")
Back to Journals » Journal of Inflammation Research » Volume 17
Mitophagy and Ferroptosis in Sepsis-Induced ALI/ARDS: Molecular Mechanisms, Interactions and Therapeutic Prospects of Medicinal Plants
Authors Cheng H , Wang X, Yao J, Yang C, Liu J
Received 26 July 2024
Accepted for publication 17 October 2024
Published 29 October 2024 Volume 2024:17 Pages 7819—7835
DOI https://doi.org/10.2147/JIR.S488655
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Huixin Cheng,1 Xuehan Wang,1 Juyi Yao,2 Chunbo Yang,3 Jian Liu1,4
1The First Clinical Medical College of Lanzhou University, Lanzhou, Gansu Province, People’s Republic of China; 2Traditional Chinese Medicine Hospital of Xinjiang Uygur Autonomous Region, Urumqi, People’s Republic of China; 3Department of Critical Medicine Center, First Affiliated Hospital of Xinjiang Medical University, Urumqi, People’s Republic of China; 4Department of Intensive Care Unit, Gansu Provincial Maternity and Child Health Hospital/Gansu Provincial General Hospital, Lan Zhou, Gansu Province, People’s Republic of China
Correspondence: Jian Liu, The First Clinical Medical College of Lanzhou University, No. 222 Tianshui South Road, Lanzhou, Gansu Province, 730013, People’s Republic of China, Email [email protected]
Abstract: Sepsis is a common critical illness characterized by high mortality rates and a significant disease burden. In the context of sepsis-induced organ dysfunction, the lungs are among the initial organs affected, which may progress to acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Recent studies have highlighted the crucial roles of mitophagy and ferroptosis in the development and progression of sepsis-induced ALI/ARDS. Identifying key convergence points in these processes may provide valuable insights for the treatment of this condition. In recent years, certain herbs and their bioactive compounds have demonstrated unique benefits in managing sepsis-induced ALI/ARDS by modulating mitophagy or ferroptosis. This review summary the mechanisms of mitophagy and ferroptosis, explores their interactions, and emphasizes their regulatory roles in the progression of sepsis-induced ALI/ARDS. Additionally, it offers a novel perspective on treatment strategies by summarizing various herbs and their bioactive compounds relevant to this condition.
Keywords: sepsis, acute lung injury, acute respiratory distress syndrome, mitophagy, ferroptosis
Sepsis is defined as a life-threatening organ dysfunction syndrome caused by a dysregulated immune response to infection (sepsis 3.0).1 The Health Institute of Metrology and Evaluation conducted an epidemiological study on the global burden of sepsis, reporting 48.9 million new cases and 1.1 million sepsis-related deaths worldwide in 2017, accounting for 19.7% (18.2%–21.4%) of global deaths.2 Despite significant progress in recent years in anti-infective therapy, fluid resuscitation, and organ support technologies, the incidence of sepsis continues to rise, posing a serious threat to human health and placing a heavy burden on the global healthcare system. In sepsis-induced organ dysfunction, the lungs are among the first organs affected, characterized by the disruption of endothelial barrier integrity and injury to alveolar epithelium, eventually leading to acute lung injury (ALI) or acute respiratory distress syndrome (ARDS).3,4 ALI and ARDS represent different stages in the progression of the same disease, with ALI indicating the early stage. Without timely and effective treatment, ALI can progress to ARDS, characterized by acute hypoxemia, bilateral pulmonary infiltrates, and potentially fatal outcomes.5 Therefore, it is essential to actively investigate the pathogenesis and potential therapeutic targets for sepsis-induced ALI/ARDS.
Cell death serves as a crucial response mechanism for the body to adapt to external environmental stress and internal abnormalities, playing a pivotal role in the processes of growth, development, and the maintenance of tissue homeostasis. Cell death can be categorized into various forms, including programmed cell death (such as apoptosis) and non-programmed cell death (such as necrosis).6 In recent years, the discovery of new forms of cell death, such as ferroptosis, cuproptosis, mitophagy, immunogenic cell death and disulfidptosis, has further expanded our understanding of the complexity of cell death mechanisms.7 Mitophagy is a crucial form of selective autophagy vital for maintaining cellular and mitochondrial homeostasis.8 Mitochondria, often referred to as the power sources of the cell, regulate numerous processes, including oxidative phosphorylation, energy production, inflammatory response, and apoptosis. However, mitochondrial damage can disrupt cellular homeostasis, increase the production of reactive oxygen intermediates, lead to oxidative stress, and promote the activation of inflammatory pathways.9 Therefore, maintaining mitochondrial homeostasis is essential for the health of both cells and organisms.10 Mitophagy servers as a primary mechanism for removing damaged mitochondria through autophagic flux and can be triggered by various stimuli, such as the accumulation of reactive oxygen species (ROS) and hypoxia.11 Mitophagy can be activated through a variety of pathways, including both ubiquitin (Ub)-dependent and Ub-independent mechanisms.12 When cells undergo pathological alterations, damaged or dysfunctional mitochondria generate excessive ROS and release pro-apoptotic factors, causing metabolic changes, cellular damage, and cell death. Nevertheless, disruption of mitophagy impairs the body’s ability to eliminate these damaged mitochondria, further contributing to the onset of disease. Recently, numerous studies suggested that the destruction of mitophagy is related to a variety of diseases, such as inflammation, neurodegenerative diseases, cardiovascular diseases, and tumors.13 The discovery of mitophagy’s role in human pathology and aging has made it a topic of considerable research interest.
Ferroptosis is an intricate form of cell death regulated by iron ions, initially proposed by Dixon in 2012. In recent years, with the deepening of research, significant progress has been made in elucidating the molecular mechanisms controlling the induction and regulation of ferroptosis.14 Studies indicate that iron and ROS generated from lipid and iron metabolism are pivotal in this process. The process involves phospholipid peroxidation of unsaturated fatty acids and the formation of lipid hydroperoxides. The generated hydroperoxides interact with iron ions, leading to the production of free radicals that subsequently drive ferroptosis.15 Therefore, previous investigations focused on cellular metabolism and revealed the intimate link between ferroptosis and metabolic pathways. In recent years, researchers have discovered the critical role of ferroptosis in tumor suppression and immune surveillance based on its potential biological functions. Additionally, excessive ferroptosis may lead to organ damage, neurodegenerative diseases, ischemia/reperfusion injury, and other disorders. Hence, ferroptosis holds promise as a potential therapeutic target for the treatment of various diseases.16
There is an important interplay between mitophagy and ferroptosis. As research advances, more scholars are connecting these processes to investigate their correlation and mechanisms. Initially, mitophagy plays a crucial role in either promoting or inhibiting ferroptosis in cells. Under mild stress and early iron overload conditions, mitophagy can sequester iron within mitochondria, reducing ROS production and thereby alleviating ferroptosis.17,18 However, extensive mitophagy may promote lipid peroxidation (LPO) and ferroptosis by increasing additional iron. Therefore, mitophagy may exact varying effects on ferroptosis depending on autophagic flux. Similarly, ferroptosis also affects mitophagy. Research indicates that during ferroptosis, increased oxidative stress levels can destabilize mitochondria, triggering mitophagy. Moreover, certain ferroptosis inhibitors may also influence mitophagy.19 In conclusion, an in-depth exploration of the mechanisms connecting these processes provides valuable insights into various diseases, including tumors, inflammation, and cardiovascular diseases, from different perspectives.
The pathogenesis of ALI/ARDS induced by sepsis is extremely complex. Initially, inflammatory cell infiltration, oxidative stress, and vascular leakage were considered key factors contributing to its onset. As research progresses, new forms of cell death such as pyroptosis, ferroptosis, and mitophagy, have been identified concerning sepsis-induced ALI/ARDS.20–22 Mitochondrial dysfunction may lead to sepsis-induced organ dysfunction, and studies have shown that the increased damaged mitochondria in alveolar epithelium can activate nuclear factor erythroid 2-related factor 2 (Nrf2), thereby inducing mitophagy and alleviating sepsis-induced ALI/ARDS.23 Jiang et al24 suggested that Sirtuin 1 (SIRT1) has the capability to enhance mitophagy in pulmonary endothelial cells, thereby limiting excessive activation of stimulator of interferon genes (STING) and NOD-like receptor thermal protein domain associated protein 3 (NLRP3), potentially serving as a therapeutic target for this disease. Additionally, ferroptosis, a form of iron-dependent programmed cell death characterized by the accumulation of LPO, plays a significant role in the development of sepsis-induced ALI/ARDS. CircEXOC5 has been found to promote ferroptosis through the insulin-like growth factor 2 mRNA binding protein 2 (IGF2BP2)/activating transcription factor 3 (ATF3) axis, exacerbating sepsis-induced ALI,25 whereas yes-associated protein 1 (YAP1) can alleviate sepsis-induced ALI by inhibiting ferroptosis mediated by ferritinophagy.26 Therefore, actively seeking potential targets to promote mitophagy and inhibit ferroptosis may provide new ways for the treatment of sepsis-induced ALI/ARDS. Traditional herbs and their bioactive compounds have been utilized for thousands of years. Recent advancements in biochemical and pharmacological techniques have facilitated more comprehensive investigations into the active components of these herbs and their mechanisms of action. Research has shown that herbs and their bioactive compounds can offer distinct advantages in treating various diseases and providing organ protection by modulating mitophagy and ferroptosis, particularly in the context of inflammatory diseases, tumors, and strokes.27–29 Currently, there is extensive focus on the role of mitophagy and ferroptosis in sepsis-induced ALI/ARDS, with some herbs demonstrating effectiveness in targeting these pathways for disease treatment.
