")
Back to Journals » Journal of Inflammation Research » Volume 17
Molecular Landscape and Prognostic Value in the Post-Translational Ubiquitination, SUMOylation and Neddylation in Osteosarcoma: A Transcriptome Study
Authors Jia C, Yao X, Dong Z, Wang L, Zhao F, Gao J, Cai T
Received 15 September 2023
Accepted for publication 25 June 2024
Published 4 July 2024 Volume 2024:17 Pages 4315—4330
DOI https://doi.org/10.2147/JIR.S440459
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Chenguang Jia,1,* Xiaowei Yao,1,* Zhaoliang Dong,1 Lianbo Wang,1 Fangchao Zhao,2,* Jianguo Gao,1,* Tao Cai3,*
1Department of Orthopedics, Hebei Chest Hospital, Shijiazhuang, Hebei, People’s Republic of China; 2Department of Thoracic Surgery, The Second Hospital of Hebei Medical University, Shijiazhuang, Hebei, People’s Republic of China; 3Hebei Chest Hospital, Shijiazhuang, Hebei, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Tao Cai, Hebei Chest Hospital, No. 372, Shengli North Street, Shijiazhuang, Hebei, People’s Republic of China, Tel/Fax +86-311-86911080, Email [email protected]
Background: Post-translational modifications (PTM) significantly influence the pathogenesis and progression of diverse neoplastic conditions. Nevertheless, there has been limited research focusing on the potential of PTM-related genes (PTMRGs) as tumor biomarkers for predicting the survival of specific patients.
Methods: The datasets utilized in this research were obtained from the TARGET and GEO repositories, respectively. The gene signature was constructed through the utilization of LASSO Cox regression method. GSEA and GO was used to identify hub pathways associated with risk genes. The functionality of risk genes in osteosarcoma (OS) cell lines was verified through the implementation of the CCK-8 assay, cell cycle analysis, and immunofluorescence assay.
Results: Two distinct PTM patterns and gene clusters were finally determined. Significant differences in the prognosis of patients were found among two different PTM patterns and gene clusters, so were in the function enrichment and the landscape of TME immune cell infiltration. Moreover, we examined two external immunotherapy cohorts and determining that patients in the low-risk group was more likely to profit from immunotherapy. In addition, we mapped the expression of the genes in the signature in distinct cells using single-cell analysis. Finally, CCK-8 assay, cell cycle analysis, and immunofluorescence assay were utilized to confirm that RAD21 was expressed and functioned in OS.
Conclusion: In conclusion, this study elucidated the potential link between PTM and immune infiltration landscape of OS for the first time and provided a new assessment protocol for the precise selection of treatment strategies for patients with advanced OS.
Keywords: post-translational modifications, osteosarcoma, immune microenvironment, immune therapy, RAD21
Introduction
Osteosarcoma (OS) is an exceedingly aggressive and malignant neoplasm of the bone, primarily afflicting the pediatric and adolescent populations.1 Over the course of recent decades, the survival outcomes for patients with OS have shown remarkable improvement, primarily attributed to the advancements in multimodal therapeutic approaches encompassing surgical interventions, chemotherapy, and radiation therapy.2 Presently, ongoing research endeavors pertaining to the treatment of OS primarily revolve around two areas: the identification of novel therapeutic targets and the enhancement of the efficacy of established treatment modalities.3 Several potential therapeutic targets for OS have been identified, encompassing receptor tyrosine kinases, immune checkpoints, and metabolic pathways.4,5 Furthermore, the progressions in genomics and transcriptomics have resulted in the discovery of several molecular subtypes of OS. These distinct subtypes may exhibit diverse responses to treatment and varying prognoses.6 Comprehending the molecular attributes of OS holds the potential to advance personalized therapeutic approaches and enhance clinical outcomes for patients.
