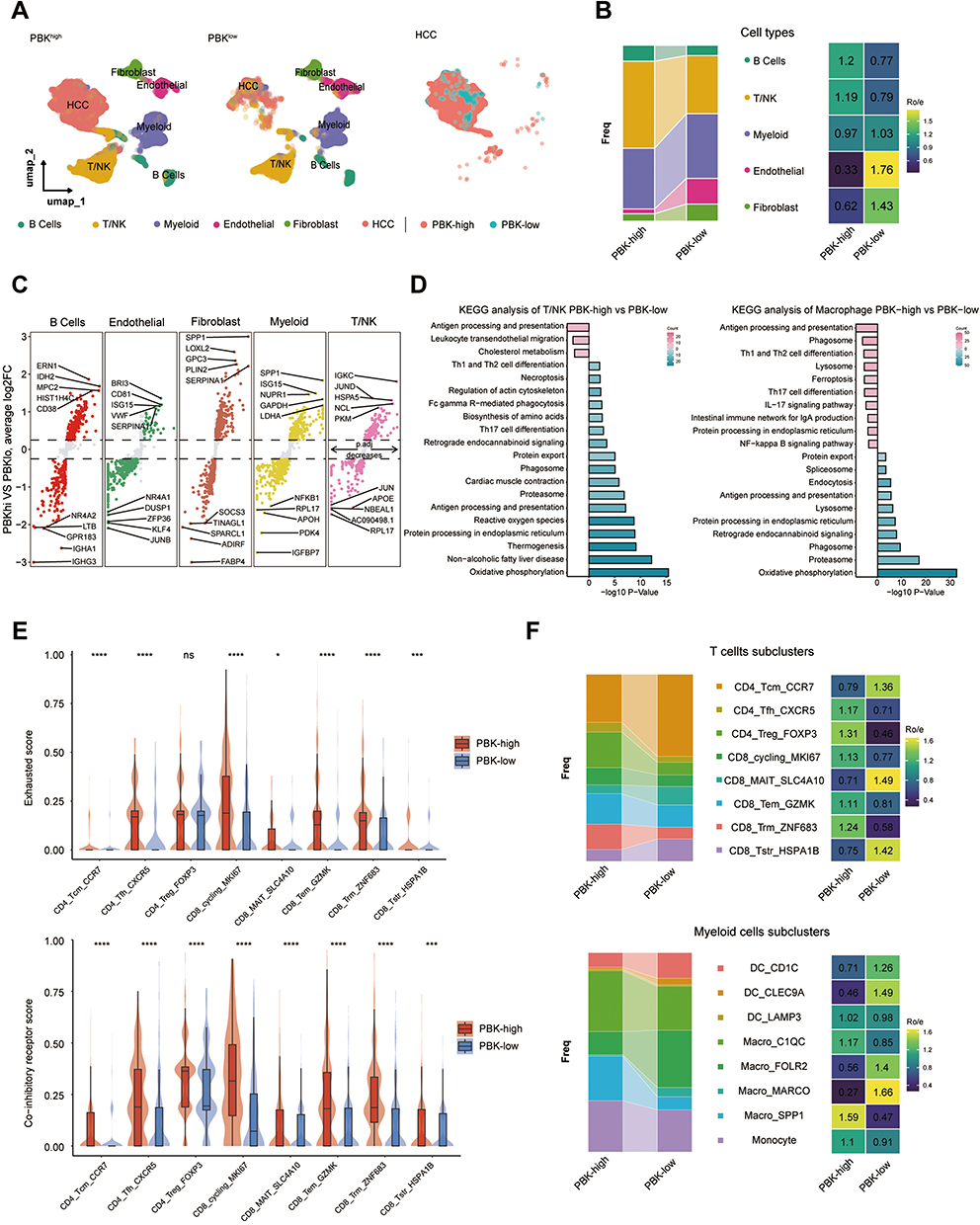 |
Figure 7 Single-cell characterization of the HCC tumor microenvironment stratified by PBK expression. (A) UMAP visualization of major TME cell populations from scRNA-seq data (24 HCC samples): Overall TME (Left), PBK-high vs PBK-low groups (Middle), Malignant cells (Right).(B) Compositional differences in major cell lineages between PBK-high and PBK-low tumors (Bar plot and Heatmap). (C) Differentially expressed genes (DEGs) between PBK-high and PBK-low tumors (Volcano plot). (D) KEGG pathway enrichment in CD8(+) T cells and macrophages.(E) Exhaustion scores and co-inhibitory receptor scores in T/NK cells (Violin plots).(F) Distribution frequencies of T cell subsets and macrophage subsets between groups (Bar plot and Heatmap). *P < 0.05, ***P < 0.001, ****P < 0.001. |
PBK as a Pan-Cancer Prognostic Biomarker
To evaluate the pan-cancer role of PBK, we analyzed its expression across 33 human malignancies. PBK was consistently overexpressed in all tumor types examined (Figure 8A and B) and its expression strongly correlated with advanced clinical stages and higher tumor grades, underscoring its prognostic relevance (Figure 8C and D). Univariate analysis of 10,071 TCGA samples revealed that elevated PBK expression was associated with increased mortality risk in cholangiocarcinoma (CHOL), renal cancers (KICH/KIRC/KIRP), low-grade glioma (LGG), HCC, lung adenocarcinoma (LUAD), mesothelioma (MESO), pancreatic adenocarcinoma (PAAD), pheochromocytoma/paraganglioma (PCPG), and sarcoma (SARC). Conversely, higher PBK levels were associated with improved survival in ovarian cancer (OV) and thymoma (THYM) (Figure 8E). Leveraging the TISIDB database, we further identified widespread significant positive correlations between PBK expression and the infiltration of activated CD4(+) T cells and Th2 cells across 30 cancers, implicating PBK as a potential regulator of tumor immune microenvironment (Figure 8F).
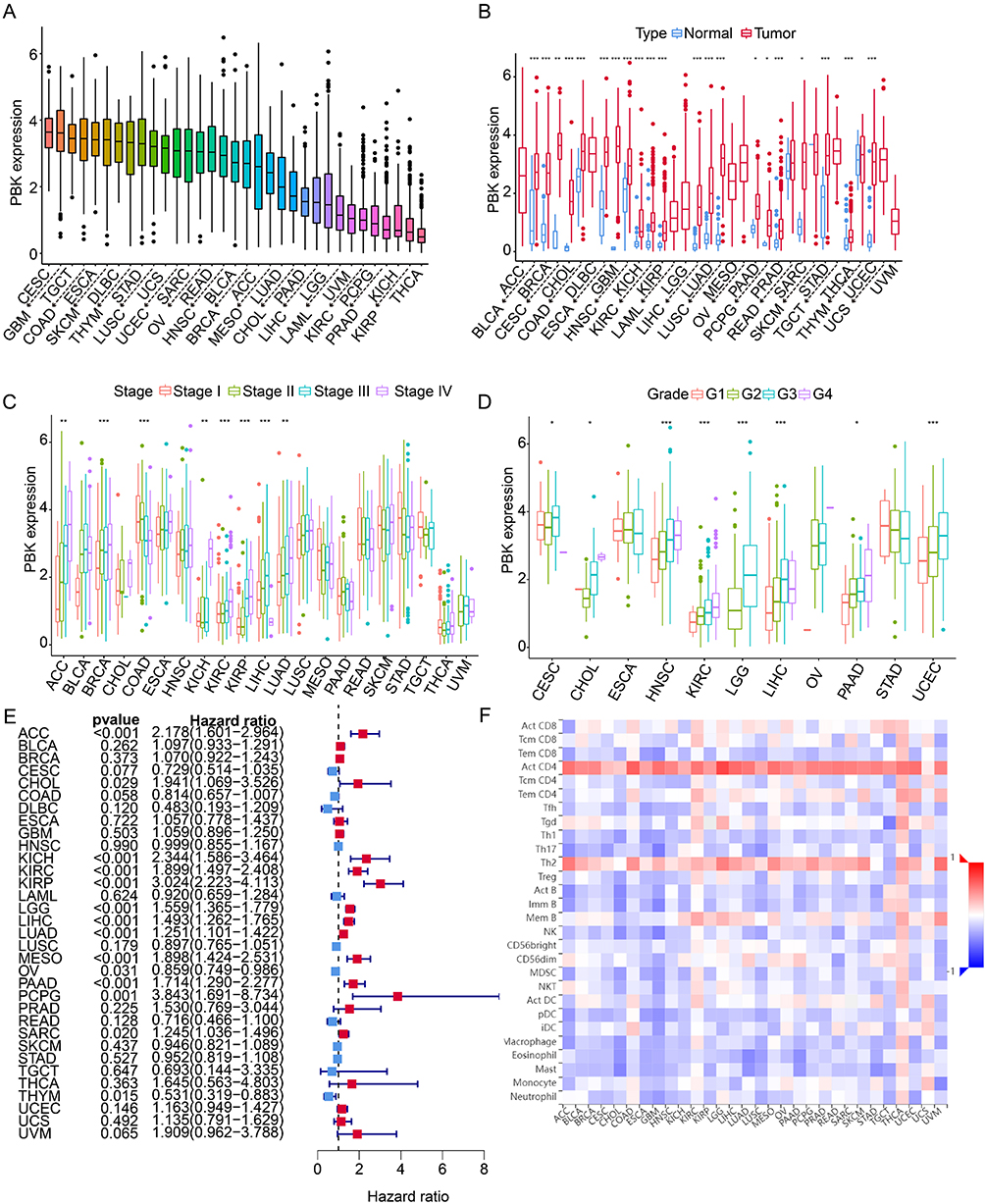 |
Figure 8 Pan-cancer analysis of PBK expression and its clinical significance. (A) PBK mRNA expression levels across 33 tumor types (TCGA).(B) Differential PBK expression between tumor and adjacent normal tissues.(C and D) Association between PBK expression and (C) tumor grade or (D) clinical stage.(E) Forest plot of univariate Cox regression analysis for overall survival. HR > 1 indicates worse prognosis with high PBK expression; dashed line indicates HR = 1.(F) Correlation between PBK expression and immune cell infiltration levels across 30 cancer types (TISIDB database). *P < 0.05, ***P < 0.001, ****P < 0.001. |
Discussion
Our integrative multiomics analysis establishes PBK as a central regulator of HCC pathogenesis, with critical roles in tumor proliferation, genomic instability, and immune microenvironment modulation. Mechanistically, PBK coordinates cell cycle progression through dual pathways: (1) activation of the polycomb repressor complex 1 (PRC1) to ensure mitotic spindle formation and chromosomal segregation,27 (2) suppression of p53-mediated tumor suppression via p21 downregulation.28 These oncogenic functions are corroborated by pathway enrichment analyses demonstrating significant activation of cell cycle-related signaling in PBK-high tumors. The therapeutic relevance of these findings is further evidenced by PBK knockdown experiments showing reduced tumor growth through p38 pathway modulation and enhanced DNA damage repair.29
Notably, PBK expression shows a significant correlation with elevated tumor mutation burden (TMB) and TP53 mutation prevalence, indicating its potential role in driving genomic instability. This association mirrors observations in lung adenocarcinoma where PBK synergizes with mutant p53 to promote carcinogenesis.30 The genomic destabilization induced by PBK likely accelerates clonal evolution through accumulation of driver mutations,31 providing a plausible mechanism for its association with aggressive HCC phenotypes.