In conclusion, the regulation of mitophagy and ferroptosis is pivotal in the pathogenesis and progression of sepsis. Promoting mitophagy and inhibiting ferroptosis to a certain extent may provide a novel approach for treating sepsis-induced ALI/ARDS. This review provides a comprehensive overview of the mechanisms and potential therapeutic targets of mitophagy, ferroptosis and their interactions in sepsis-induced ALI/ARDS. The aim is to establish a theoretical foundation for further investigations into the interplay between mitophagy and ferroptosis in sepsis, facilitating a nuanced understanding of the intricate network mechanisms involved. Additionally, this article delves into the therapeutic potential of herbs and their bioactive compounds that target mitophagy and ferroptosis in sepsis-induced ALI/ARDS, aiming to offer a fresh perspective on its treatment.
Mechanism of Mitophagy
Mitophagy is essential for controlling the quantity of mitochondria in cells and preserving their proper function. Since the concept was first proposed, researchers have been actively exploring its regulatory mechanism. Currently, the mechanisms of mitophagy are generally categorized into two main pathways: Ub-dependent pathway and Ub-independent pathway (Figure 1). In this section, we will focus on elucidating the molecular mechanisms underlying these two distinct types of mitophagy.
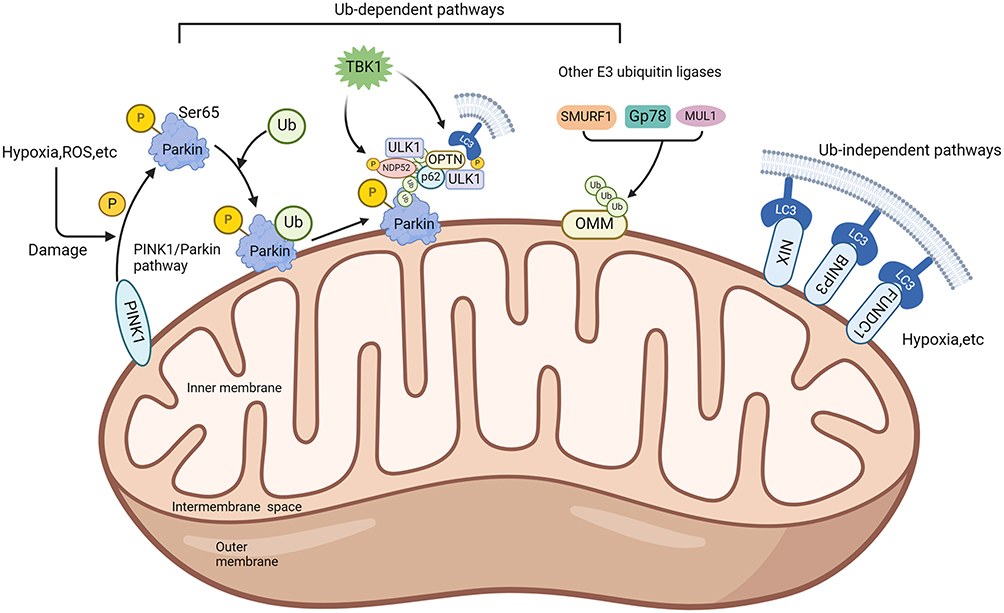 |
Figure 1 Overview of the mitophagy mechanisms. The mechanisms of mitophagy are generally categorized into two main pathways: Ub-dependent pathway and Ub-independent pathway. The black arrow indicates activation. Created in BioRender. Cheng, H. (2024) BioRender.com/g89m244. Abbreviations: ROS, reactive oxygen species; PINK1, PTEN-induced putative kinase 1; Ub, ubiquitin; TBK1, TANK-binding kinase 1; ULK1, UNC51-like kinase-1; OPTN, optineurin; NDP52, nuclear dot protein 52; SMURF1, smad ubiquitination regulation factor-1; GP78, glycoprotein 78; MUL1, mitochondrial E3 ubiquitin ligase 1; NIX, Nip3-like protein X; BNIP3, BCL2/adenovirus E1B 19kDa interacting protein-3; FUNDC1, FUN14 domain containing 1; OMM, outer mitochondrial membrane; LC3, microtubule-associated protein 1A/1B-light chain 3. |
Ub-Dependent Pathway
The Ub-dependent pathway facilitates mitophagy by ubiquitinating various mitochondrial surface proteins. The PTEN-induced kinase 1 (PINK1) /Parkin (PARK2) pathway represents the most extensively researched mechanism for eliminating damaged mitochondria in mammals. PINK1, encoded by the PARK6 gene, is typically continuously translocated to the inner mitochondrial membrane, where it is subsequently released into the cytoplasm for degradation by the ubiquitin proteasome system.30 When the mitochondrial membrane potential is compromised, PINK1 accumulates on the outer mitochondrial membrane through the translocase of the outer membrane.31 The accumulation activates and phosphorylates ubiquitin at serine 65 (Ser65), thereby recruiting Parkin and activating mitochondrial quality control pathways. Parkin is a kind of E3 ubiquitin ligase by PARK2 gene encoding, responsible for attaching ubiquitin to substrate proteins, when the S65 residue of Parkin is phosphorylated by PINK1, it functions downstream of PINK1. Damage to mitochondria can induce a conformational change in Parkin, converting it into an active E3 ubiquitin ligase.32 Research indicates that in the process of PINK1/Parkin-induced mitophagy, p62 facilitates the accumulation of damaged mitochondria via its PB1 oligomeric domain. Notably, knockout of p62 hinders the final elimination of damaged mitochondria without affecting Parkin’s recruitment to mitochondria.33 In addition, the ubiquitin chains assembled by Parkin can recruit the autophagy receptor TANK-binding kinase 1 (TBK1), thereby directly or indirectly mediating the phosphorylation of autophagy receptors.34 Apart from the PINK1/Parkin pathway, PINK1 can directly recruit autophagy receptors such as optineurin (OPTN) and nuclear dot protein 52 (NDP52) to mitochondria through ubiquitin phosphorylation, thereby enhancing mitophagy induction.35 Furthermore, research indicated that E3 ubiquitin ligases such as smad ubiquitination regulatory factor-1 (SMURF1), mitochondrial E3 ubiquitin ligase 1 (MUL1), and glycoprotein 78 (Gp78) participate in ubiquitinating mitochondrial proteins and induce mitophagy.36–39
Ub-Independent Pathway
Unlike the Ub-dependent pathway, the outer mitochondrial membrane hosts numerous autophagy receptors containing microtubule-associated protein 1A/1B-light chain 3 (LC3) interacting domains, enabling direct binding with LC3 to initiate mitophagy. Examples include BCL2/adenovirus E1B 19kDa interacting protein-3 (BNIP3), Nip3-like protein X (NIX)/BNIP3 like (BNIP3L) receptor, FUN14 domain-containing 1 (FUNDC1) receptor, and others. Studies indicated that conditions such as hypoxia and increased ROS induce BNIP3L expression, thereby activating mitophagy.40 Similar to BNIP3L, BNIP3 also contains a BH3 domain structure. The study found that knockout BNIP3 significantly reduces mitophagy. Unlike NIX, which primarily regulates physiological levels of mitophagy, BNIP3 excessively activates mitophagy and leads to cell death.41 FUNDC1, a kind of complete outer mitochondrial membrane protein, induces mitophagy under hypoxia conditions by interacting with LC3.42
Mechanism of Ferroptosis
The concept of ferroptosis was initially proposed in 2012 and is defined as an iron-dependent form of non-apoptotic cell death. Recent advancements have significantly enhanced our understanding of the mechanisms underlying ferroptosis. Research has identified its complex relationships with iron, amino acid, and lipid metabolism, resulting in LPO, elevated ROS levels, and iron accumulation. In this section, we focus on elucidating the mechanism of ferroptosis (Figure 2).