Protein post-translational modifications (PTM) encompass a diverse array of chemical alterations, involving the covalent attachment of functional groups to proteins, proteolytic cleavage of regulatory subunits, or even the complete degradation of entire proteins.7 These modifications encompass phosphorylation, glycosylation, ubiquitination, acetylation, methylation, and numerous others. These modifications are intricately regulated by sophisticated mechanisms, frequently altered in the context of cancer.8,9 Given the pivotal significance of these modifications and their extensive deregulation in cancer, an increasing focus is being directed towards the advancement of therapeutic strategies that precisely target the ubiquitin and ubiquitin-like pathways, particularly SUMO and Nedd8, in the context of solid tumors.10,11 The dysregulation of three PTMs - namely SUMOylation, ubiquitination, and neddylation - collectively referred to as SUN, exerts various influences on the neoplastic transformation and progression. These effects encompass critical processes such as epithelial-mesenchymal transition, adaptation to hypoxia, modulation of tumor suppressors and oncogenic mediators, and the development of drug resistance.12,13 As a result, these PTMs present themselves as promising candidates for therapeutic intervention.14
In this study, we conducted an investigation into the PTM patterns of OS. Our approach involved classifying OS patients into two distinct PTM patterns based on the expression levels of post-translational modification-related genes (PTMRGs). Subsequently, we conducted survival analyses to examine the implications of these patterns. Moreover, we delved into the hallmark pathways and characteristics of TME immune cell infiltration associated with each PTM pattern. Notably, our findings shed light on the potential of these PTM patterns to serve as predictive indicators for immunotherapy and targeted therapy responsiveness in OS patients.
Materials and Methods
Dataset and Preprocessing
Downloaded the transcriptomic data sourced from the TARGET-OS cohort and GSE21257 dataset, excluding repeated sequencing, samples with no survival status, and those with a total survival time of less than one day. The final inclusion encompassed 84 samples from the TARGET-OS cohort, along with 53 samples from the GSE21257 cohort. Regarding Bulk transcriptome data, normalization was performed using log2(x+1).
Furthermore, we downloaded the GSE162454 dataset from the single-cell repository TISCH, encompassing six OS samples. We examined the annotations of the major-lineage entry in the metadata information from the TISCH database. In brief, after quality control of single-cell data, we analyzed the top 2000 variably expressed genes in each normalized sample, utilizing variance-stabilizing transformations. Subsequently, based “Seurat” packages we employed the “IntegrateData” function to amalgamate the data, followed by using the “ScaleData” function to effectuate scaling. Lastly, we invoked the “RunPCA” function to reduce the dimensions of principal component analysis (PCA). In the end, cell clustering was accomplished by employing the FindNeighbors and FindClusters functions.
Reactome is a database of biological pathway knowledge. We sequentially queried the genes related to the processes of SUMOylation, ubiquitination, and neddylation. After eliminating certain redundant genes, the identified 498 genes were designated as PTMRGs.
Consensus Clustering
In Bulk transcriptome data, unsupervised consistent cluster analysis was conducted based on prognostic PTMRGs. PCA was employed to ascertain the relative independence of each subtype from others. Using the R package “consensusClusterPlus”, we ascertained the optimal number of clusters by performing 100 repetitions with a resampling of 0.8 to validate the stability of subtypes.
Immune Correlation Analysis
The abundance of immune cells in different samples was estimated using the CIBERSORT algorithm and single-sample gene set enrichment analysis (ssGSEA) algorithm.
Construction and Validation of Prognostic Model
In the TARGET-OS queue, the adoption of the least absolute shrinkage and selection operator (LASSO) model was employed to eliminate superfluous genes. Based on the coefficients of the multifactorial Cox regression, we constructed a risk model and validated it using GSE21257 as the validation dataset. The model’s performance was assessed using Kaplan-Meier survival analysis. Furthermore, from previous literature, we downloaded two cohorts that received immune checkpoint blockade (ICB) therapy, namely IMvigor210 and GSE78220.15,16 These cohorts were utilized to generate a risk score, which was then validated within the immunotherapy cohort using the same formula to assess its efficacy.
Enrichment Analysis
Using the “limma” package, we conducted an analysis of differentially expressed genes among various subtypes (adj. p < 0.05, |logFC| > 1). Upon identifying the differentially expressed genes (DEGs), we employed the “clusterProfiler” package for Gene Ontology (GO) analysis, where pathways with a P-value < 0.05 and q-value < 0.05 were considered significantly enriched. Furthermore, “h.all.v7.5.1.symbols.gmt” served as the preconceived gene set, employing the “GSVA” package for conducting gene set variation analysis (GSVA) to investigate the heterogeneity of various biological processes. The t-values indicated discrepancies in pathway activities between the high-risk and low-risk groups. The enrichment analysis results were visualized using the “ggplot2” and “GseaVis” packages.
