")
Back to Journals » International Journal of General Medicine » Volume 18
Molecular Subtyping of Hepatocellular Carcinoma via Lysosome-Related Genes for Prognosis and Therapy Prediction
Authors Yao Y, Zhu T, Shen X , Ma J, Zhu X , Jiang J
Received 26 November 2024
Accepted for publication 11 June 2025
Published 16 July 2025 Volume 2025:18 Pages 3933—3950
DOI https://doi.org/10.2147/IJGM.S490019
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Kenneth Adler
Yiyang Yao,1,* Tong Zhu,2,* Xiaoyi Shen,3 Junyong Ma,4 Xudong Zhu,5,6 Jie Jiang7
1Department of Gastroenterology, Qidong People’s Hospital, Nantong, People’s Republic of China; 2Department of Breast Surgery, Panjin Central Hospital, Panjin, People’s Republic of China; 3Department of General Practice, Tongren Hospital, Shanghai Jiao Tong University, School of Medicine, Shanghai, People’s Republic of China; 4Department of Hepatic Surgery, Eastern Hepatobiliary Surgery Hospital, Navy Medical University, Shanghai, People’s Republic of China; 5Department of General Surgery, Cancer Hospital of China Medical University, Cancer Hospital of Dalian University of Technology, Liaoning Cancer Hospital & Institute, Shenyang, People’s Republic of China; 6Liaoning Provincial Key Laboratory of Precision Medicine for Malignant Tumors, Shenyang, People’s Republic of China; 7Department of Gastroenterology, Nanjing Drum Tower Hospital, The Affiliated Hospital of Nanjing University Medical School, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xudong Zhu, Department of General Surgery, Cancer Hospital of China Medical University, Cancer Hospital of Dalian University of Technology, Liaoning Cancer Hospital & Institute, Liaoning Provincial Key Laboratory of Precision Medicine for Malignant Tumors, Shenyang, 110042, People’s Republic of China, Tel +86 13354204706, Email [email protected] Jie Jiang, Department of Gastroenterology, Nanjing Drum Tower Hospital, The Affiliated Hospital of Nanjing University Medical School, Nanjing, 210008, People’s Republic of China, Tel +86 13162999914, Email [email protected]
Background: Lysosomes play an important role in the pathological processes of cancer development. However, its effects on the prognosis and tumor microenvironment of hepatocellular carcinoma (HCC) remain unclear. Therefore, we aim to explore the novel molecular subtypes of HCC via lysosome-related genes (LRGs) for prognosis and therapy prediction in this study.
Methods: Using the data of TCGA, differential expression and survival analyses were performed. Consequently, 109 key LRGs were obtained and 374 HCC samples were clustered into two groups: C1 and C2. A three-gene prognostic prediction nomogram was constructed using WCGNA and Cox regression analyses. Furthermore, pathway enrichment conditions, immune infiltration, immune checkpoint expression, and drug sensitivity were analyzed for the two subtypes. RT-qPCR was also used to validate the expression of the selected key LRGs.
Results: Key LRGs were highly expressed in the C1 subtype, and their prognosis was worse. The degree of immune cell infiltration and pathway enrichment results were also significantly different between the two subtypes. Furthermore, the three-gene prognostic prediction nomogram including LAPTM4B, PRKCD and LPCAT1, had a relatively high prognostic prediction ability. Meanwhile, the expression of immune checkpoints, human leukocyte antigen, and TIDE score were higher in the C1 subtype, suggesting that immune evasion was more likely to occur in this subtype. Drug sensitivity analysis showed that several drugs were more sensitive to C1 subtypes and might serve as drug candidates for these patients.
Conclusion: We identified two novel molecular subtypes of HCC based on LRGs, and found that the LRGs related subtypes demonstrated significant efficacy in predicting the prognosis and therapeutic outcomes for patients with HCC. Moreover, a novel prognostic prediction nomogram was also developed, which possessed excellent prognostic prediction capabilities. We hope the novel LRG-related subtypes and nomogram of HCC would provide new insights and guidelines for clinical practice in the future.
Keywords: hepatocellular carcinoma, lysosomes, tumor immune, prognosis, tumor microenvironment, molecular subtypes
Introduction
Liver cancer ranks sixth in morbidity and third in mortality among all kinds of malignant tumors worldwide, with almost 906,000 new cases and 83,000 new deaths each year, making it a serious threat to human’s health.1 Hepatocellular carcinoma (HCC) is the most common primary liver cancer and accounts for 80% of all cases. Its risk factors include hepatitis B virus (HBV) or hepatitis C virus (HCV) infection, alcohol ingestion, metabolic syndrome, and cirrhosis.2–4 In terms of treatment, it is essential to develop an individualized and multidisciplinary approach based on the clinical stages and systemic conditions of specific patients with HCC. Liver resection and transplantation can be performed in patients with early-stage disease. For patients with unresectable advanced HCC, transarterial chemoembolization (TACE) and radiofrequency ablation (RFA) are performed.5,6 Immunotherapy, systemic chemotherapy, and targeted therapy are widely used to treat advanced HCC. Several studies have reported that immunotherapy in combination with targeted therapy is superior to single-agent targeted therapy in terms of patient prognosis, which has become the preferred treatment for advanced HCC.6–10 However, the median overall survival (OS) and progression-free survivals (PFS) are only 19.2 months and 6.8 months respectively for patients with HCC.11 This may be attributed by the high heterogeneity of HCC. Therefore, it is necessary to differentiate patients with HCC more precisely at the molecular level.