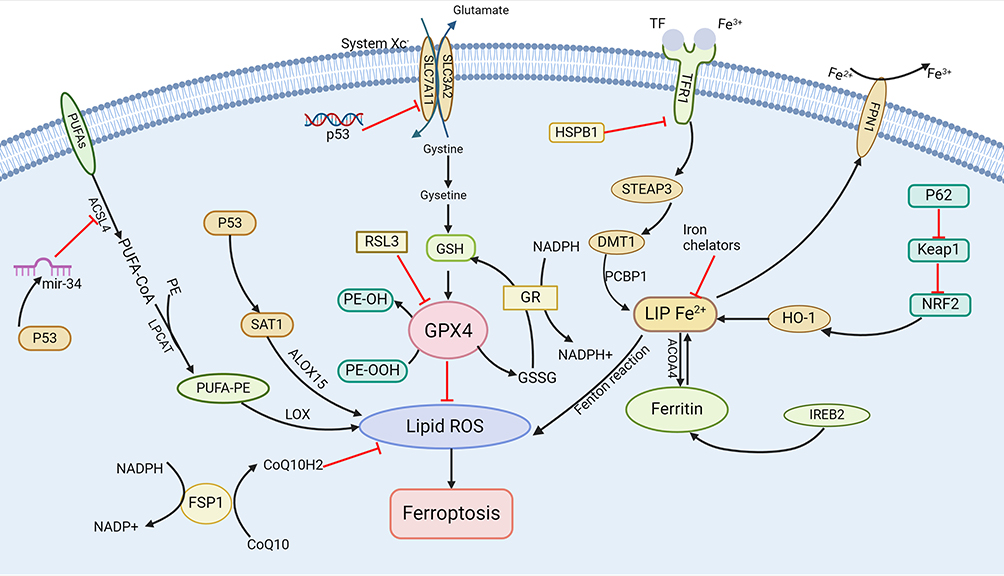 |
Figure 2 Overview of the ferroptosis mechanisms. The regulation of ferroptosis involves several pathways, including GSH/GPX4 regulation pathways, FSP1, mechanisms related to iron and lipid metabolism, and the regulatory axis involving p53. The black arrow indicates activation and the red arrow suppression indicates inhibition. Created in BioRender. Cheng, H. (2024) BioRender.com/w14d139. Abbreviations: PUFAs, polyunsaturated fatty acids; ACSL4, Acyl-CoA synthetase long-chain family member 4; PE, phosphatidylethanolamine; GPX4, glutathione Peroxidase 4; SLC7A11, solute carrier family 7 member 11; GSH, glutathione; SAT1, spermidine/spermine N1-acetyltransferase 1; ALOX15, arachidonate-5-lipoxygenase; FSP1, ferroptosis suppressor protein 1; ROS, reactive oxygen species; GSSG, oxidized glutathione; TFR1, transferrin Receptor 1; HSPB1, heat shock protein family B (Small) member 1; STEAP3, six-transmembrane epithelial antigen of prostate 3; SLC3A2, solute carrier family 3 member 2; DMT1, divalent metal transporter 1; PCBP2, poly (rC) binding protein 2; NRF2, nuclear factor erythroid-2 related factor 2; Keap1, Kelch like ECH associated protein 1; HO-1, heme oxygenase-1, NCOA4, nuclear receptor coactivator 4; IREB2, iron-responsive element binding protein 2; LPCAT, lysophosphatidylcholine acyltransferase. |
The Ferroptosis Pathway Regulated by Glutathione Peroxidase 4 (GPX4)
GPX4 is a selenoprotein recognized as a critical regulator of ferroptosis, catalyzing the converting glutathione (GSH) to oxidized glutathione (GSSG) and reducing cytotoxic lipid peroxides (L-OOH) to their corresponding lipid alcohols (L-OH).43 GPX4 can prevent ferroptosis by eliminating intracellular peroxide, thereby sustaining cellular viability. Conversely, inhibiting GPX4 activity leads to the accumulation of LPO, initiating ferroptosis. The System Xc - / GSH/GPX4 axis is called the core pathway, pivotal in mitigating LPO-induced ferroptosis. Consequently, blocking the System Xc−/GSH/GPX4 axis can effectively inhibit ferroptosis.44,45 In addition, research identifies certain triggers of ferroptosis, such as RAS-selective lethal 3 (RSL3), which potentially acts by directly inhibiting GPX4 activity, reducing cellular antioxidant capacity, promoting ROS accumulation, and ultimately causing ferroptosis.46
Ferroptosis Suppressor Protein 1 (FSP1)
FSP1 is a glutathione-independent inhibitor of ferroptosis that functions similarly to GPX4.47 Studies have shown that FSP1 can consume nicotinamide adenine dinucleotide (NADH)/ nicotinamide adenine dinucleotide phosphate (NADPH) to convert coenzyme Q10 into coenzyme Q10H2. Coenzyme Q10 is a lipophilic molecule primarily located in the inner mitochondrial membrane, while coenzyme Q10H2 is a lipophilic antioxidant, capable of inhibiting ferroptosis by trapping free radicals and preventing LPO.48 In addition to the GSH-independent FSP1-CoQ10-NAD(P)H axis, FSP1 also effectively suppresses ferroptosis via the non-canonical vitamin K redox cycle. Research demonstrates that FSP1 consumes NAD(P)H to reduce vitamin K to VKH2, which scavenges free radicals and inhibits LPO.49
Iron Metabolism
Iron is a vital cofactor for numerous enzymes involved in redox control; however, its presence can result in the generation of oxygen radicals that may damage cellular components. Hence, regulating iron levels in tissues is crucial. Studies have found that transferrin (TF), ferritins, hepcidin, and ferroportin (FPN) as key players in maintaining body iron homeostasis.50 Ferritin and hepcidin are integral regulators of cellular function and play a significant role in controlling cell death, which is associated with redox processes. Notably, ferritin deficiency has been shown to trigger ferroptosis by downregulating solute carrier family 7 member 11 (SLC7A11).51 In addition, inhibiting important transcription factors like iron-responsive element binding protein 2 (IREB2) can significantly increase the expression of ferritin light chain 1 (FTL1) and ferritin heavy chain 1 (FTH1), thereby reducing the occurrence of ferroptosis.52
Regulation of Lipid Metabolic Pathways
Lipid metabolism is intricately linked to ferroptosis, with polyunsaturated fatty acids (PUFAs) constitute a fundamental component of this process owing to their vulnerability to LPO. Free PUFAs act as substrates for the synthesis of lipid signaling transduction mediators but must be esterified into membrane phospholipids and oxidized to transmit ferroptosis signals. Arachidonic acid (AA) is one of the most abundant PUFAs in animal cells. Research indicates that phosphatidylethanolamine (PE) containing AA or its derivatives is a crucial phospholipid in the induction of ferroptosis.53 Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) participate in the biosynthesis and remodeling of PE, activating PUFAs and affecting their transmembrane properties. Consequently, downregulating ACSL4 and LPCAT3 increase unesterified AA levels while decrease the accumulation of lipid peroxide substrates, thereby inhibiting ferroptosis. However, under the catalysis of lipoxygenase (LOX), PUFA-PE can induce oxidative effects that lead to ferroptosis.54 Studies have demonstrated that the ACSL4/LPCAT3/15-LOX and p53/SLC7A11/12-LOX pathways can trigger LPO production during ferroptosis.55
P53-Mediated Ferroptosis
The tumor suppressor gene TP53, also known as p53, responds to various stresses, including metabolic dysregulation, DNA damage, and oncogene activation. Recent research has shown that p53 influence ferroptosis through classical and non-classical pathways.56,57 SLC7A11, a pivotal component of the cysteine-glutamate antiporter system, is a direct target of p53 inhibition. Investigations have found that P53 modulates the activity of GPX4 by suppressing the expression of SLC7A11, which leads to a decreased antioxidant capacity, an accumulation of ROS, and the promotion of ferroptosis.58 Furthermore, P53 can transcriptionally activate spermidine/spermine N1-acetyltransferase 1 (SAT1), inducing LPO and subsequent ferroptosis through the upregulation of arachidonate-5-lipoxygenase (ALOX15) expression.59 However, some studies suggested that P53 may also delay or inhibit ferroptosis. For instance, the p53/miR-34/ACSL4 axis participates in ferroptosis regulation, with ACSL4 serving as a key regulator of PUFAs generation in the cell membrane. P53 can activate miR-34 to downregulate the ACSL4 expression, thereby limiting the occurrence of LPO and ferroptosis.60 Consequently, P53 may regulate ferroptosis through a bidirectional pathway, highlighting the necessity for further investigation into its mechanisms in future studies.
Mechanism of Cross-Talk Between Mitophagy and Ferroptosis
There is a close relationship between mitophagy and ferroptosis (Figure 3), with potential intersections in redox and iron homeostasis. Research indicates that ROS play a pivotal role in mediating the interaction between these two pathways. Primarily produced by mitochondria, ROS is implicated in the onset and progression of various diseases. The accumulation of ROS, particularly mitochondrial ROS (mtROS), can trigger mitophagy, however, reducing mtROS levels can safeguard cells against severe oxidative stress damage.61 ROS are a primary trigger of ferroptosis, with mtROS induced ferroptosis being recognized as a pivotal mechanism underlying sepsis-induced cardiac dysfunction.62 Research indicates that the interaction between FUNDC1 and GPX4 facilitates the recruitment of GPX4 from the cytoplasm to the mitochondria via the mitochondrial protein import system. This process subsequently enhances the degradation of GPX4 through mitophagy, thereby promoting ferroptosis.63 It is important to note that BNIP3 and NIX mediated mitophagy can inhibit ferroptosis by downregulating mtROS.64 Peng et al demonstrated that acute paraquat poisoning enhances FUNDC1-mediated mitophagy, subsequently inhibiting c-Jun N-terminal kinase (JNK) expression while promoting the expression of nuclear receptor coactivator 4 (NCOA4). This cascade results in the activation of ferritinophagy, thereby influencing ferroptosis.65 One study has demonstrated that mitophagy can activate ferritinophagy, resulting in the release of ferrous iron from ferritin and an increase in intracellular ferrous ion levels. These unstable ferrous ions can react with hydrogen peroxide via the Fenton reaction, generating hydroxyl radicals. Additionally, a decrease in GSH and GPX4 levels may lead to the peroxidation of membrane phospholipids, ultimately resulting in ferroptosis.66
 |
Figure 3 Overview of the cross-talk mechanism between mitophagy and ferroptosis. The black arrow indicates activation and the red arrow suppression indicates inhibition. Created in BioRender. Cheng, H. (2024) BioRender.com/z57u952. Abbreviations: ROS, reactive oxygen species; FUNDC1, FUN14 domain-containing 1; BNIP3, BCL2/adenovirus E1B 19kDa interacting protein-3; AMPK, Adenosine 5′-monophosphate activated protein kinase; GPX4, Glutathione peroxidase; PINK1, PTEN-induced kinase 1; PARK2, Parkin; JNK, c-Jun N-terminal kinase; NCOA4, nuclear receptor coactivator 4; GSH, glutathione; GSSG, oxidized glutathione; Nrf2, nuclear factor erythroid 2-related factor 2; HO-1, heme oxygenase-1. |
The PINK1/Parkin pathway is a crucial mechanism in mediating mitophagy. Recent investigations have revealed that deficiency in PARK2 results in elevated ROS, reduced GSH, iron release, upregulation of heme oxygenase-1 (HO-1), and depletion of GPX4, ultimately resulting in ferroptosis. Consequently, PINK1/Parkin mediated mitophagy may potentially mitigate ferroptosis through the ROS/HO-1/GPX4 signaling pathway.67 Adenosine 5′-monophosphate activated protein kinase (AMPK) serves as a guardian of metabolism and mitochondrial homeostasis. It has been shown to selectively eliminate damaged mitochondria and plays a pivotal role in the activation of mitophagy through phosphorylation.68,69 Furthermore, AMPK is a crucial regulator of ferroptosis; research indicates that it can inhibit ferroptosis in degenerated chondrocytes through the Nrf2/HO-1 pathway. Thus, AMPK may serve as a key molecule in the interaction between these processes.70
Role of Mitophagy and Ferroptosis in Sepsis-Induced ALI/ARDS
Role of Mitophagy in Sepsis-Induced ALI/ARDS
Upon the onset of sepsis, the balance between pro-inflammatory mediators and anti-inflammatory mediators regulates the progression of inflammatory response. Maintenance of this balance is crucial for controlling infection within the patient’s body. However, disruption of this homeostasis, results in the release of pro-inflammatory mediators into the bloodstream, precipitating a more extensive inflammatory cascade and organ dysfunction.71 Recent advancements have been achieved in understanding the mechanism of mitophagy in sepsis-induced organ dysfunction, as research in this area has increasingly deepened. Research indicates that sepsis can result in a reduction of mitochondrial membrane potential. Under normal conditions, this potentially facilitates the engulfment of damaged mitochondria by autophagic bodies, followed by their degradation through lysosomal fusion. This process enhances the recovery of septic patients with organ dysfunction.72 Consequently, the alterations in mitochondria induced by sepsis may serve as the pathophysiological mechanism that leads to organ failure in septic patients.