In vitro Assays
The hFOB1.19 osteoblastic cell line and the U2-OS osteosarcoma cell line were cultured in DMEM medium containing 10% fetal bovine serum and 1% penicillin-streptomycin, at a temperature of 37°C with 5% CO2. When the cellular density converged to 75–85%, passaging was conducted. U2-OS cells and their corresponding transfected cells were cultured in Gibco BRL’s Dulbecco’s Modified Eagle Medium (DEME) containing 10% fetal bovine serum. The cells were seeded at a density of 2×103 cells per well in a 96-well plate with triplicate samples. Cell proliferation was assessed using the CCK-8 method. Logarithmically growing U2-OS cells were seeded into a 6-well plate at a cell density of 2×105. Upon reaching 50% confluence, the cells were transfected. According to the instructions of the LipofectamineTM 2000 transfection kit, untreated cells were employed as the control group, and substituted with fresh complete growth medium six hours post-transfection. After 24 hours of cultivation, the expression of RAD21 in the cells was assessed using RT-qPCR, with the plasmid transfection originating from the esteemed company, Jima. Following the manufacturer’s instructions, total RNA was extracted from the cell line using TRIzol, and cDNA was synthesized using the PrimeScriptT-MRT reagent kit. Using the SYBR Green Master Mix for RT-qPCR, the mRNA expression levels were normalized to GAPDH mRNA levels. The expression levels were calculated using the 2−ΔΔCt method, and the primers were obtained from the Qinko Corporation.
Results
Classification of PTM-Related Tumor Clusters
Reactome is a database encompassing knowledge of biological pathways. In sequence, we queried genes associated with the processes of ubiquitination, SUMOylation, and neddylation. Seventy-nine, one hundred eighty-seven, and two hundred forty-five genes were respectively associated with ubiquitination, SUMOylation, and neddylation. Noteworthy was the fact that BRCA1, BIRC5, and VHL collectively partook in the processes of SUMOylation and neddylation; PCNA was involved in SUMOylation and ubiquitination processes; UBE2D1, UBE2D3, UCHL3, UBE2D2, RPS27A, UBC, VCP, UBB, and UBA52 participated in the processes of ubiquitination and neddylation. Therefore, after eliminating certain duplicated genes, we designated the identified 498 genes as PTMRGs (Figure 1A).
 |
Figure 1 Consensus clustering analysis of 40 PTMRGs. (A) Venn plot of SUMOylation-related genes, ubiquitination-related genes, and neddylation-related genes. (B) Univariate Cox regression of PTMRGs for the screening of prognosis-related genes. (C) Heatmap of the consensus matrices for k = 2. (D) The CDF curves for clusters at k = 2 to 9. (E) Kaplan–Meier curves based on two clusters in the TARGET cohort. (F) PCA plot for the two clusters in the TARGET cohort. (G) Heatmap and the clinical parameters of the two clusters. (H) Kaplan–Meier curves based on two clusters in the GSE21257 cohort. |
The univariate Cox regression analysis identified 40 prognostically relevant PTMRGs (Figure 1B). In the TARGET queue, employing the expression profile formed by the aforementioned forty prognostic-associated PTMRGs, we partitioned all tumor samples into k subtypes using the R software package “ConsensusClusterPlus”. Based on the heat map (Figure 1C) and the CDF curve (Figure 1D) consensus scores, it was concluded that k = 2 represented the optimal grouping. Survival analysis indicated that the overall survival rate of B subtype was markedly lower than that of A subtype (Figure 1E). In addition, PCA showed that the above two subtypes had obvious transcriptome heterogeneity (Figure 1F), suggesting that there may be two different PTM modes in OS. We demonstrated the differential expression of two subtypes and PTMRGs in the TARGET cohort. Upon examination, it was deduced that the existence of two distinct PTM patterns might arise from the differential expression of PTMRGs. For instance, in the B subtype, there were elevated expressions of NOP58, SATB2, DCUN1D5, and RPS27A, whereas in the A subtype, HDAC7, PML, CISH, UBE2Z, and BCL10 exhibited heightened expressions (Figure 1G). Finally, in order to further validate the robustness of this classification, we conducted verification within the GSE21257 cohort, and the Results equally demonstrated a poorer prognosis for subtype B (Figure 1H).