Tumor microenvironment analysis further reveals PBK’s immunosuppressive influence, characterized by enrichment of SPP1(+) macrophages (marked by pro-angiogenic signatures and MHC-II suppression) and exhausted CD8(+) T cells. This immunoevasive landscape presents a notable therapeutic paradox, as PBK-high tumors nevertheless exhibit enhanced responsiveness to immune checkpoint blockade (ICB). We hypothesize that PBK’s cancer-testis antigen properties may bolster tumor immunogenicity transiently, while its long-term effects induce chronic immunosuppressive remodeling, creating a TME primed for ICB-mediated reinvigoration of exhausted T cells. However, the precise molecular crosstalk between PBK and immune checkpoint regulation warrants further investigation.
Several limitations merit consideration: First, the signaling networks linking PBK to immune cell polarization remain elusive. Second, while bioinformatics analyses and in vitro models provide valuable mechanistic insights, preclinical validation in immunocompetent HCC models is essential. Third, the predictive value of PBK expression for ICB response requires prospective validation in dedicated clinical cohorts. Finally, the functional heterogeneity of PBK across HCC molecular subtypes warrants systematic exploration.
Conclusion
Our multiomics framework defines PBK as both a key driver of HCC oncogenesis and a factor that correlates with immune checkpoint responsiveness. By simultaneously coupling mitotic regulation with TME remodeling, PBK emerges as a promising target for precision immunotherapy in HCC. Future studies should prioritize the mechanistic dissection of PBK’s immunomodulatory functions and the clinical validation of its biomarker potential to realize its translational relevance.
Abbreviations
PBK, PDZ-binding kinase; HCC, hepatocellular carcinoma; CTA, cancer/testis antigen; MAGEA,melanoma-associated antigen A ;MAGEC2,melanoma-associated antigen C2;NY-ESO-1,the New York esophageal squamous cell carcinoma; qRT‒PCR, reverse transcription polymerase chain reaction; MAPKK, mitogen-activated protein kinase; TCGA, The Cancer Genome Atlas; WGCNA, weighted gene co-expression network analysis; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; GSEA, Gene Set Enrichment Analysis; GSVA, Gene Set Variation Analysis; ESTIMATE, Estimation of Stromal and Immune cells in Malignant Tumor Tissues using Expression data; TMB, tumor mutation burden; OS, overall survival, PFS, progression-free survival; ICB, immune checkpoint blockade; HAVCR2, hepatitis A Virus Cellular Receptor 2; ENTPD1, ectonucleoside triphosphate diphosphohydrolase 1 gene; LAYN, layilin, ; LAG3, lymphocyte activation gene 3; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; CTLA-4, cytotoxic T lymphocyte-associated antigen 4; Tcm, central memory T cells; Tfh, follicular helper T cells; Treg, regulatory T cells; MAIT, mucosal-associated invariant T cells; Tem, effector memory T cells; Trm, tissue-resident Memory T cells; Tstr, stressed T cells; DC, dendritic cells; Macro, macrophages; ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast carcinoma; CESC, cervical squamous cell carcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; DLBC, diffuse large B cell lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; LGG, low-grade glioma; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukaemia; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma.
Data Sharing Statement
Data will be made available on request.
Ethics
This study was conducted in accordance with the Declaration of Helsinki. Utilization of the hepatocellular carcinoma tissue microarray (TMA) received approval from the Institutional Review Board of Zhongshan Hospital, Fudan University (Approval No. B2021-065R).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Juan Zhang, Maopei Chen, Guohe Song were supported by National Natural Science Foundation of China [81400768 & 81802360 & 82203643]. Guohe Song was supported by Science and Technology Innovation Plan of Shanghai Science and Technology Commission [22YF1407200]. Xiaojian Ni was supported by Shanghai Municipal Commission of Science and Technology General Project[22ZR1457900] and Natural Science Foundation of Fujian Province(2022J05328). Boheng Zhang was supported by the Foundation of the Natural Science Foundation of Fujian Province [2022J011428].