Mitochondria play a critical role in regulating Ca2+ homeostasis, determine cell fate through triggering apoptosis/necrosis and generating ROS. Additionally, mtROS and mtDNA can activate the NLRP3 inflammasome, thereby connecting mitochondria dysfunction to the pathogenesis of sepsis and associated organ injury. Suliman et al73 observed widespread activation of mitophagy in the alveolar region in a mouse model of Staphylococcus aureus-induced pneumonia. This quality control process facilitated the removal and replacement of damaged mitochondria in lung cells, thereby enhancing cell survival and supporting alveolar function. Nrf2 is a transcription factor that regulates mitochondrial quality, cytoprotective gene expression, oxidative stress repair, and anti-inflammatory and anti-apoptotic processes, influencing mitochondrial function through the transcription regulation of downstream factors such as HO-1, B-cell lymphoma-2 (Bcl-2), and B-cell lymphoma-extra-large (Bcl-XL).74 Nrf2 activation has been shown to resist mitochondrial toxins and regulate the production of reduced GSH, thereby reducing mtROS production. However, Nrf2 deficiency may lead to impaired mitochondrial fatty acid oxidation and decreased adenosine triphosphate (ATP) production.23 The downregulation of classic autophagy during sepsis has been identified as a contributing factor to lung inflammation, while the activation of Nrf2 can promote mitophagy in the alveolar area, selectively removing damaged mitochondria promoting tissue repair and enhancing cell survival, thereby mitigating sepsis-induced ALI/ARDS. Therefore, promoting mitophagy may be an effective treatment for sepsis-induced ALI/ARDS.23 Protein kinase C-alpha (PRKCA), a member of the serine-threonine-specific protein kinase C family, is involved in mediating cell growth and inflammatory responses. Research suggested that PRKCA/miR-15a-5p/ pyruvate dehydrogenase kinase 4 (PDK4) axis can restrain sepsis-induced ALI/ARDS by promoting mitophagy and inhibiting anti-inflammatory response.75 OPTN is a multifunctional protein, and researchers have found that in the cecal ligation and puncture (CLP) induced ALI, melatonin regulates the mitophagy associated with OPTN. This findings suggests that melatonin may serve as a therapeutic approach to mitigate sepsis-induced ALI.76
In summary, mitophagy is closely related to the pathogenesis and progression of ALI/ARDS in sepsis. Modulating mitophagy during the disease is expected to ameliorate the inflammatory response and oxidative stress in sepsis, thereby improving lung function and the long-term prognosis of patients. In recent years, significant progress has been made in the targeted regulation of mitophagy in sepsis-induced ALI/ARDS. Certain targets and drugs that regulate mitophagy are expected to become effective therapeutic approaches for sepsis.
Role of Ferroptosis in Sepsis-Induced ALI/ARDS
Ferroptosis is a form of programmed cell death characterized by iron-dependence LPO-mediated membrane damage. In recent years, the research on the role of ferroptosis in sepsis and sepsis-related organ dysfunction has progressed rapidly. Studies has highlighted the importance of macrophages, TF secretion, antioxidant molecules in airway epithelium and cilia in maintaining lung iron homeostasis.22 In sepsis, dysregulated inflammation can cause widespread damage to alveolar epithelial and microvascular endothelial cells, resulting in pulmonary edema and excessive neutrophil infiltration. Once these protective mechanisms are compromised, iron accumulation resulting from iron metabolism disorders can lead to oxidative stress, inflammation, and mitochondrial dysfunction, thereby exacerbating lung injury.77 Consequently, the association between ferroptosis and ALI/ARDS in sepsis highlights the necessity for further investigation and discourse on its implications for sepsis-induced ALI/ARDS.
Zhang et al26 discovered that overexpressing YAP1 can inhibit the degradation of ferritin, leading to the accumulation of Fe2+ by disrupting the NCOA4-FTH1 interaction. This disruption subsequently blocks the transport of cytosolic Fe2+ to mitochondria through the sideroflexin 1 (SFXN1), resulting in a reduction of mtROS production. As a result, YAP1 can effectively inhibit ferroptosis and alleviate sepsis-induced ALI. AU-rich element RNA binding factor 1 (AUF1), an mRNA binding protein, acts as a crucial regulator in turning off inflammatory response and alleviating sepsis. The study identified NRF2 and ATF3 as two critical factors involved in ferroptosis. The findings showing that AUF1 can reversely regulate the expression of NRF2 and ATF3. Furthermore, upregulation of NRF2 and downregulation of ATF3 can respectively increase the expression levels of SLC7A11 and GPX4, thereby inhibiting ferroptosis.78 Brg/Brm-associated factor 155 (Srg3/BAF155) was found to be significantly upregulated in sepsis-induced ALI, promoting ferroptosis by regulating the activation of the NF-κB signaling pathway. In addition, Srg3 can be transcriptional activated by interferon regulatory factor 7 (Irf7), and specific inhibition of Irf7 can significantly improve ALI symptoms.79 Protectin conjugates in tissue regeneration 1 (PCTR1), a specialized pro-resolving mediator, has been shown in previous research to increase serum superoxide dismutase (SOD) and GPX4 levels in mice, reducing ROS production and improving multiple organ injuries associated with sepsis.80 Lv et al81 found that PCTR1 can decrease the Fe2+, prostaglandin-endoperoxide synthase 2 (PTGS2), and ROS production, while increasing the expression levels of GSH and GPX4, thereby alleviating mitochondrial ultrastructure damage and contributing to lung protection. In addition, studies have shown that extracellular cold-inducible RNA-binding protein (eCIRP) can induce ferroptosis in sepsis-induced ALI by downregulating GPX4 expression and elevating lipid ROS levels. This suggests that targeting eCIRP to regulate ferroptosis could present a novel therapeutic approach for sepsis-induced ALI.82
In conclusion, ferroptosis is intricately related to sepsis-induced ALI/ARDS, with its regulation involving complex mechanisms, such as iron accumulation, LPO, and GPX4 regulation. Although numerous studies have made discoveries and advancements in this field, the specific mechanisms require further investigation. Nonetheless, we believe that the regulation of ferroptosis may emerge as an effective treatment for sepsis-induced ALI/ARDS.
Role of Crosstalk Between Mitophagy and Ferroptosis in Sepsis-Induced ALI/ARDS
Mitophagy and ferroptosis represent two critical forms of cell death marked by complex regulatory mechanisms. Research has demonstrated that mitochondrial dysfunction can significantly impact both mitophagy and ferroptosis. The NEET protein family member CISD3 (CDGSH iron sulfur domain 3) plays a crucial role in maintaining mitochondrial homeostasis and regulating ROS metabolism. Knockdown of CISD3 induces mitochondria fragmentation, leading to the accumulation of free iron and lipid peroxides, thereby promoting ferroptosis. In contrast, activation of mitophagy has been shown to mitigate CISD3-related ferroptosis.18 Tang et al identified that the GTPase Dynamin-related Protein 1 (Drp1), a pivotal enzyme in mitochondrial fission regulation, is essential in controlling cell death. Knockdown of Drp1 mitigates damage to the mitochondrial membrane potential, thereby safeguarding mitochondrial integrity, maintaining redox homeostasis, and alleviating the adverse effects of ferroptosis on cells.83 Furthermore, numerous studies have elucidated the significant interaction between mitophagy and ferroptosis. Research indicates that mitophagy can exert varying effects on ferroptosis depending on the level of autophagic flux.19 Current investigations have identified key nodes within the interaction network between mitophagy and ferroptosis. For instance, Ji et al demonstrate that inhibiting thioredoxin-interacting protein (TXNIP) expression enhances PINK1-mediated mitophagy and concurrently inhibits ferroptosis.84 Additionally, protein O-GlcNAcylation serves as a critical nutrient sensor of glucose flux; its inhibition leads to mitochondrial fragmentation and promotes mitophagy, resulting in an increase in labile iron and heightened sensitivity of cells to ferroptosis.17 Furthermore, other studies have highlighted the essential roles of BNIP3 and NIX in regulating mitophagy and ferroptosis, suggesting that these proteins modulate the levels of mtROS and protect cells from ferroptosis through their involvement in mitophagy.64
The crosstalk mechanism between mitophagy and ferroptosis is involved in the occurrence and progression of sepsis and sepsis-induced ALI/ARDS. Mitophagy is a process of selective autophagy targeting damaged mitochondria and plays a critical regulatory role in maintaining mitochondrial homeostasis. It may exert inhibitory effects on the release of ROS, consequently influencing the ferroptosis pathway. The Kelch repeat and BTB domain-containing protein 7 (KBTBD7), a member of the Kelch protein family, is crucial in the regulation of inflammatory factors. Knockdown of KBTBD7 decreases mtROS production and improves mitochondrial dysfunction, thereby decreasing the incidence of ferroptosis and alleviating sepsis-induced ALI.85 The CDGSH iron-sulfur domain-containing protein 1 (CISD1), a member of the NET family localized to the outer mitochondrial membrane, has been shown to mitigate mitochondrial dysfunction and ferroptosis by reducing ROS accumulation in Lipopolysaccharides (LPS)-induced ALI.86 Olaparib has been found to effectively target and mitigate markers associated with ferroptosis while improving mitochondrial quality, suggesting its protective mechanism may be attributed to the enhancement of mitophagy and mitochondrial integrity, particularly in sepsis-induced organ dysfunction scenarios.87 It is concluded that mitophagy and ferroptosis are paramount in the pathophysiology of sepsis-induced ALI/ARDS, with their crosstalk likely occurring through iron homeostasis, oxidative stress, and common target proteins and pathways. Currently, there is limited research on the interaction between mitophagy and ferroptosis in the context of sepsis-induced ALI/ARDS. However, elucidating the mechanisms of their interaction and identifying key cross-targets may provide novel therapeutic strategies for managing sepsis-induced ALI and ARDS.