Lysosomes are unique organelles that were first discovered by Prof. Christian de Duve in the 1950s.12 Since their discovery, extensive research has revealed that lysosomes are not merely the cell’s recycling center but also play central roles in cellular degradation, metabolic regulation, and signal transduction.13,14 They are critical for processes such as autophagy, immunoregulation, and nutrient sensing, and their dysfunction is implicated in both tumor and non-tumor diseases.15,16 In cancers, lysosomes regulate tumor cell proliferation, invasion, and treatment resistance, making them a focal point for therapeutic exploration. For instance, Pechincha et al demonstrated that the lysosomal enzyme trafficking factor (LYSET) is essential for energy uptake in nutrient-deficient conditions, a common feature of the tumor microenvironment (TME).17 Notably, the growth of various tumors is significantly inhibited by LYSET knockout, highlighting the potential of targeting lysosomal pathways as a promising strategy for cancer treatment.18 These findings underscore the importance of investigating lysosome-related genes (LRGs) in HCC, as they may reveal novel molecular subtypes and provide valuable insights for personalized therapeutic approaches.
The TME is a dynamic and complex ecosystem that plays pivotal roles in cancer progression. Composed of immune cells (eg, macrophages, T lymphocytes, dendritic cells), cancer-associated fibroblasts (CAFs), vascular networks, the extracellular matrix (ECM), and a myriad of signaling molecules, the TME influences key cancer hallmarks such as apoptosis, evasion, proliferation, invasion, and metastasis.19–22 Importantly, the TME has emerged as a promising therapeutic target, with numerous drugs now designed to modulate its components, including immune checkpoint inhibitors and anti-angiogenic agents.23–27 Lysosomes are intricately linked to the TME, regulating critical biological processes such as the polarization of M2 macrophages, which promote tumor progression,28 and the antigen-presenting capacity of dendritic cells, which shape anti-tumor immune responses.29 Despite these advances, the specific mechanisms by which LRGs interact with the TME to drive HCC progression remain poorly understood. Elucidating these mechanisms is crucial, as it could uncover new therapeutic targets and pave the way for individualized treatment strategies for patients with HCC in the future.
In this study, bulk-RNA data for HCC and adjacent normal tissues were obtained from public databases. Differentially expressed LRGs were analyzed, and COX regression analysis was used to screen prognosis-related genes in HCC. Clustering analysis was used to identify two novel molecular subtypes based on the genes related to lysosomes in HCC. Furthermore, we analyzed pathway enrichment conditions, immune infiltration, immune checkpoint expression, and drug sensitivity in the two subtypes. In addition, we constructed a novel multiple-factors prognosis prediction model based on the screened LRGs. Finally, we hope that our results will provide new insights and guide clinical practice in the future.
Materials and Methods
Data Collection and Processing
Bulk-RNA data and clinical information of 374 patients with HCC were obtained from liver hepatocellular carcinoma (LIHC) cohort in The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov). Data from the validation sets were downloaded from the International Cancer Genome Consortium (ICGC) database (https://docs.icgc-argo.org/docs/data-access/icgc-25k-data) and GSE database (GSE14520). 869 LRGs were acquired from Molecular Signature Database v7.5.1 (MSigDB, https://www.gsea-msigdb.org/gsea/msigdb/), which were shown in Supplementary Table 1. The entire analytical process of this study is presented in this flowchart (Figure 1).
 |
Figure 1 Flow chart of this study. |
Identification of Differentially Expressed LRGs
In this study, differentially expressed analysis was utilized to screen LRGs related to HCC with the “edgR” R package. To identify prognosis-associated LRGs, Cox proportional hazard regression analysis was performed using the prognosis data from the LIHC cohort of TCGA and the “survival” R package.
ConsensusClusterPlus Analysis of LRGs
The “ConsensusClusterPlus” R package was used to perform Consensus Cluster Plus analysis of clinical samples in TCGA-LIHC. The intersection of differentially expressed LRGs and prognosis-associated LRGs was selected as the key LRGs, which were mapped to HCC samples. By subsampling the proportion of features from these genes, an agglomerative hierarchical clustering algorithm was used to partition each sample into k clusters. The clustering process was repeated nine times. The clustering proportion of the key LRGs groups was defined by pairwise consensus values, which were calculated and integrated into the consensus matrix (CM), and the HCC samples were divided into k clusters. These clusters were defined as the consensus clusters (HCC subtypes). Moreover, the number of clusters and confidence in cluster sample qualifications were calculated based on quantitative and visual stability evidence. Principal component analysis (PCA) was also performed to minimize bias resulting from the categorical variable analysis.