Immune Microenvironment Differences of Different PTM-Related Tumor Clusters
The distribution of immune cells emphasizes the distinct tumor immune microenvironment (TIME) in the two OS subtypes. Through the implementation of ssGSEA analysis, we conducted a more in-depth examination of the composition of diverse immune cell populations in distinct samples. The utilization of box plots effectively highlighted and elucidated the disparities in immune cell content among various subtypes. In all types of immune cells, The proportion of Activated B cell, Immature B cell, MDSC, Natural killer cell, Regulatory T cell and Type1 T helper cell in B subtype was significantly higher than that in A subtype (Figure 2A). The A subtype possessed a greater capacity for stimulating the adaptive immune system, as evidenced by their elevated immune matrix score (Figure 2B) and human leukocyte antigen (HLA) activity (Figure 2C). This could potentially account for the superior survival prognosis observed in this subtype. In our investigation, we conducted a comparative analysis of the expression patterns of immune checkpoint inhibitors (ICIs) across various subtypes. Notably, subtype A exhibited notably elevated mRNA expression levels in a majority of ICIs, including but not limited to PDCD1LG2, TNFRSF18, LAIR1, and CD27 (Figure 2D).
 |
Figure 2 Analysis of immune cell content and immune checkpoint. (A) Different immune cell infiltrations were compared between two clusters. (B) Immune score and stromal score of the subclasses generated by the “ESTIMATE” algorithm. (C) HLAs expression level between two clusters. (D) Differences in the abundance of immune-checkpoint-related genes between two clusters. *P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant. |
Chemotherapy Sensitivity and Pathway Differences
Next, we explored the correlation between two subtypes and the response to chemotherapy. Our investigation revealed a significant association between subtype A and a decreased half maximal inhibitory concentration (IC50) for AP24534 and AS601245, indicating enhanced sensitivity to chemotherapy, but ABT263 and BAY613606 were just the reverse (Figure 3A).
 |
Figure 3 Chemotherapy sensitivity and pathway enrichment analysis. (A) Differences in IC50 of chemotherapy drugs between two clusters. (B) Volcano plot of DEGs, red for high expression and blue for low expression. (C) GO enrichment results of DEGs. (D) KEGG enrichment results between two clusters. |
Upon evaluating the DEGs in both clusters (|DEG_logFC| > 1, DEG_p value < 0.05), a total of 831 genes were identified to exhibit significant differential expression across two clusters (Figure 3B). The application of GO analysis demonstrated a notable enrichment of DEGs in processes associated with extracellular matrix organization and extracellular matrix structural constituent (Figure 3C). Upon KEGG analysis, it was evident that subtype B was mainly involved in the biological synthesis and metabolism of substances, whereas the A subtype was associated with immunity, encompassing the interplay between the MAPK signaling pathway and cytokines and receptors (Figure 3D).
Building a Predictive Signature
We constructed a prognostic model employing eleven PTMRGs, identified through univariate Cox regression analysis in the TARGET cohort, which exhibited statistically significant correlation with overall survival. The research employed LASSO Cox regression and multivariate Cox regression analyses to ascertain a final set of six genes (Figure 4A and B). The risk score was calculated by taking into account the coefficients and expression patterns of six crucial genes integrated into the model (Figure 4C). 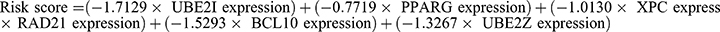 .
.
 |
Figure 4 Construction and validation of PTMRGs prognostic model. (A and B) Six genes were selected for multivariate regression analysis using Lasso regression. (C) Multivariate Cox regression analysis of PTMRGs was shown by forest plot. (D) The relationship between model genes and survival. (E) Correlation analysis between model genes and risk score. (F and G) Kaplan-Meier survival analysis of risk score in the TARGET and GSE21257 cohorts. |
Figure 4D illustrated the influence of individual genes on survival. The research outcomes indicated that certain genes such as BCL10, PPARG, UBE2I, UBE2Z, and XPC displayed a protective effect, suggesting that attenuating their expression might hold promise in enhancing overall survival. Conversely, RAD21 emerged as a gene associated with heightened risk, as increased expression correlated with a reduction in patient survival. The crucial aspect was that the expression levels of these six genes and their correlation with the risk score were validated (Figure 4E). According to the median risk score, patients were categorized into high-risk and low-risk subgroups. Subsequent survival analysis confirmed that the overall survival period of the high-risk subgroup was significantly shorter than that of the low-risk subgroup in both the TARGET and GSE21257 cohorts (Figure 4F and G).