Herbs and Their Bioactive Compounds Affect ALI/ARDS by Targeting Mitophagy and Ferroptosis
Traditional Chinese medicine (TCM) emphasizes the harmony and balance of the body, showcasing its unique advantage in the treatment of sepsis. Studies have uncovered the capacity of herbs and their bioactive compounds to inhibit platelet aggregation, regulate inflammation and immune response, and improve microcirculation. These effects contribute to prevention of sepsis onset and progression, thereby improving patient prognosis.88 Studies also indicated that the synergistic effect of herbs, compound preparations, and their active ingredients with antibiotics can significantly decrease the incidence of drug-resistant bacterial infection in patients with sepsis, while also mitigating and treating organ damage caused by sepsis.89 In addition, during the widespread COVID-19 pandemic, herbal medicines like “Xuebijing”, Shenfu injection, “Shengmai injection” and others played a crucial role in managing ARDS. Therefore, understanding the clinical therapeutic efficacy, mechanism, and pharmacokinetics of herbs and their bioactive compounds in the treatment of sepsis and related organ dysfunction is crucial. This section of the paper summarizes the research on how herbs and their bioactive compounds influence ALI/ARDS by targeting mitophagy or ferroptosis, in order to provide a reference from different perspectives for subsequent treatment for the disease (Table 1).
 |
Table 1 Herbs and Their Bioactive Components for the Treatment of ALI/ARDS by Targeted Regulation of Mitophagy and Ferroptosis |
Herbs and Their Bioactive Compounds Affect ALI/ARDS by Targeting Mitophagy
Resveratrol, a phytoalexin produced by plants in response to stimuli, was first identified from the root of Veratrum grandiflorum o. Loes (baiwulu) in 1939. This compound exhibits properties including anti-inflammatory, antioxidant, cardiovascular protection, and anti-cancer. Resveratrol is identified in two isomeric forms, with trans-isomer demonstrating higher bioavailability and stability compared to the cis form.97 Findings from in vivo and in vitro experiments demonstrate that resveratrol exhibits anti-inflammatory properties by inhibiting the production of pro-inflammatory factors. Specifically, resveratrol reduces the mRNA expression and protein secretion of interleukin(IL)-17, and inhibits the production of IL-6, IL-1, and tumor necrosis factor-α (TNF-α) in a dose-dependent manner.98 Another study confirmed the efficacy of administering 40 mg of resveratrol daily in diminishing the levels of pro-inflammatory factors, including TNF-α, IL-6, and C-reactive protein (CRP).99 Therefore, resveratrol has great potential in the treatment of inflammatory diseases. In the context of sepsis-induced ALI/ARDS, resveratrol showcases its unique advantages. Studies have identified that mitochondrial dysfunction and mitophagy mediated by phospholipid scramblase 3 (PLSCR-3) are key factors in lung injury caused by CLP.100 After resveratrol treatment, an improvement in mitochondrial function was observed, and the levels of autophagy-related (ATG) proteins and LC3-I/LC3-II significantly decreased, while the expression level of P62 markedly increased. Therefore, resveratrol can ameliorate sepsis-induced ALI/ARDS by regulating PLSCR-3 levels to improve mitochondrial dysfunction and mediate mitophagy. Moreover, studies further highlight resveratrol’s potential in mitigating lung injury and inflammation through inhibits the oligomerization of the caspase recruitment domain (ASC) and the NLRP3 activation. Resveratrol has been shown to reduce the content of inflammation factors in serum and bronchoalveolar lavage fluid, such as IL-1β and IL-1, while simultaneously upregulating the expression of Pink1, Parkin, Beclin1, ATG5 and LC3B - II proteins, thereby enhancing mitophagy.90 These effects suggest that resveratrol may improve ALI in mice via its activation of the PINK1/Parkin pathway, the promotion of mitophagy, and the inhibition of NLRP3 inflammasome activation. In conclusion, these findings suggest that resveratrol may become a valuable addition to therapeutic strategies for treating sepsis-induced ALI/ARDS in the future.
Berberine, a quaternary ammonium isoquinoline alkaloid isolated from several traditional Chinese herbs, is widely utilized in treating infectious diseases such as gastroenteritis, colitis, and dysentery due to its anti-inflammatory, antibacterial, and regulation of glucose and lipid metabolism properties.101,102 Research has shown that berberine is oxidized to oxyberberine, which enhances safety and exhibits superior anti-inflammatory, antimicrobial, and anti-arrhythmic activities.103 Recent studies have demonstrated that oxyberberine can improve sepsis-induced ALI, thereby attracting significant attention to its underlying mechanism.104 Researchers found that in an in-vivo model, oxyberberine can inhibit the accumulation of pro-inflammatory cytokines and alleviate LPS-induced lung injury and pulmonary edema. In vitro study indicate that oxyberberine suppresses the inflammatory response in LPS-induced ALI by inhibiting PINK1/Parkin mediated mitophagy.91 Therefore, oxyberberine may present unique pharmacological applications in the prevention and treatment of ALI.
Kahweol (KA) is a natural diterpene extracted from coffee beans that has been found to have deleterious effects on serum lipid levels under certain circumstances, thereby increasing the potential risk of cardiovascular diseases. However, studies have also shown that Kahweol exhibits various beneficial pharmacological activities, such as anti-inflammatory, antitumor, anti-diabetic, and anti-angiogenesis effects.105 In recent years, research has highlighted Kahweol emerged as a potential agent in alleviating ALI caused by sepsis. Li et al indicate that kahweol mitigates LPS-induced ALI in septic mice by reduce inflammation and oxidative stress. Furthermore, kahweol promotes the activation of the Calcium-calmodulin (CaM)-dependent protein kinase II (CaMKKII)/AMPK-dependent mitophagy pathway, which is crucial for maintaining mitochondrial homeostasis and alleviating sepsis-induced ALI.92 Therefore, kahweol may serve as a promising plant-based therapeutic for the treatment of sepsis-induced ALI.
Herbs and Their Bioactive Compounds Affect ALI/ARDS by Targeting Ferroptosis
Puerarin is a flavonoid monomer isolated from the pueraria lobata. With ongoing research into its pharmacological properties, puerarin has become widely utilized in the treatment of cardiovascular and cerebrovascular diseases, inflammation, diabetes, Parkinson’s disease, and cancer.106 Studies have demonstrated that puerarin exerts several beneficial effects, including the inhibition of calcium influx, improvement of microcirculation, reduction of insulin resistance, and scavenging of oxygen free radicals. At present, three main formulations of puerarin are extensively employed in clinical practice.107 Previous research has shown that puerarin can ameliorate LPS-induced lung injury by suppressing inflammatory responses.108 Recently, Xv et al93 used LPS-induced human alveolar epithelial cells (A549) to construct an ALI model of sepsis. Their findings revealed that puerarin reduced the expression of ROS, malondialdehyde (MDA), and GSH in LPS-induced A549 cells, thereby alleviating cell damage and inflammatory response. In addition, the study observed that LPS exposure increased total and ferrous iron levels in A549 cells and altered ferroptosis-related proteins, including a decrease in the expression of SLC7A11, GPX4, and FTH1, alongside an increase in nicotinamide adenine dinucleotide phosphate oxidase 1 (NOX1) expression. Following puerarin intervention, there was an observed increase in the expression of total iron, ferrous iron, SLC7A11, GPX4 and FTH1, and a decrease in NOX1 expression in A549 cells. Following puerarin intervention, the aforementioned changes were reversible, suggesting that puerarin may mitigate sepsis-induced ALI by inhibiting LPS-induced ferroptosis in A549 cells.