Survival and Pathway Enrichment Analysis of Different Molecular Subtypes
Firstly, to compare the prognoses of the two subtypes, Kaplan-Meier (KM) survival analysis was performed using the “Kaplan-Meier survival” and “survminer” R packages. The Kaplan Meier survival curve was used to show the survival difference of the two subtypes. Then, with “Cluster profiler” R package, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) function and pathway enrichment analysis were performed to further explore downstream signaling pathways which key LRGs were mostly enriched in. Gene Set Variation Analysis (GSVA) was used to obtain differentially expressed gene sets in “h.all.v7.4. symbols.gmt” among the subtypes. Gene set enrichment analysis (GSEA) was also conducted on the selected genes expressing in both subtypes and the top 5 hallmark pathways were shown.
TME and Immune Infiltration Analysis
To analyze the differences of TME among different subtypes, Estimation of STromal and Immune cells in MAlignant Tumors using Expression data (ESTIMATE) analysis was performed by utilizing the R package “estimate”. The infiltration degree of various immune cells in different subtypes was also calculated by Cell-type Identification By Estimating Relative Subsets Of RNA Transcripts (CIBERSORT) algorithm.30–33 Single-Sample Gene Set Enrichment Analysis (ssGSEA) was used to evaluate and quantify the enrichment levels of immune-related sets in different subtypes.
Identification of Alternative Genes by Weighted Gene Correlation Network Analysis (WGCNA)
To further screen for alternative genes to construct a prognosis prediction model, WGCNA was performed. After determining the fittest soft-thresholding power for each subtype, the key LRGs were clustered into several modules. The degree of immune infiltration was considered a clinical phenotype for further co-expression analysis, and a module-trait relationship heatmap was created to find the module most associated with the degree of immune cell infiltration. The intersection of the results for each subtype was identified as an alternative gene used to construct the prognosis prediction model.
Independent Prognostic Analysis, Assessment of Clinical Relevance, and Construction of a Nomogram
To further assess the prognostic value of these alternative genes, a multivariate Cox regression analysis was performed. Subsequently, a prognostic model was constructed according to the formula (Risk Score = 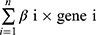 ), which could be utilized to predict the prognosis of patients with HCC. In this formula, “n” represented OS-associated LRGs, and “β” represented the coefficient of each OS-associated LRG. To further assess the clinical relevance of the prognostic model and OS-associated LRGs, gene expression heatmaps, survival status scatter plots, and risk-score distribution plots were constructed. Simultaneously, the AUC curves were used to test the accuracy of this prognostic mode using the R package “timeROC”. A nomogram was constructed to intuitively display the relationships between OS-associated LRGs and prognosis. The regression model was fitted and the nomogram was depicted utilizing the R package “rms”. The 1-year survival, 3-year survival, and 5-year survival rates could be illustrated clearly based on the expression of the selected key LRGs.
), which could be utilized to predict the prognosis of patients with HCC. In this formula, “n” represented OS-associated LRGs, and “β” represented the coefficient of each OS-associated LRG. To further assess the clinical relevance of the prognostic model and OS-associated LRGs, gene expression heatmaps, survival status scatter plots, and risk-score distribution plots were constructed. Simultaneously, the AUC curves were used to test the accuracy of this prognostic mode using the R package “timeROC”. A nomogram was constructed to intuitively display the relationships between OS-associated LRGs and prognosis. The regression model was fitted and the nomogram was depicted utilizing the R package “rms”. The 1-year survival, 3-year survival, and 5-year survival rates could be illustrated clearly based on the expression of the selected key LRGs.
RT-qPCR Validation of Selected Key LRGs
Total RNA was isolated from ten pairs of HCC and normal tissues using TRIzol solution (Solarbio). The isolated RNA was reverse transcribed using a cDNA synthesis kit (Vazyme). RT-qPCR was performed using SYBR Green PCR Master Mix (Vazyme) and primers binding to the target genes, and GAPDH. The primer sequences were listed in Table 1. The cycling protocol was performed in accordance with the manufacturer’s instructions. The relative mRNA levels were calculated using the 2−ΔΔCt method. These specimens were obtained from Liaoning Cancer Hospital and Institute, China. This study was approved by the Institutional Review Board of our institution.
 |
Table 1 The Sequences of the Primers |
Immune Checkpoints Analysis, Human Leukocyte Antigen (HLA) Analysis, and Tumor Immune Dysfunction and Exclusion (TIDE) Analysis
To study the immune mechanisms of each subtype, immune checkpoint and HLA analyses were performed. Using the TIDE analysis tool (http://tide.dfci.harvard.edu/), the immune escape phenomenon and overall response rate to immunotherapy in each subtype were also investigated.
Drug-Sensitive Analysis
The pRRophetic (version 0.5) algorithm in the R package was used to identify the most suitable targeted or chemotherapeutic drug. The half-maximal inhibitory concentration (IC50) was calculated to assess the sensitivity of HCC cells to drugs using the Genomics of Drug Sensitivity in Cancer (GDSC, https://www.cancerrxgene.org).