The Correlation Between Risk Score and Immunotherapy Response and Immune Microenvironment
In this study, a comprehensive analysis of the immune microenvironment was conducted, employing the CIBERSORTx method to precisely assess the infiltration levels of 22 distinct immune cell types. The resulting immune landscape of the TARGET-OS samples was visually represented in Figure 5A. Figure 5B illustrated that the low-risk group exhibited significantly activated NK cells, whereas there were no apparent discrepancies in the levels of other immune cells between the two risk subgroups. In our investigation, we conducted an assessment of the correlation between the six genes proposed in the model and the abundance of immune cells. Notably, a substantial and statistically significant correlation was detected between the expression levels of these six genes and the presence of various immune cell types (Figure 5C).
 |
Figure 5 Immunological landscape of the two risk subgroups. (A) The heatmap displayed the proportion of immune cell infiltration in each OS sample. (B) Differences in immune cell infiltration between low- and high-risk groups. (C) Correlations between immune cell content and model genes. (D and E) Sensitivity prediction of different subgroups to the two immune checkpoint inhibitors. *P < 0.05, **P < 0.01, ***P < 0.001. |
The advent of immunotherapy, exemplified by PD-1/PD-L1 checkpoint inhibitors, marked a momentous breakthrough in the management of neoplastic conditions. Considering the potential therapeutic effects of immunotherapy, specifically PD-1 and PD-L1 immune checkpoint inhibitors, in diverse malignancies, encompassing OS (osteosarcoma), we conducted additional analysis to evaluate the prognostic significance of the risk score within the IMvigor210 and GSE78220 cohorts. We conducted an evaluation of the association between the risk score and the response to immune therapy in the IMvigor210 cohort, which comprised individuals diagnosed with advanced urothelial carcinoma receiving PD-L1 blockade treatment. Patients belonging to the low-risk category demonstrated pronounced survival benefits as opposed to those in the high-risk cohort. The risk score of patients within the progressive disease (PD)/stable disease (SD) cohort exhibited a notably elevated magnitude in contrast to the partial response (PR)/complete response (CR) cohort. It is important to highlight that the prevalence of PR/CR in the high-risk cohort exhibited a notably lower occurrence compared to the low-risk cohort. Nevertheless, the proportion of PD/SD patients demonstrated an opposing trend, indicating that risk score could unveil patients’ response to ICB therapy (Figure 5D). A similar analysis was undertaken within the GSE78220 cohort, which consisted of individuals afflicted with melanoma and undergoing PD-1 blockade therapy. In contrast to patients categorized as high-risk, those classified as low-risk demonstrated prominent survival benefits. The risk score of the PD cohort exhibited a statistically significant elevation when contrasted with the group of PR/CR. The remarkable observation in this study was that the low-risk subtype accounted for the majority (80%) of cases within the PR/CR group, whereas the high-risk subtype was prominently represented (65%) in the PD group. This findings suggested that the risk score exhibited potential as a promising prognostic marker for assessing the immunotherapeutic response in patients with OS (Figure 5E).
Analysis of Enrichment
To delve further into exploring the alterations in pathways among patients from different risk groups, we performed GO enrichment analysis on DEGs between high and low risk groups. The findings demonstrated that biological processes (BP) primarily encompassed lymphocyte differentiation, mononuclear cell differentiation, and the regulation of T cell activation. Regarding cellular component (CC), the external surface of the plasma membrane, the immunological synapse, and the T cell receptor complex exhibited the highest levels of representation. The primary categories observed in molecular function (MF) were immune receptor activity, G protein-coupled purinergic nucleotide, and receptor activity cytokine receptor binding (Figure 6A).
 |
Figure 6 Enrichment analysis and functional annotation. (A) GO enrichment analysis. (B) GSVA shows the enrichment of hallmark gene sets in different risk groups. (C) GSEA enrichment method showed pathway differences between high- and low-risk groups. |
Analysis of hallmark pathway gene signatures underscored that the high and low-risk groups exhibited some distinctions. The top 5 enriched signatures in the high-risk group included coagulation, xenobiotic metabolism, complement, apoptosis, and pancreas beta cells. The top 5 enrichment features in the low-risk group included estrogen response early, cholesterol homeostasis, estrogen response late, Wnt β-catenin signaling and unfolded protein response (Figure 6B).
GSEA was performed on both the high- and low-risk cohorts, as depicted in Figure 6C. The GSEA enrichment analysis results revealed that OS patients with a high-risk profile exhibited significant enrichment in processes related to antigen processing and presentation, as well as the cytosolic DNA sensing pathway. On the other hand, OS patients classified as low-risk displayed enrichment in processes associated with primary immunodeficiency and the chemokine signaling pathway.