Ginseng is a renowned medicinal plant extensively used in traditional medicine across Far Eastern countries such as China, South Korea, and Japan.109 Panaxydol (PX), a polyacetylene compound, is the primary component of ginseng exhibiting various biological activities such as anti-tumor, anti-inflammatory, anti-oxidation, and neuroprotection effects.110 These properties highlight its potential for preventing and treating multiple diseases. Studies have shown that PX can inhibit LPS-induced ALI, improve lung pathological changes, reduce pulmonary edema and inflammation, as well as inhibit ferroptosis in mice models. In addition, PX has been found to suppress ferroptosis and inflammation in the human bronchial epithelial cells (BEAS-2B) through the Keap1 (Kelch like ECH associated protein 1)-Nrf2/HO-1 signaling pathway. Therefore, PX may exert a protective effect by inhibiting ferroptosis and could represent a new strategy for ALI treatment in the future.94
Exocarpium Citri Grandis (ECG) is the dried immature or near-mature outer peel of the Huazhou pomelos which belongs to the orange citrus group and is known as “huajuhong” in China. ECG contains active flavonoids, polysaccharides, volatile oils, coumarin, and other compounds. As a precious TCM with a history of hundreds of years, ECG has been demonstrated to possess anti-inflammatory, antioxidant, immunomodulatory, and regulatory effects on lipid and glucose metabolism. These properties have facilitated its clinical application in the treatment of pneumonia, diabetes, hyperlipidemia, and related diseases.111 Previous research found that ECG can significantly ameliorate organ and tissue damage caused by iron overload and ROS accumulation. Xu et al reported that ECG can alleviate pulmonary inflammatory responses by inhibiting the Toll-like receptor 4 (TLR4)/ myeloid differentiation factor 88 (MyD88)/NF-κB p65 and JAK/STAT (signal transducers and activators of transcription) signaling pathway. Furthermore, ECG mitigates the activation of the NLRP3 inflammasome mediated by the TXNIP/NLRP3 pathway and regulates ferroptosis through the Nrf2 / GPX4 pathway in a mouse model of LPS-induced ALI.95
Lonicera japonica Thunb (LJT) belongs to Caprifoliaceae family, because of the unique pharmacological properties, are widely applied to various disease.112 Modern pharmacology studies show that LJT and its active ingredients exhibit extensive effects, such as antibacterial, anti-inflammatory, antiviral, endotoxin inhibition, lipid-lowering, and antipyretic activities.113,114 Studies have found that both in vivo (mice) and in vitro (BEAS-2B cells) models of LPS-induced ALI, LJT extract and its active component luteoloside, can alleviate lung injury and inflammation via the NF-κB signaling pathway.115 Xiong et al96 discovered that different parts of LJT have pharmacological activities, such as ferroptosis inhibition, antioxidative stress, and anti-inflammatory. Luteolin-7-O-rutinoside, apigenin-7-O-rutinoside, and amentoflavone are potentially crucial ingredients of LJT that exhibit inhibitory effects on ferroptosis. Furthermore, LJT shows potential in alleviate LPS-induced ALI by suppressing ferroptosis through activation of the Nrf2/GPX4 axis. Therefore, these discoveries present a promising therapeutic candidate for managing ALI.
Conclusion and Prospects
Mitophagy and ferroptosis are crucial in the pathogenesis and progression of sepsis-induced ALI/ARDS. Certain drugs that promote mitophagy or inhibit ferroptosis have emerged as potential therapeutic strategies. In recent years, the investigation of interactions among various modes of cell death has emerged as a current research hotspot and a promising direction for future studies.116 Research on the mechanisms underlying the interaction between mitophagy and ferroptosis is still in its early stages. A significant challenge is the limited foundational studies and the absence of large-scale multicenter clinical research. Furthermore, ALI and ARDS resulting from sepsis represent different stages of the same disease, yet the differential effects of mitophagy and ferroptosis at these stages remain unexplored. Therefore, further studies are necessary to elucidate the effects of the interaction mechanisms on the progression and severity of various diseases. Research has demonstrated that mitochondrial dysfunction resulting in increased ROS generation is a significant factor driving ferroptosis. Regulating mitophagy levels can suppress ferroptosis and mitigate sepsis-induced ALI/ARDS by decreasing mtROS production. It is important to note that mtROS is not the sole trigger for inducing ferroptosis. Although enhancing mitophagy alone may not completely prevent the occurrence of ferroptosis, drugs targeting both processes simultaneously hold significant therapeutic promise for sepsis-induced ALI/ARDS. Therefore, elucidating mitophagy and the intricate interplay with ferroptosis and identifying their key intersections are crucial for guiding the development and application of novel treatments for ALI/ARDS in sepsis. In recent years, certain herbs and their bioactive compounds have demonstrated advantages in treating sepsis-induced ALI/ARDS by regulating mitophagy and ferroptosis. However, the application of these herbs and their bioactive compounds may encounter certain issues. Firstly, current research primarily investigates the underlying mechanisms of these drugs, however, it lacks evidence regarding their long-term therapeutic effects and potential side effects. As a result, there routine use in clinical practice is limited. Second, in contrast to chemical drugs and biological agents, the components of herbs are complex and may act through multiple pathways and targets. This complexity complicates the definition of safe dose ranges and identification of toxic components. Finally, herbs derived from natural sources, has complex components and production processes, which may result in unavoidable differences between batches, potentially leading to instability in treatment effects. Future research demands extensive in vitro and in vivo investigations to elucidate the mechanisms and active components of these potentially efficacious therapeutic candidates, as well as rigorous completion of clinical trials to mitigate potential toxicities and adverse effects. To sum up, this review examines the mechanisms and interactions between mitophagy and ferroptosis, highlighting their significant roles in the pathogenesis of sepsis-induced ALI/ARDS. Furthermore, certain herbs and their bioactive compounds may emerge as potential therapeutic options for these conditions in the future.
Funding
This work was supported by China International Medical Foundation (Z-2018-35-2101) and Foundation of Lanzhou Science and Technology Bureau (2023-2-107).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801. doi:10.1001/jama.2016.0287
2. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395(10219):200–211. doi:10.1016/S0140-6736(19)32989-7
3. Rello J, Valenzuela-Sánchez F, Ruiz-Rodriguez M, Moyano S. Sepsis: a Review of Advances in Management. Adv Ther. 2017;34(11):2393–2411. doi:10.1007/s12325-017-0622-8
4. Nakamori Y, Park EJ, Shimaoka M. Immune Deregulation in Sepsis and Septic Shock: reversing Immune Paralysis by Targeting PD-1/PD-L1 Pathway. Front Immunol. 2021;11:624279.
5. Qiao X, Yin J, Zheng Z, Li L, Feng X. Endothelial cell dynamics in sepsis-induced acute lung injury and acute respiratory distress syndrome: pathogenesis and therapeutic implications. Cell Commun Signaling. 2024;22(1):22. doi:10.1186/s12964-023-01466-w
6. Yuan J, Ofengeim D. A guide to cell death pathways. Nat Rev Mol Cell Biol. 2024;25(5):379–395. doi:10.1038/s41580-023-00689-6
7. Newton K, Strasser A, Kayagaki N, Dixit VM. Cell death. Cell. 2024;187(2):235–256. doi:10.1016/j.cell.2023.11.044
8. Green douglas R, Levine B. To Be or Not to Be? How Selective Autophagy and Cell Death Govern Cell Fate. Cell. 2014;157(1):65–75. doi:10.1016/j.cell.2014.02.049
9. D’Arcy MS. Mitophagy in health and disease. Molecular mechanisms, regulatory pathways, and therapeutic implications. Apoptosis. 2024;29(9–10):1415–1428. doi:10.1007/s10495-024-01977-y
10. Yao R-Q, Ren C, Xia Z-F, Yao Y-M. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy. 2020;17:385–401. doi:10.1080/15548627.2020.1725377
11. Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2022;24:167–185. doi:10.1038/s41580-022-00542-2
12. Duan Y, Yao R-Q, Ling H, et al. Organellophagy regulates cell death: a potential therapeutic target for inflammatory diseases. J Adv Res. 2024;24:167–185.
13. Bharath LP, Agrawal M, McCambridge G, et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020;32:44–55. doi:10.1016/j.cmet.2020.04.015
14. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171:273–285. doi:10.1016/j.cell.2017.09.021
15. Guo R, Fang X, Shang K, Wen J, Ding K. Induction of ferroptosis: a new strategy for the control of bacterial infections. Microbiol Res. 2024;284:127728.
16. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–282. doi:10.1038/s41580-020-00324-8
17. Yu F, Zhang Q, Liu H, et al. Dynamic O-GlcNAcylation coordinates ferritinophagy and mitophagy to activate ferroptosis. Cell Discovery. 2022;8:8. doi:10.1038/s41421-021-00358-y
18. Li Y, Wang X, Huang Z, et al. CISD3 inhibition drives cystine-deprivation induced ferroptosis. Cell Death Dis. 2021;13:12. doi:10.1038/s41419-021-04464-3
19. Li J, Jia Y-C, Ding Y-X, Bai J, Cao F, Li F. The crosstalk between ferroptosis and mitochondrial dynamic regulatory networks. Int J Bio Sci. 2023;19(9):2756–2771. doi:10.7150/ijbs.83348
20. Liu F, Yang Y, Peng W, et al. Mitophagy-promoting miR-138-5p promoter demethylation inhibits pyroptosis in sepsis-associated acute lung injury. Inflam Res. 2022;72:329–346. doi:10.1007/s00011-022-01675-y
21. Jiang J, Huang K, Xu S, Garcia JGN, Wang C, Cai H. Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biol. 2020;36:101638. doi:10.1016/j.redox.2020.101638
22. Wang Y, Zhao Z, Xiao Z. The Emerging Roles of Ferroptosis in Pathophysiology and Treatment of Acute Lung Injury. J Inflamm Res. 2023;16:4073–4085. doi:10.2147/JIR.S420676
23. Chang AL, Ulrich A, Suliman HB, Piantadosi CA. Redox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsis. Free Radic Biol Med. 2015;78:179–189. doi:10.1016/j.freeradbiomed.2014.10.582
24. Jiang T, Liu E, Li Z, et al. SIRT1-Rab7 axis attenuates NLRP3 and STING activation through late endosomal-dependent mitophagy during sepsis-induced acute lung injury. Int J Surg. 2024;110(5):2649–2668. doi:10.1097/JS9.0000000000001215
25. Wang W, Xu R, He P, et al. CircEXOC5 Aggravates Sepsis-Induced Acute Lung Injury by Promoting Ferroptosis Through the IGF2BP2/ATF3 Axis. J Infect Dis. 2024;229(2):522–534. doi:10.1093/infdis/jiad337
26. Zhang J, Zheng Y, Wang Y, et al. YAP1 alleviates sepsis-induced acute lung injury via inhibiting ferritinophagy-mediated ferroptosis. Front Immunol. 2022;13:884362.