Statistical Analysis
In this study, only a two-tailed P value < 0.05 was considered statistically different. R version 3.6.2 software and GraphPad Prism v. 8.01 were used for statistical analysis and visualizations. The Student’s t-test was performed to analyze the data obeying a normal distribution. The Mann–Whitney U-test was used to evaluate non-parametric data.
Results
Identification of Novel Molecular Subtypes of HCC Based on LRGs
The flowchart of the study was shown in Figure 1. A total of 109 genes were selected based on the interaction between the results of differential expression analysis and univariate Cox regression analysis (Figure 2A). These genes were used for the cluster analysis of 374 HCC samples. According to the consensus matrix plot, tSNE scatter plot, and PCA scatter plot (Figure 2B–D), HCC samples were divided into two subtypes, C1 and C2 (k=2). As shown in the heatmap, the expression of the selected LRGs in C1 subtype was significantly higher than that in C2 subtype (Figure 2E). In addition, the clinicopathological features of C1 and C2, including tumor size and pathological heterogeneity, were significantly different (Figure 2E). Therefore, C1 subtype could be regarded as a specific subtype with a high correlation with lysosome-related genes. The survival outcome of the C1 subtype was remarkably poorer than that of C2, according to the KM survival curve (Figure 2F). Subsequently, external validation was conducted using the ICGC and GSE databases and the same conclusions were obtained (Supplementary Figure 1). These results suggest that key LRGs are strongly linked to the prognosis of patients with HCC and deserve more in-depth analysis.
 |
Figure 2 Identification of novel molecular subtypes of HCC based on LRGs. (A) The forest plot displayed the 109 key LRGs and their respective hazard ratios. (B) Cluster analysis by ConsensusClusterPlus. (C) Principal component analysis by tSNE. (D) Principal component analysis by PCA. (E) The heatmap showed HCC samples were divided into 2 subtypes, C1 subtype and C2 subtype based on the expression of the 109 key LRGs. (F) K-M survival curves showed different prognosis of the two HCC subtypes in TCGA. |
Pathway Enrichment Analysis Between C1 and C2 Subtypes
Based on differential expression profiles, the two subtypes were significantly different at the molecular level. The differentially expressed genes were then used for pathway enrichment analysis of subtypes C1 and C2. We firstly performed GO analysis using TCGA, ICGC, and GSE databases and found that the immune response processes (immune response-regulating cell surface receptor signaling pathway involved in phagocytosis, inflammatory response, etc), positive regulation of cell communication, and positive regulation of signaling mainly differed between the two subtypes (Figure 3A–C). Moreover, GSEA-HALLMARK analysis was performed to avoid bias induced by the enrichment analysis. Using TCGA, ICGC, and GSE databases, HALLMARK analysis showed significant differences in the G2M_Checkpoint, E2F_Targets, Angiogenesis, KRAS signaling pathway, MTORC1 signaling pathway, and other important cancer-related pathways between the two subtypes (Figure 3D–F). Furthermore, using TCGA, ICGC, and GSE databases, we performed KEGG analysis and found that the two groups were mainly different in the cell cycle, ECM-receptor interaction, fructose and mannose metabolism, cellular senescence, proteoglycans in cancer, PPAR signaling pathway, fatty acid metabolism, p53 signaling pathway, biosynthesis of amino acids, ubiquinone, and other terpenoid-quinone biosynthesis pathways (Figure 3G–I). The results of the GSVA analysis also showed corresponding results (mTORC1_signaling, PI3K_AKT_mTOR_signaling, Wnt_beta-catenin_signaling, etc). (Supplementary Figure 2). These results suggest that the immune response, metabolic processes, and cancer-related signaling pathways play vital roles in shaping the two subtypes of HCC based on LRGs.
 |
Figure 3 Pathway enrichment analysis between C1 and C2 subtypes. (A–C) Results of GO enrichment analysis based on TCGA, ICGC and GSE databases. (D–F) Results of GSEA-HALLMARK enrichment analysis based on TCGA, ICGC and GSE databases. (G–I) Results of KEGG pathway enrichment analysis based on TCGA, ICGC and GSE databases. |
Immune Microenvironment Analysis of The Two Subtypes
To explore the differences in the immune microenvironment between subtypes C1 and C2, ESTIMATE, CIBERSORT algorithm, and ssGSEA analysis were further conducted. TCGA database was used as the test set, and the ICGC and GSE databases were used as validation sets. According to the ESTIMATE results, the immune Score of C1 subtype was significantly higher than that of C2 subtype (Figure 4A). Moreover, CIBERSORT and ssGSEA analyses showed that the proportions of immune cells between subtypes C1 and C2 were also different at the overall level (Figure 4B and C). Other cytokines, interferons and receptors, interleukins and receptors, chemokines and receptors involved in related intercellular communication pathways have also been investigated. The results suggested that the expression of cell communication molecules in C1 subtype was significantly higher than that in C2 subtype (Supplementary Figure 3A). The results were validated using ICGC and GSE databases (Supplementary Figure 3B and C). Communication molecules are strongly related to immune cells, suggesting that C1 subtype is closely related to immune cells, which also means that the degree of key LRGs expression in HCC patients may affect the efficacy of immunotherapy by regulating the level of immune infiltration.