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. 2024;74(3):229–263. doi:10.3322/caac.21834
2. Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380(15):1450–1462. doi:10.1056/NEJMra1713263
3. Altaf S, Saleem F, Sher AA, et al. Potential therapeutic strategies to combat HCC. Curr Mol Pharmacol. 2022;15(7):929–942. doi:10.2174/1874467215666220103111009
4. der Bruggen P V, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254(5038):1643–1647. doi:10.1126/science.1840703
5. Xu Y, Xu X, Ni X, et al. Gene-based cancer-testis antigens as prognostic indicators in hepatocellular carcinoma. Heliyon. 2023;9(3). doi:10.1016/j.heliyon.2023.e17506
6. Cheng YH, Wong EW, Cheng CY. Cancer/testis (CT) antigens, carcinogenesis and spermatogenesis. Spermatogenesis. 2011;1(3):209–220. doi:10.4161/-spmg.1.3.17990
7. Almeida LG, Sakabe NJ, deOliveira AR, et al. CTdatabase: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 2009;37(suppl_1):D816–D819. doi:10.1093/nar/gkn673
8. Gordeeva O. Cancer-testis antigens: unique cancer stem cell biomarkers and targets for cancer therapy. In: Seminars in Cancer Biology. Vol. 53. Elsevier;2018:75–89. doi:10.1016/j.semcancer.2018.08.006
9. Kobayashi Y, Higashi T, Nouso K, et al. Expression of MAGE, GAGE and BAGE genes in human liver diseases: utility as molecular markers for hepatocellular carcinoma. J Hepatol. 2000;32(4):612–617. doi:10.1016/S0168-8278(00)80223-8
10. Suzuki K, Tsujitani S, Konishi I, et al. Expression of MAGE genes and survival in patients with hepatocellular carcinoma. Int J Oncol. 1999;15(6):1227–1259. doi:10.3892/ijo.15.6.1227
11. Luo G, Huang S, Xie X, et al. Expression of cancer-testis genes in human hepatocellular carcinomas. Cancer Immun. 2002;2(1):1.
12. Xu H, Gu N, Liu ZB, et al. NY-ESO-1 expression in hepatocellular carcinoma: a potential new marker for early recurrence after surgery. Oncol Lett. 2012;3(1):39–44. doi:10.3892/ol.2011.441
13. Qiu G, Fang J, He Y. 5′ CpG island methylation analysis identifies the MAGE-A1 and MAGE-A3 genes as potential markers of HCC. Clin Biochem. 2006;39(3):259–266. doi:10.1016/j.clinbiochem.2006.01.014
14. Riener MO, Wild PJ, Soll C, et al. Frequent expression of the novel cancer testis antigen MAGE-C2/CT-10 in hepatocellular carcinoma. Int J Cancer. 2009;124(2):352–357. doi:10.1002/ijc.23966
15. Esfandiary A, Ghafouri-Fard S. MAGE-A3: an immunogenic target used in clinical practice. Immunotherapy. 2015;7(6):683–704. doi:10.2217/imt.15.29
16. Cui Z, Li Y, Gao Y, et al. Cancer-testis antigen lactate dehydrogenase C4 in hepatocellular carcinoma: a promising biomarker for early diagnosis, efficacy evaluation and prognosis prediction. Aging. 2020;12(19):19455. doi:10.18632/aging.103879
17. Ren X, Zhang L, Zhang Y, et al. Insights gained from single-cell analysis of immune cells in the tumor microenvironment. Annu Rev Immunol. 2021;39(1):583–609. doi:10.1146/annurev-immunol-110519-071134
18. Kurachi M. CD8+ T cell exhaustion. Semin Immunopathol. 2019;41(3):327–337. doi:10.1007/s00281-019-00744-5
19. Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–548. doi:10.1038/nature25501
20. Lu Y, Yang A, Quan C, et al. A single-cell atlas of the multicellular ecosystem of primary and metastatic hepatocellular carcinoma. Nat Commun. 2022;13(1):4594. doi:10.1038/s41467-022-32283-3