Validation of the Signature Genes Expression and Biological Function in OS
Utilizing the annotation data available in the TISCH database, we successfully categorized the single-cell dataset into eight distinct cell types: plasma cells, conventional CD4 T cells (CD4 Tconv), exhausted CD8 T cells (CD8 Tex), endothelial cells, malignant cells, fibroblasts, monocytes/macrophages (mono/macro), and osteoblasts (Figure 7A). Following this, we conducted an investigation into the expression of key genes implicated in the risk recognition system. The results revealed a notable upregulation of RAD21 in malignant cells (Figure 7B). Furthermore, the RNA-seq data from diverse cellular lineages in the HPA database reaffirmed the ubiquitous expression of RAD21 in mesenchymal cell lines (Figure 7C).
 |
Figure 7 Validation of the expression of the signature genes in OS. (A) Cells were annotated into eight different cell types. (B) Expression of different risk genes in different cells. (C) The expression of RAD21 in RNA-seq data of different cell lines in HPA database. (D) Cellular localization of RAD21, taking U2-OS cells as an example. (E) The relationship between RAD21 expression and cell cycle. (F) Analysis of RAD21 mRNA expression in human osteosarcoma cell U2-OS versus normal human osteoblasts hFOB1.19. (G) The transfection efficacities of three siRNAs in human osteosarcoma cell U2-OS. (H) U2-OS cell proliferation after anti-RAD21 siRNA transfection was measured using CCK-8 assays. **P < 0.01, ****P < 0.0001. |
Subsequently, we conducted an analysis of the U2-OS osteosarcoma cell line, and fluorescence staining revealed that RAD21 might be localized within the cell nucleus (Figure 7D). Subsequent cell cycle analysis suggested that cells in the S-phase exhibited diminished expression of RAD21 (Figure 7E). Subsequently, RT-qPCR was employed to assess the mRNA expression levels of RAD21 in OS cell lines (U2-OS) and normal osteoblast cell line (hFOB1.19) (Figure 7F). The findings revealed a notable up-regulation of RAD21 expression in OS cells when compared to osteoblast hFOB1.19. Three different siRNAs were assessed for their transfection efficiency, and it was observed that siRNA significantly inhibited the proliferation of OS cell (Figure 7G). The transfection of the corresponding siRNAs into U2-OS cells resulted in a notable reduction in cell viability, as evidenced by the findings from the CCK-8 assay (Figure 7H). The findings of that study indicated that RAD21 possibly promotes tumorigenesis of OS, although the precise underlying mechanism warranted further investigation.
Discussion
In this literary piece, we embarked upon an exploration of genes associated with the processes of ubiquitination, SUMOylation, and neddylation within the Reactome database. We have effectively ascertained a total of 498 PTMRGs. Subsequently, univariate Cox regression analysis was employed to identify 40 PTMRGs demonstrating significant prognostic value. Employing unsupervised clustering based on the expression levels of these genes, we successfully categorized the patients into two discernible genomic subtypes, denoted as subtype A and B. Notably, patients falling under subtype A exhibited a notably favorable survival outcome in comparison.
The research results revealed a noteworthy association between subtype A and elevated levels of immune cell infiltration and expression of ICIs. Significantly, our findings indicate that subtype A exhibited a noteworthy level of sensitivity towards AP24534 and AS601245 treatment. In contrast, subtype B displayed lower predicted IC50 values in response to ABT263 and BAY613606 treatment. In our investigation, we conducted a KEGG enrichment analysis comparing the two distinct gene clusters. The results revealed that subtype B exhibited a substantial enrichment in pathways associated with metabolism and substance biosynthesis, while gene subtype A showed a notable enrichment in pathways related to immune response.
Moreover, in light of the distinct individual variations and intricate nature of PTM, an essential undertaking was to investigate the underlying PTM patterns present in each individual. Consequently, we developed a comprehensive risk scoring system aimed at quantifying the PTM profile of individual patients suffering from OS. Patients in the high-risk category exhibited a diminished life expectancy. To elucidate this phenomenon, we conducted a comparative analysis of the tumor microenvironment (TME) between the high-risk and low-risk groups. It is crucial to acknowledge that OS have historically been classified as “cold” tumors, exhibiting a notable level of immunosuppression. This phenomenon may be attributed to our limited comprehension of the TME in patients with OS. Additional investigation into the TME of OS is imperative to establish a robust groundwork for the development and application of immunotherapeutic approaches. Surprisingly, we did not observe any significant disparities in TME immune cell infiltration between the high and low-risk groups. Additionally, most immune cells showed no evident variations in distribution between the low and high-risk groups. Nevertheless, we discerned a noteworthy correlation between model genes and immune cell infiltration. In our study, we conducted a comprehensive examination of the pathway enrichment in both high- and low-risk groups, with the aim of elucidating the fundamental pathways and biological mechanisms underlying PTM patterns. Our findings revealed that the high-risk group exhibited significant enrichment in pathways closely associated with cell proliferation and tumorigenesis, aligning well with the observed unfavorable prognosis.