 |
Figure 4 Immune infiltration analysis of the two subtypes. (A) The box plots showed the stromal score, immune score and ESTIMATES score of both subtypes in TCGA, ICGC and GSE databases based on ESTIMATES. (B) The box plots showed percentage of several immune cells of both subtypes in TCGA, ICGC and GSE databases based on CIBERSORT algorithm. (C) The heatmaps showed the expression level of several immune cell gene sets of both subtypes in TCGA, ICGC and GSE databases based on ssGSEA. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Abbreviation: ns, non-significant. |
Identification of Immune-Related LRGs by WCGNA
WGCNA was performed to further investigate the relationships between the key LRGs expression and immune cell infiltration. For the C1 subtype, the soft threshold was equal to 4, R2 was equal to 0.87, and mean connectivity was equal to 6.16 (Figure 5A and B). Using the above parameters, a cluster dendrogram was constructed, showing key LRGs in the C1 subtype, which were clustered into three different gene modules marked by diverse colors (Figure 5C and D). The module-trait relationship heatmap illustrated that the turquoise module was strongly associated with different immune cells in the C1 subtype. The same method was used to analyze C2 subtype, with a soft threshold of 4. Then R2 was equal to 0.90, and the mean connectivity was equal to 5.41 (Figure 5E and F). Key LRGs in C2 subtype can be clustered into four different gene modules, marked by diverse colors. The turquoise module in C2 subtype was relatively less related to immune cells (Figure 5G and H). Subsequently, the intersection of the turquoise module in subtypes C1 and C2 was performed, and alternative prognostic LRGs were identified for further analysis, including PRKCD, CPNE1, AGRN, P2RX4, PYGB, HRG, LAPTM4B, FTH1, SRC, DSN1, TUBB, LPCAT1, TTR, CKAP4, HSP90AB1, and GLA. The results also indicated that these identified genes were potentially critical players in the malignant development and prognostic prediction of the novel HCC subtypes C1 and C2, offering opportunities for deeper mechanistic insights, biomarkers identifying, and targeted therapeutic strategies development.
 |
Figure 5 Identification of immune-related LRGs by WCGNA. (A–B) Analysis of network topology for soft powers in C1 subtype. (C) Dendrogram and genes module colors in C1 subtype. (D) The module-trait relationships displayed the correlations between different gene modules and immune cells in C1 subtype. (E–F) Analysis of network topology for soft powers in C2 subtype. (G) Dendrogram and genes module colors in C2 subtype. (H) The module-trait relationships displayed the correlations between different gene modules and immune cells in C2 subtype. |
Construction of the Multiple Genes Prognosis Prediction Model
Multivariate Cox regression analysis was performed to reduce the dimensions of the input genes obtained from WGCNA analysis, PRKCD, LAPTM4B and LPCAT1 were selected as OS-associated LRGs and were used to construct a multiple gene prognosis prediction model. Using this model, all HCC samples were divided into high-risk and low-risk groups based on the median risk scores of all samples. Consistent with Figure 2, the high-risk group was similar to the C1 subtype and the low-risk group was similar to the C2 subtype (Figure 6A and B). The fishbone diagram also suggested that the risk score of C1 subtype was significantly higher than that of C2 subtype (Figure 6C). The heatmap showed that the expression of these three genes was remarkably higher in C1 subtype than that in subtype C2 (Figure 6D). The nomogram illustrates the survival outcomes of patients with HCC based on the expression of RKCD, LAPTM4B, and LPCAT1 in TCGA. With this help, the 1-year survival, 3-year survival, and 5-year survival rates could be intuitively predicted (Figure 6E). The AUC curve of ROC in TCGA was 0.730 at 1 year, 0.674 at 3 years and 0.660 at 5 years (Figure 6F); The AUC curve of ROC in ICGC was 0.778 at 1 year, 0.837 at 3 years and 0.729 at 5 years (Figure 6G); The AUC curve of ROC in GSE was 0.673 at 1 year, 0.672 at 3 years and 0.670 at 5 years (Figure 6H). The AUC values suggested that the model had good specificity and sensitivity.
 |
Figure 6 Establishment of multiple factors prognosis prediction model. (A) Risk score distribution plots of C1 subtype and C2 subtype based on TCGA. (B) Survival status scatter plots of C1 subtype and C2 subtype based on TCGA. (C) The fishbone diagram showed the differences in risk score of both subtypes based on TCGA. (D) The heatmap showed the expression condition of OS-associated LRGs (PRKCD, LAPTM4B, and LPCAT1) in both subtypes. (E) Nomogram developed based on the expression of PRKCD, LAPTM4B, and LPCAT1. (F) ROC curves for OS-related LRGs based on TCGA database. (G) ROC curves for OS-related LRGs based on ICGC database. (H) ROC curves for OS-related LRGs based on GSE database. ***P<0.001. |
The Expression of Selected Three Genes Were Significantly Increased in HCC Tissues Compared with Normal Tissues
We further validated the expression of three selected genes, PRKCD, LAPTM4B, and LPCAT1 in ten pairs of fresh HCC and normal tissues using RT-qPCR analysis. We found that the expression levels of PRKCD, LAPTM4B, and LPCAT1 were significantly higher in HCC tissues than those in normal tissues (P<0.05, Figure 7A–C). These results indicated that they may serve as potential therapeutic targets for patients with HCC.