27. Shen Z, Zhao L, Yoo S-A, et al. Emodin induces ferroptosis in colorectal cancer through NCOA4-mediated ferritinophagy and NF-κb pathway inactivation. Apoptosis. 2024;29(9–10):1810–1823. doi:10.1007/s10495-024-01973-2
28. Kong L, Liu Y, Li J, et al. Ginsenoside Rg1 alleviates chronic inflammation-induced neuronal ferroptosis and cognitive impairments via regulation of AIM2 - Nrf2 signaling pathway. J Ethnopharmacol. 2024;330:118205.
29. Li L, Song J-J, Zhang M-X, et al. Oridonin ameliorates caspase-9-mediated brain neuronal apoptosis in mouse with ischemic stroke by inhibiting RIPK3-mediated mitophagy. Acta Pharmacol Sin. 2022;44:726–740. doi:10.1038/s41401-022-00995-3
30. Wang N, Zhu P, Huang R, et al. PINK1: the guard of mitochondria. Life Sci. 2020;259:118247.
31. Park S, Choi S-G, Yoo S-M, et al. Pyruvate stimulates mitophagy via PINK1 stabilization. Cell Signalling. 2015;27(9):1824–1830. doi:10.1016/j.cellsig.2015.05.020
32. Riley BE, Lougheed JC, Callaway K, et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun. 2013;4:1982.
33. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2014;6:1090–1106. doi:10.4161/auto.6.8.13426
34. Heo JM, Ordureau A, Swarup S, et al. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci Adv. 2018;4:443.
35. Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. doi:10.1038/nature14893
36. Lu Y, Li Z, Zhang S, Zhang T, Liu Y, Zhang L. Cellular mitophagy: mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics. 2023;13:736–766. doi:10.7150/thno.79876
37. Orvedahl A, Sumpter R, Xiao G, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480(7375):113–117. doi:10.1038/nature10546
38. Igarashi R, Yamashita SI, Yamashita T, et al. Gemcitabine induces Parkin-independent mitophagy through mitochondrial-resident E3 ligase MUL1-mediated stabilization of PINK1. Sci Rep. 2020;10:10. doi:10.1038/s41598-019-56089-4
39. Fu M, St-Pierre P, Shankar J, et al. Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. ?mol Biol Cell. 2013;24:1153–1162. doi:10.1091/mbc.e12-08-0607
40. Schweers RL, Zhang J, Randall MS, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci. 2007;104(49):19500–19505. doi:10.1073/pnas.0708818104
41. Shi RY, Zhu SH, Li V, Gibson SB, Xu XS, Kong JM. BNIP3 Interacting with LC3 Triggers Excessive Mitophagy in Delayed Neuronal Death in Stroke. CNS Neurosci Ther. 2014;20(12):1045–1055. doi:10.1111/cns.12325
42. Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177–185. doi:10.1038/ncb2422
43. Li J, Cao F, H-l Y, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11:88.
44. F-J L, Long H-Z, Zhou Z-W, Luo H-Y, S-G X, Gao L-C. System Xc−/GSH/GPX4 axis: an important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front Pharmacol. 2022;13:910292.
45. Lewerenz J, Hewett SJ, Huang Y, et al. The Cystine/Glutamate Antiporter System xc−in x c − in Health and Disease: from Molecular Mechanisms to Novel Therapeutic Opportunities. Antioxid Redox Signaling. 2013;18(5):522–555. doi:10.1089/ars.2011.4391
46. Yang Wan S, SriRamaratnam R, Welsch Matthew E, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell. 2014;156(1–2):317–331. doi:10.1016/j.cell.2013.12.010
47. Li W, Liang L, Liu S, Yi H, Zhou Y. FSP1: a key regulator of ferroptosis. Trends Mol Med. 2023;29(9):753–764. doi:10.1016/j.molmed.2023.05.013
48. Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–698. doi:10.1038/s41586-019-1707-0
49. Mishima E, Ito J, Wu Z, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022;608(7924):778–783. doi:10.1038/s41586-022-05022-3
50. Rochette L, Dogon G, Rigal E, Zeller M, Cottin Y, Vergely C. Lipid Peroxidation and Iron Metabolism: two Corner Stones in the Homeostasis Control of Ferroptosis. Int J Mol Sci. 2022;24(1):24. doi:10.3390/ijms24010024
51. Fang X, Cai Z, Wang H, et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ Res. 2020;127(4):486–501. doi:10.1161/CIRCRESAHA.120.316509
52. Gammella E, Recalcati S, Rybinska I, Buratti P, Cairo G. Iron-Induced Damage in Cardiomyopathy: oxidative-Dependent and Independent Mechanisms. Oxid Med Cell Longev. 2015;2015:1–10. doi:10.1155/2015/230182
53. Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. 2022;82(12):2215–2227. doi:10.1016/j.molcel.2022.03.022
54. Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2016;13:81–90. doi:10.1038/nchembio.2238
55. Chu B, Kon N, Chen D, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21(5):579–591. doi:10.1038/s41556-019-0305-6
56. Levine AJ. p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer. 2020;20(8):471–480. doi:10.1038/s41568-020-0262-1
57. Liu Y, Tavana O, Gu W. p53 modifications: exquisite decorations of the powerful guardian. J Mol Cell Biol. 2019;11(7):564–577. doi:10.1093/jmcb/mjz060
58. Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62. doi:10.1038/nature14344
59. Ou Y, Wang S-J, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci. 2016;113:E6806–E6812. doi:10.1073/pnas.1607152113
60. Liu Y, Gu W. p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 2022;29(5):895–910. doi:10.1038/s41418-022-00943-y
61. Zhong Y, Xia S, Wang G, et al. The interplay between mitophagy and mitochondrial ROS in acute lung injury. Mitochondrion. 2024;78:101920.
62. Li N, Wang W, Zhou H, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. 2020;160:303–318. doi:10.1016/j.freeradbiomed.2020.08.009
63. Bi Y, Liu S, Qin X, et al. FUNDC1 interacts with GPx4 to govern hepatic ferroptosis and fibrotic injury through a mitophagy-dependent manner. J Adv Res. 2024;55:45–60. doi:10.1016/j.jare.2023.02.012
64. Yamashita S-I, Sugiura Y, Matsuoka Y, et al. Mitophagy mediated by BNIP3 and NIX protects against ferroptosis by downregulating mitochondrial reactive oxygen species. Cell Death Differ. 2024;31(5):651–661. doi:10.1038/s41418-024-01280-y
65. Peng H, Fu S, Wang S, et al. Ablation of FUNDC1-dependent mitophagy renders myocardium resistant to paraquat-induced ferroptosis and contractile dysfunction. Biochim Biophys Acta Mol Basis Dis. 2022;1868(9):166448. doi:10.1016/j.bbadis.2022.166448
66. Singh LP, Yumnamcha T, Devi TS. Mitophagy, Ferritinophagy and Ferroptosis in Retinal Pigment Epithelial Cells Under High Glucose Conditions: implications for Diabetic Retinopathy and Age-Related Retinal Diseases. JOJ Ophthalmol. 2021;8(5):77–85.
67. Lin Q, Li S, Jin H, et al. Mitophagy alleviates cisplatin-induced renal tubular epithelial cell ferroptosis through ROS/HO-1/GPX4 axis. Int J Bio Sci. 2023;19(4):1192–1210. doi:10.7150/ijbs.80775
68. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2017;19:121–135. doi:10.1038/nrm.2017.95
69. Laker RC, Drake JC, Wilson RJ, et al. AMPK phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun. 2017;8(1):8. doi:10.1038/s41467-017-00021-9
70. Wan Y, shen K, Yu H, Fan W. Baicalein limits osteoarthritis development by inhibiting chondrocyte ferroptosis. Free Radic Biol Med. 2023;196:108–120. doi:10.1016/j.freeradbiomed.2023.01.006
71. Bone RC. Immunologic Dissonance: a Continuing Evolution in Our Understanding of the Systemic Inflammatory Response Syndrome (SIRS) and the Multiple Organ Dysfunction Syndrome (MODS). Ann Internal Med. 1996;125:680–687.
72. Pan P, Wang X, Liu D. The potential mechanism of mitochondrial dysfunction in septic cardiomyopathy. J Int Med Res. 2018;46(6):2157–2169. doi:10.1177/0300060518765896
73. Suliman HB, Kraft B, Bartz R, Chen L, Welty-Wolf KE, Piantadosi CA. Mitochondrial quality control in alveolar epithelial cells damaged by S. aureus pneumonia in mice. Am J Physiol Lung Cell Mol Physiol. 2017;313(4):L699–L709. doi:10.1152/ajplung.00197.2017
74. Vomund S, Schäfer A, Parnham M, Brüne B, von Knethen A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int J Mol Sci. 2017;19(1):18. doi:10.3390/ijms19010018
75. Zhu Q-J, Wang J, Li Y, et al. PRKCA Promotes Mitophagy through the miR-15a-5p/PDK4 Axis to Relieve Sepsis-Induced Acute Lung Injury. Infect Immun. 2023;91:465.
76. Ling J, Yu S, Xiong F, Xu T, Li S. Melatonin Attenuates Sepsis-Induced Acute Lung Injury via Inhibiting Excessive Mitophagy. Drug Des Devel Ther. 2023;17:2775–2786. doi:10.2147/DDDT.S423264
77. Johnson ER, Matthay MA. Acute Lung Injury: epidemiology, Pathogenesis, and Treatment. J Aerosol Med Pulm Drug Delivery. 2010;23(4):243–252. doi:10.1089/jamp.2009.0775
78. Wang Y, Chen D, Xie H, et al. AUF1 protects against ferroptosis to alleviate sepsis-induced acute lung injury by regulating NRF2 and ATF3. Cel lMol Life Sci. 2022;79(2):79. doi:10.1007/s00018-021-04029-9
79. Ling X, Wei S, Ling D, et al. Irf7 regulates the expression of Srg3 and ferroptosis axis aggravated sepsis-induced acute lung injury. Cell Mol Biol Lett. 2023;28:91.