Over the past decade, there has been a rapid development of immunotherapies involving ICBs specifically directed against PD-1 and PD-L1, demonstrating significant progress in the treatment of various solid tumors. The utilization of atezolizumab,17 nivolumab,18 and pembrolizumab19 in clinical trials has yielded encouraging findings, propelling immunotherapy into an emerging therapeutic option for individuals afflicted with advanced-stage and metastatic conditions.20 Nevertheless, the efficacy of ICB therapy in treating patients with OS remains limited, with only around 20% of patients exhibiting positive responses due to the intricate molecular heterogeneity of the disease.21 Therefore, there is a pressing need to identify specific biomarkers and genomic signatures that can reliably predict and discern patients who are likely to experience favorable outcomes from immunotherapies. Subsequently, we conducted an in-depth validation of the correlation between the risk score and the responsiveness to immunotherapy. This validation was based on the data obtained from both the IMvigor 210 cohort and the GSE78220 cohort. Unsurprisingly, our investigation revealed a notable correlation between low-risk status and enhanced responsiveness to immunotherapy. Consequently, our findings demonstrate that PTM patterns play a noteworthy role in discerning distinct TME. Consequently, these PTM patterns hold significant promise as effective biomarkers for identifying patients who are suitable candidates for immunotherapy.
Nevertheless, it is crucial to acknowledge the presence of various inadequacies and constraints within our study. Firstly, the identification of numerous novel PTM-related proteins has emerged as a noteworthy development. However, it is crucial to acknowledge that the pool of PTMRGs we assembled and employed for our analyses may not be entirely exhaustive. This potential limitation could introduce a degree of bias into our research investigations. Secondly, the present omics datasets solely offer insights into mRNA levels, whereas the PTM mechanisms are reliant on proteins. This discrepancy may introduce certain inaccuracies in the analyses. Thirdly, our study is constrained by a limited number of clinical samples, we lack verification from additional clinical data sets beyond the publicly available data. The inclusion of such data would be invaluable in bolstering the validity of our conclusions. Furthermore, the role of PTM in other types of cancer remains unverified. Hence, we are fully poised to procure additional clinical samples to corroborate our findings comprehensively and to evaluate the significance of PTM in the context of other malignant bone tumors. Finally, the precise underlying mechanisms governing the interplay between PTM patterns and the infiltration of immune cells within the TME have not been fully elucidated, demanding additional comprehensive investigations.
Conclusion
In Conclusion, our study has successfully discerned two distinct PTM patterns within OS patients, exhibiting divergent contrasting immune features and prognostic outcomes. The findings of our study present a novel reference point for devising management approaches targeting advanced OS.
Study Approval
All datasets in the present study were downloaded from public databases. These public databases allowed researchers to download and analyze public datasets for scientific purposes. The current research follows the TCGA data access policies and publication guidelines. Users can download relevant data for free, our study is based on open-source data, there are no ethical issues and other conflicts of interest. Inclusion of the Human cell lines (purchased from the Cell Repository of Shanghai Institute of Cell Science, Chinese Academy of Sciences) in the study were approved by Hebei Chest Hospital review board.
Acknowledgments
We express our profound appreciation for the data supplied by repositories such as TARGET and GEO. We would like to acknowledge reviewers and editors for their helpful comments on this paper.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Ritter J, Bielack SS. Osteosarcoma. Ann Oncol. 2010;21(Suppl 7):vii320–vii325. doi:10.1093/annonc/mdq276.
2. Cole S, Gianferante DM, Zhu B, Mirabello L. Osteosarcoma: a surveillance, epidemiology, and end results program-based analysis from 1975 to 2017. Cancer. 2022;128(11):2107–2118. doi:10.1002/cncr.34163.