 |
Figure 7 Validation of the high expression of PRKCD (A), LAPTM4B (B), and LPCAT1 (C) in HCC and normal samples via RT-qPCR analysis. *P<0.05. |
Immune Checkpoints and TIDE Analysis
Furthermore, we explored the landscape of immune checkpoints for both subtypes. Compared to the C2 subtype, all types of immune checkpoints and HLA were highly and ubiquitously expressed in the C1 subtype using the data (Figure 8A and B), which was also validated in the ICGC and GSE databases (Figure 8C–F). Meanwhile, the bar chart and circular chart suggested that the immunologic response rate in C1 subtype was significantly lower than that in C2 subtype based on data from the ICGC, and GSE databases, expect the data in TCGA databases (Supplementary Figure 4). The violin chart also showed that the TIDE score of the C1 subtype was significantly higher than that of the C2 subtype, suggesting that the possibility of the immune escape phenomenon in the C1 subtype may be higher than that of the C2 subtype based on the data from the above databases (Figure 8G–I).
 |
Figure 8 Immune checkpoints, HLA and TIDE analysis of the two subtypes. (A) The results of immune checkpoints analysis in both subtypes based on TCGA. (B) The results of HLA analysis in both subtypes based on TCGA. (C) The results of immune checkpoints analysis in both subtypes based on ICGC. (D) The results of HLA analysis in both subtypes based on ICGC. (E) The results of immune checkpoints analysis in both subtypes based on GSE. (F) The results of HLA analysis in both subtypes based on GSE. (G–I) The results of TIDE analysis in both subtypes based on TCGA, ICGC and GSE databases. *P<0.05, **P<0.01, ***P<0.001. Abbreviation: ns, non-significant. |
Drug-Sensitive Analysis in Multiple Databases
Based on data from the GDSC website, drug sensitivity responsiveness was also assessed between subtypes C1 and C2. We found that the responsiveness of candidate drugs between subtypes C1 and C2 was relatively different and that C1 subtype had higher drug sensitivity responsiveness. We found that the IC50 values of the Akt inhibitor A.443654, PLK1 inhibitor BI.2536, Gemcitabine, SGK1 inhibitor GSK.650394, Pyrimethamine, Eg5 inhibitor S.Trityl.L.cysteine in C1 subtype were much lower in the C1 subtype. These drugs might provide more efficient treatments for patients with HCC (Figure 9A–C).
 |
Figure 9 Drug sensitivity analysis. (A) The results of drug sensitivity analysis in both subtypes based on TCGA. (B) The results of drug sensitivity analysis in both subtypes based on ICGC. (C) The results of drug sensitivity analysis in both subtypes based on GSE. |
Discussion
HCC is the most common primary liver cancer,34–36 and is treated with multidisciplinary management, including hepatectomy, liver transplantation, radiofrequency ablation, chemotherapy, immunotherapy, and targeted therapy at present.37–40 However, tumor recurrence, metastasis, and chemotherapy resistance greatly affect the survival outcomes of patients with HCC.41,42 Researchers have constructed several molecular subtypes to guide HCC therapies and improve the prognosis of patients with HCC. The molecular subtypes include metabolism, immunity, polyposis, cuproptosis, ferroptosis, glycolysis, and so on.43–50 Although various molecular subtypes have been identified, the survival status of patients is still not very satisfied. Thus, the development of novel and accurate molecular subtypes to improve HCC survival is urgently required.
Lysosomes play an important role in various diseases including various kinds of cancers. Zeng et al reported that lysosomal degradation was targetable in osteosarcoma lung metastasis.51 Alejandro et al found that the lysosomal pathway contributed to cisplatin-induced cancer cell death.52 Therefore, it may be useful to predict and improve the survival outcomes of patients with HCC by constructing novel molecular subtypes based on the LRGs. In this study, 869 LRGs were downloaded from MSigDB, and differential expression analysis was performed based on the Bulk-RNA sequence data of HCC and normal tissues from TCGA database. After differential expression and survival analyses, 109 screened genes were used for cluster analysis. Using consensus clustering analysis, 374 HCC samples were clustered into two subtypes based on the 109 prognostic LRGs. Remarkably, the survival time in the C1 subtype was significantly shorter than that in the C2 subtype, indicating that the LRG-related molecular type could significantly distinguish the survival outcomes of patients with HCC. The prognostic differences between subtypes C1 and C2 also suggest that LRGs play a vital role in HCC. Thus, we further conducted GO, GSEA, KEGG, and GSVA enrichment analyses to explore the differences between the downstream signaling pathways of both subtypes. The differentially modulated genes were significantly linked to cancer-related pathways, including hallmark_G2M_checkpoint, hallmark_E2F_targets, p53 signaling, mTORC1 signaling, PI3K_AKT_mTOR_signaling, Wnt_beta_catenin_signaling, Notch signaling, TNFA_signaling_via_NFκB, and the inflammatory_response. In addition, glycolysis, fructose and mannose metabolism, cholesterol homeostasis, and fatty acid metabolism related to solid cancers were also altered in the enrichment analysis. Moreover, previous studies have reported that hallmark_G2M_checkpoint, hallmark_E2F_targets, mTORC_signaling pathway, and PI3K_Akt_mTOR signaling pathway are strongly related to tumor proliferation and metastasis.53–56 Glycolysis, cholesterol homeostasis, and fatty acid metabolism were also closely associated with the metastasis and invasion of solid tumors.57–59 In addition, the pathways related to anti-tumor immune responses, including leukocyte migration, phagocytosis, inflammatory response,60 were also significantly enriched between these two subtypes, suggesting that the two subtypes might have differences in the infiltration degree of several immune cells. The following analyses further support this hypothesis.