21. Ma L, Heinrich S, Wang L, et al. Multiregional single-cell dissection of tumor and immune cells reveals stable lock-and-key features in liver cancer. Nat Commun. 2022;13(1):7533. doi:10.1038/s41467-022-35291-5
22. Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. doi:10.1126/science.1254257
23. Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16(12):1289–1296. doi:10.1038/s41592-019-0619-0
24. Zhang L, Yu X, Zheng L, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564(7735):268–272. doi:10.1038/s41586-018-0694-x
25. Aibar S, González-Blas CB, Moerman T, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–1086. doi:10.1038/nmeth.4463
26. Wu T, Hu E, Xu S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2(3):100141. doi:10.1016/j.xinn.2021.100141
27. Huang H, Lee MH, Liu K, et al. PBK/TOPK: an effective drug target with diverse therapeutic potential. Cancers. 2021;13(9):2232. doi:10.3390/cancers13092232
28. He F, Yan Q, Fan L, et al. PBK/TOPK in the differential diagnosis of cholangiocarcinoma from hepatocellular carcinoma and its involvement in prognosis of human cholangiocarcinoma. Human Pathol. 2010;41(3):415–424. doi:10.1016/j.humpath.2009.05.016
29. Ayllón V, O’Connor R. PBK/TOPK promotes tumor cell proliferation through p38 MAPK activity and regulation of the DNA damage response. Oncogene. 2007;26(24):3451–3461. doi:10.1038/sj.onc.1210142
30. Lei B, Qi W, Zhao Y, et al. PBK/TOPK expression correlates with mutant p53 and affects patients’ prognosis and cell proliferation and viability in lung adenocarcinoma. Human Pathol. 2015;46(2):217–224. doi:10.1016/j.humpath.2014.07.026
31. Zlobec I, Molinari F, Kovac M, et al. Prognostic and predictive value of TOPK stratified by KRAS and BRAF gene alterations in sporadic, hereditary and metastatic colorectal cancer patients. Br J Cancer. 2010;102(1):151–161. doi:10.1038/sj.bjc.6605452
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Decaprenyl Diphosphate Synthase Subunit 1 (PDSS1): A Potential Prognostic Biomarker and Immunotherapy-Target for Hepatocellular Carcinoma
Yang Y, Li J, Tang M, Nie B, Huang W
Cancer Management and Research 2022, 14:1627-1639
Published Date: 3 May 2022
Identification of KRBA1 as a Potential Prognostic Biomarker Associated with Immune Infiltration and m6A Modification in Hepatocellular Carcinoma
Liu Y, Fu B, Yu Z, Song G, Zeng H, Gong Y, Ding Y, Huang D
Journal of Hepatocellular Carcinoma 2022, 9:497-516
Published Date: 31 May 2022
Low MARCO Expression is Associated with Poor Survival in Patients with Hepatocellular Carcinoma Following Liver Transplantation
Zhang Q, Wei Y, Li Y, Jiao X
Cancer Management and Research 2022, 14:1935-1944
Published Date: 11 June 2022
Histological Severity of Cirrhosis Influences Surgical Outcomes of Hepatocellular Carcinoma After Curative Hepatectomy
Liang BY, Gu J, Xiong M, Zhang EL, Zhang ZY, Lau WY, Wang SF, Guan Y, Chen XP, Huang ZY
Journal of Hepatocellular Carcinoma 2022, 9:633-647
Published Date: 23 July 2022
Bioinformatics Analysis of the Key Genes and Pathways in Multiple Myeloma

Sheng X, Wang S, Huang M, Fan K, Wang J, Lu Q
International Journal of General Medicine 2022, 15:6999-7016
Published Date: 5 September 2022