3. Dean DC, Shen S, Hornicek FJ, Duan Z. From genomics to metabolomics: emerging metastatic biomarkers in osteosarcoma. Cancer Metastasis Rev. 2018;37(4):719–731. doi:10.1007/s10555-018-9763-8.
4. Dana PM, Sadoughi F, Asemi Z, Yousefi B. Molecular signaling pathways as potential therapeutic targets in osteosarcoma. Curr Med Chem. 2022;29(25):4436–4444. doi:10.2174/0929867329666220209110009.
5. Harrison DJ, Geller DS, Gill JD, Lewis VO, Gorlick R. Current and future therapeutic approaches for osteosarcoma. Expert Rev Anticancer Ther. 2018;18(1):39–50. doi:10.1080/14737140.2018.1413939.
6. Rickel K, Fang F, Tao J. Molecular genetics of osteosarcoma. Bone. 2017;102:69–79. doi:10.1016/j.bone.2016.10.017.
7. Lee JM, Hammarén HM, Savitski MM, Baek SH. Control of protein stability by post-translational modifications. Nat Commun. 2023;14(1):201. doi:10.1038/s41467-023-35795-8.
8. Pan S, Chen R. Pathological implication of protein post-translational modifications in cancer. Mol Aspects Med. 2022;86:101097. doi:10.1016/j.mam.2022.101097.
9. Hermann J, Schurgers L, Jankowski V. Identification and characterization of post-translational modifications: clinical implications. Mol Aspects Med. 2022;86:101066. doi:10.1016/j.mam.2022.101066.
10. Salas-Lloret D, González-Prieto R. Insights in post-translational modifications: Ubiquitin and SUMO. Int J Mol Sci. 2022;23(6):3281. doi:10.3390/ijms23063281.
11. Gâtel P, Piechaczyk M, Bossis G. Ubiquitin, SUMO, and Nedd8 as therapeutic targets in cancer. Adv Exp Med Biol. 2020;1233:29–54. doi:10.1007/978-3-030-38266-7_2.
12. Pellegrino NE, Guven A, Gray K, et al. The next Frontier: translational development of ubiquitination, SUMOylation, and NEDDylation in cancer. Int J Mol Sci. 2022;23(7):3480. doi:10.3390/ijms23073480.
13. Gallo LH, Ko J, Donoghue DJ. The importance of regulatory ubiquitination in cancer and metastasis. Cell Cycle. 2017;16(7):634–648. doi:10.1080/15384101.2017.1288326.
14. Krueger KE, Srivastava S. Posttranslational protein modifications: current implications for cancer detection, prevention, and therapeutics. Mol Cell Proteomics. 2006;5(10):1799–1810. doi:10.1074/mcp.R600009-MCP200.
15. Necchi A, Joseph RW, Loriot Y, et al. Atezolizumab in platinum-treated locally advanced or metastatic urothelial carcinoma: post-progression outcomes from the Phase II IMvigor210 study. Ann Oncol. 2017;28(12):3044–3050. doi:10.1093/annonc/mdx518
16. Hugo W, Zaretsky JM, Sun L, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2017;168(3):542. doi:10.1016/j.cell.2017.01.010
17. Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, Phase 2 trial. Lancet. 2016;387(10031):1909–1920. doi:10.1016/S0140-6736(16)00561-4
18. Sharma P, Callahan MK, Bono P, et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicentre, open-label, two-stage, multi-arm, Phase 1/2 trial. Lancet Oncol. 2016;17(11):1590–1598. doi:10.1016/S1470-2045(16)30496-X
19. Balar AV, Kamat AM, Kulkarni GS, et al. Pembrolizumab monotherapy for the treatment of high-risk non-muscle-invasive bladder cancer unresponsive to BCG (KEYNOTE-057): an open-label, single-arm, multicentre, phase 2 study. Lancet Oncol. 2021;22(7):919–930. doi:10.1016/S1470-2045(21)00147-9
20. Witjes JA, Bruins HM, Cathomas R, et al. European Association of Urology Guidelines on muscle-invasive and metastatic bladder cancer: summary of the 2020 guidelines. Eur Urol. 2021;79(1):82–104. doi:10.1016/j.eururo.2020.03.055
21. Wen Y, Tang F, Tu C, Hornicek F, Duan Z, Min L. Immune checkpoints in osteosarcoma: recent advances and therapeutic potential. Cancer Lett. 2022;547:215887. doi:10.1016/j.canlet.2022.215887
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.