Tumor occurrence, invasion, metastasis, and progression are significantly affected by tumor microenvironment components such as various immune cells, stromal cells, and cytokines.61,62 Immune cells play an important role in this process.63,64 Studies have reported that Tregs act as anti-inflammatory cells, inhibit the immune response, and promote the malignant progression of tumors by secreting anti-inflammatory cytokines.65 Neutrophils also play a vital role in accelerating tumor occurrence and metastasis by secreting anti-inflammatory cytokines to regulate TME.66 In addition, cytotoxic immune cells, such as CD8+ and γδ T cells, secrete pro-inflammatory cytokines to exert anti-tumor effects.67,68 Therefore, immune infiltration analysis was performed in HCC using ssGSEA, CIBERSORT, ESTIMATE, and WGCNA methods. M0 macrophages, Tregs, dendritic cells, and neutrophils were present in a higher proportion in the C1 subtype, whereas CD8+ T cells and γδ T cells had a relatively lower percentage in the C1 subtype. These results indicated that compared with C2 subtype, C1 presented an immunoinhibitory phenotype, which was consistent with the worse survival outcomes of patients in C1 subtype.
Using WGCNA, we further screened for immune-related LRGs at the intersection of immune-related modules in both subtypes. Multivariate Cox regression analysis was performed based on the screened LRGs, and three OS-associated LRGs were identified. OS-associated LRGs include lysosomal protein transmembrane 4 beta (LAPTM4B), protein kinase C delta (PRKCD), and lysophosphatidylcholine acyltransferase 1 (LPCAT1). The multifactor prognosis model has good prediction performance and an excellent ability to distinguish the prognosis of patients with HCC. Prognostic scores can also be visually acquired using nomogram. LAPTM4B is regarded as a diagnostic biomarker and potential therapeutic target for HCC. Wang et al found that LAPTM4B significantly promoted the proliferation and autophagy of HCC.69 Wang et al found that LAPTM4B contributed to malignant proliferation of HCC stem cells and migration of myeloid-derived suppressor cells (MDSCs).70 Meanwhile, under the positive action of the transcription factor AP4, LAPTM4B remarkably promoted the invasion and metastasis of HCC and reduced sensitivity to chemotherapy.71 Moreover, using clinical samples, Zhai et al identified that allele*2 of LAPTM4B may be significantly related to the genetic susceptibility to HCC and is considered a prognostic risk factor for patients with HCC.72 Li et al found that PRKCD could serve as a key messenger in the interaction between HCC cells and platelets and play a crucial role in the process of platelet-induced tumor progression.73 Xu et al also found that high expression of PRKCD predicted poor survival outcomes in patients with HCC and was involved in tumor immune escape.74 LPCAT1 also contributes to malignant development of HCC. Sun et al found that silencing of LPCAT1 remarkably inhibited the proliferation and metastasis of HCC by targeting S100A11 and Snail.75 Zhang H and He RQ et al also found upregulated expression of LPCAT1 in HCC samples compared to non-tumor samples, which may serve as an independent prognostic predictor for the survival ofpatients with HCC. In their research, knocking down of LPCAT1 remarkably inhibited the proliferation, migration, and metastasis of HCC cells.76,77 In our study, we validated the high expression of PRKCD, LAPTM4B, and LPCAT1 in 10 pairs of fresh HCC and normal samples using RT-qPCR analysis. All results indicate that our nomogram may have a relatively high predictive ability for the prognosis of patients with HCC.