80. Y-j L, Li H, Tian Y, et al. PCTR1 ameliorates lipopolysaccharide-induced acute inflammation and multiple organ damage via regulation of linoleic acid metabolism by promoting FADS1/FASDS2/ELOV2 expression and reducing PLA2 expression. Lab Invest. 2020;100(7):904–915. doi:10.1038/s41374-020-0412-9
81. Lv Y, Chen D, Tian X, et al. Protectin conjugates in tissue regeneration 1 alleviates sepsis-induced acute lung injury by inhibiting ferroptosis. J Transl Med. 2023;21(1):21. doi:10.1186/s12967-022-03859-w
82. Shimizu J, Murao A, Nofi C, Wang P, Aziz M. Extracellular CIRP Promotes GPX4-Mediated Ferroptosis in Sepsis. Front Immunol. 2022;13:903859.
83. Tang S, Fuß A, Fattahi Z, Culmsee C. Drp1 depletion protects against ferroptotic cell death by preserving mitochondrial integrity and redox homeostasis. Cell Death Dis. 2024;15:626.
84. Ji H, Zhao Y, Ma X, et al. Upregulation of UHRF1 Promotes PINK1-mediated Mitophagy to Alleviates Ferroptosis in Diabetic Nephropathy. Inflammation. 2023;47:718–732. doi:10.1007/s10753-023-01940-0
85. Li X, Lin Z, Xu S, Zhang N, Zhou J, Liao B. Knockdown of KBTBD7 attenuates septic lung injury by inhibiting ferroptosis and improving mitochondrial dysfunction. Int Immunopharmacol. 2024;133:112129.
86. Zhang X, Peng T, Li C, et al. Inhibition of CISD1 alleviates mitochondrial dysfunction and ferroptosis in mice with acute lung injury. Int Immunopharmacol. 2024;130:111685.
87. Liu R, Li F, Hao S, et al. Low-dose Olaparib improves septic cardiac function by reducing ferroptosis via accelerated mitophagy flux. Pharmacol Res. 2024;200:107056.
88. Cheng C, Yu X. Research Progress in Chinese Herbal Medicines for Treatment of Sepsis: pharmacological Action, Phytochemistry, and Pharmacokinetics. Int J Mol Sci. 2021;23(1):22. doi:10.3390/ijms23010022
89. Zhao X, Lv C, Chen Y, Yu X, Wang Z. Chinese expert consensus on early prevention and intervention of sepsis. Asian Pac J Trop Med. 2020;13:335–349.
90. Wu D, Zhang H, Li F, et al. Resveratrol alleviates acute lung injury in mice by promoting Pink1/Parkin-related mitophagy and inhibiting NLRP3 inflammasome activation. Biochim Biophys Acta Gen Subj. 2024;1868(7):130612. doi:10.1016/j.bbagen.2024.130612
91. Zhao R, Wang B, Wang D, et al. Oxyberberine Prevented Lipopolysaccharide-Induced Acute Lung Injury through Inhibition of Mitophagy. Oxid Med Cell Longev. 2021;2021(1):1–12. doi:10.1155/2021/6675264
92. Li G, Fu T, Wang W, et al. Pretreatment with Kahweol Attenuates Sepsis‐Induced Acute Lung Injury via Improving Mitochondrial Homeostasis in a CaMKKII/AMPK‐Dependent Pathway. Mol Nutr Food Res. 2023;67:2300083.
93. Xu B, Wang H, Chen Z. Puerarin Inhibits Ferroptosis and Inflammation of Lung Injury Caused by Sepsis in LPS Induced Lung Epithelial Cells. Front Pediatrics. 2021;9. doi:10.3389/fped.2021.706327
94. Li J, Lu K, Sun F, et al. Panaxydol attenuates ferroptosis against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1 pathway. J Transl Med. 2021;19:19. doi:10.1186/s12967-020-02676-3
95. Xu Z, Li J, Zhou K, et al. Exocarpium Citri Grandis ameliorates LPS-induced acute lung injury by suppressing inflammation, NLRP3 inflammasome, and ferroptosis. J Ethnopharmacol. 2024;329:118162.
96. Xiong L, Liu Y, Wang Y, et al. The protective effect of Lonicera japonica Thunb. against lipopolysaccharide-induced acute lung injury in mice: modulation of inflammation, oxidative stress, and ferroptosis. J Ethnopharmacol. 2024;331:118333.
97. Singh AP, Singh R, Verma SS, et al. Health benefits of resveratrol: evidence from clinical studies. Med Res Rev. 2019;39(5):1851–1891. doi:10.1002/med.21565
98. Fuggetta MP, Bordignon V, Cottarelli A, et al. Downregulation of proinflammatory cytokines in HTLV-1-infected T cells by Resveratrol. J Exp Clin Cancer Res. 2016;35(1). doi:10.1186/s13046-016-0398-8.
99. Sebai H, Ben-Attia M, Sani M, Aouani E, Ghanem-Boughanmi N. Protective effect of resveratrol in endotoxemia-induced acute phase response in rats. Arch Toxicol. 2008;83:335–340. doi:10.1007/s00204-008-0348-0
100. Wang C, Yuan J, Du J. Resveratrol alleviates acute lung injury through regulating PLSCR-3-mediated mitochondrial dysfunction and mitophagy in a cecal ligation and puncture model. Eur J Pharmacol. 2021;913:174643.
101. Song D, Hao J, Fan D. Biological properties and clinical applications of berberine. Fronti Medi. 2020;14(5):564–582. doi:10.1007/s11684-019-0724-6
102. Dou Y, Huang R, Li Q, et al. Oxyberberine, an absorbed metabolite of berberine, possess superior hypoglycemic effect via regulating the PI3K/Akt and Nrf2 signaling pathways. Biomed Pharmacother. 2021;137:111312.
103. C-L L, Tan L-H, Wang Y-F, et al. Comparison of anti-inflammatory effects of berberine, and its natural oxidative and reduced derivatives from Rhizoma Coptidis in vitro and in vivo. Phytomedicine. 2019;52:272–283. doi:10.1016/j.phymed.2018.09.228
104. Chen B, Gong S, Li M, et al. Protective effect of oxyberberine against acute lung injury in mice via inhibiting RhoA/ROCK signaling pathway. Biomed Pharmacother. 2022;153:113307.
105. Ren Y, Wang C, Xu J, Wang S. Cafestol and Kahweol: a Review on Their Bioactivities and Pharmacological Properties. Int J Mol Sci. 2019;21(1):20. doi:10.3390/ijms21010020
106. Wong KH, Li GQ, Li KM, Razmovski-Naumovski V, Chan K. Kudzu root: traditional uses and potential medicinal benefits in diabetes and cardiovascular diseases. J Ethnopharmacol. 2011;134(3):584–607. doi:10.1016/j.jep.2011.02.001
107. Zhou YX, Zhang H, Peng C. Puerarin: a Review of Pharmacological Effects. Phytother Res. 2013;28:961–975. doi:10.1002/ptr.5083
108. Wang X, Yan J, Xu X, et al. Puerarin prevents LPS-induced acute lung injury via inhibiting inflammatory response. Microb Pathogenesis. 2018;118:170–176. doi:10.1016/j.micpath.2018.03.033
109. Kim M-Y, Jeong B, Lee G-S, et al. Panaxydol extracted from Panax ginseng inhibits NLRP3 inflammasome activation to ameliorate NASH-induced liver injury. Int Immunopharmacol. 2024;128:111565.
110. Kim JY, Yu S-J, Oh HJ, Lee JY, Kim Y, Sohn J. Panaxydol induces apoptosis through an increased intracellular calcium level, activation of JNK and p38 MAPK and NADPH oxidase-dependent generation of reactive oxygen species. Apoptosis. 2010;16:347–358. doi:10.1007/s10495-010-0567-8
111. Kong F, Ding Z, Zhang K, et al. Optimization of extraction flavonoids from Exocarpium Citri Grandis and evaluation its hypoglycemic and hypolipidemic activities. J Ethnopharmacol. 2020;262:113178.
112. Yang X, Yu A, Hu W, et al. Extraction, Purification, Structural Characteristics, Health Benefits, and Application of the Polysaccharides from Lonicera japonica Thunb.: a Review. Molecules. 2023;29:28. doi:10.3390/molecules29010028
113. Shang X, Pan H, Li M, Miao X, Ding H. Lonicera japonica Thunb.: ethnopharmacology, phytochemistry and pharmacology of an important traditional Chinese medicine. J Ethnopharmacol. 2011;138:1–21. doi:10.1016/j.jep.2011.08.016
114. Li RJ, Kuang XP, Wang WJ, Wan CP, Wx L. Comparison of chemical constitution and bioactivity among different parts of Lonicera japonica Thunb. J Sci Food Agric. 2019;100:614–622. doi:10.1002/jsfa.10056
115. Jia Q, wen J, Yang Q, et al. Lonicera japonica Thunb extract ameliorates lipopolysaccharide-induced acute lung injury associated with luteolin-mediated suppression of NF-κB signaling pathway. J Inflam. 2023;20:20. doi:10.1186/s12950-023-00345-y
116. Huang D, Song X, Luo Z, Zou K. Research progress and hotspot evolution of ferroptosis and pyroptosis in acute lung injury and acute respiratory distress syndrome over the past decade. Asian J Surg. 2024;47:4167–4168. doi:10.1016/j.asjsur.2024.05.087
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.