In addition to the level of immune cell infiltration, the influence of immune checkpoints on the prognosis of patients with HCC cannot be neglected. Immune checkpoints are a crucial part of immune reactions and can inhibit certain anti-tumor immune responses, which are indispensable for regulating and maintaining immune homeostasis.78–80 For example, immune checkpoint B and T lymphocyte attenuators (BTLA) can cause immunosuppression by inhibiting the activation and proliferation of B and T cells.81 A previous study also reported that high expression of BTLA was positively related to the expression of PD-L1 and indicated poorer prognosis in patients with non-small cell lung cancer.82 In addition, classical Cytotoxic T lymphocyte associated protein-4 (CTLA-4) plays a vital role in inhibiting T cell activation and serves as a biomarker for poor prognosis in several solid cancers including HCC,83–85 nasopharyngeal carcinoma,86 breast cancer,87 and osteosarcoma.88 In our study, we found that the expression of various immune checkpoints genes in C1 subtype was significantly higher than that in C2 subtype, such as BTLA, lymphocyte activating 3(LAG-3), CTLA-4 and so on. Meanwhile, based on the TIDE analysis, the score of C1 subtype was much higher than that of C2 subtype, which meant that immune evasion was more likely to happen in C1 subtype. Moreover, the higher proportion of non-responders in the C1 subtype indicated that the efficiency of immunotherapy in patients with HCC was poorer. Thus, the above results might explain why patients with HCC in the C1 subtype had a worse survival outcome. In addition, for patients who cannot undergo curative resection, systemic chemotherapy and targeted therapy are alternative methods.89–92 To explore the drug sensitivity of both subtypes, drug-sensitivity analysis was performed. We found that A.443654, BI.2536, Gemcitabine, GSK.650394, pyrimethamine, and S. Trityl. L. cysteine were more sensitive in patients with C1 subtype than with C2 subtype. These drugs may serve as drug candidates for treating patients with C1 subtypes and deserve further research.
This study also has some limitations. Firstly, data from public databases were limited, and the clinical information of some samples might miss, resulting in biases and errors. Secondly, our conclusion was mainly obtained from bioinformatics, and more validations are needed via in vivo and in vitro assays. Thirdly, this study was conducted based on retrospective data, and it is necessary to design a prospective clinical trial with a large sample size to prove our hypothesis.
Conclusions
In conclusion, our study revealed the heterogeneity of HCC. We not only defined two new molecular subtypes of HCC based on differentially expressed LRGs, but also studied the correlations between molecular subtypes and prognosis by bioinformatic analysis of bulk sequencing data. We further explored the differences in the signaling pathways, immune cell infiltration, and sensitivity to drug therapy between the two subtypes. Additionally, we constructed a novel multiple-factor prognosis prediction model based on the screened LRGs. Finally, we hope the LRG-related subtypes of HCC will provide new insights and guidelines for future clinical practice.
Data Sharing Statement
These data of this manuscript can be available from the corresponding author upon reasonable request.
Ethics Approval and Informed Consent
The specimens used in this study were obtained from Liaoning Cancer Hospital and Institute, and were approved by the Institutional Review Board (approval number: KY20240413). Meanwhile, informed consent obtained from the study participants prior to study commencement. This study complies with the Declaration of Helsinki.
Acknowledgments
Thanks for the help of Dr Dingbao Liang, Dr Yiting Qian, Dr Xinkun Zhang, Dr Bingde Hu, Dr Bo Sun, and Dr Jie Xiong in the references searching and preparing of the original figures.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was funded by Doctoral Start-up Foundation of Liaoning Province (2023-BS-048) and “5310” Talent Strategy Project Funding Plan of Liaoning Cancer Hospital & Institute.
Disclosure
The authors declare no potential conflicts of interest regarding the research, authorship, or publication of this article.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Konyn P, Ahmed A, Kim D. Current epidemiology in hepatocellular carcinoma. Expert Rev Gastroenterol Hepatol. 2021;15(11):1295–1307. doi:10.1080/17474124.2021.1991792
3. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314. doi:10.1016/S0140-6736(18)30010-2
4. Wang B, Hao X, Yan J, Li X, Zhao M, Han T. A bibliometric analysis of immune-related adverse events in cancer patients and a meta-analysis of immune-related adverse events in patients with hepatocellular carcinoma. J Transl Int Med. 2024;12(3):225–243. doi:10.2478/jtim-2024-0003
5. Hartke J, Johnson M, Ghabril M. The diagnosis and treatment of hepatocellular carcinoma. Semin Diagn Pathol. 2017;34(2):153–159. doi:10.1053/j.semdp.2016.12.011
6. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. doi:10.1038/s41572-020-00240-3
7. Niizeki T, Tokunaga T, Takami Y, et al. Comparison of efficacy and safety of atezolizumab plus bevacizumab and lenvatinib as first-line therapy for unresectable hepatocellular carcinoma: a propensity score matching analysis. Target Oncol. 2022;17(6):643–653. doi:10.1007/s11523-022-00921-x
8. Yang TK, Yu YF, Tsai CL, et al. Efficacy and safety of combined targeted therapy and immunotherapy versus targeted monotherapy in unresectable hepatocellular carcinoma: a systematic review and meta-analysis. BMC Cancer. 2022;22(1):1085. doi:10.1186/s12885-022-10174-6
9. Gao Y, Li J, Ma M, et al. Prognostic prediction of m6A and ferroptosis-associated lncRNAs in liver hepatocellular carcinoma. J Transl Int Med. 2024;12(5):526–529. doi:10.1515/jtim-2024-0023
10. Moller K, Safai Zadeh E, Gorg C, et al. Focal liver lesions other than hepatocellular carcinoma in cirrhosis: diagnostic challenges. J Transl Int Med. 2022;10(4):308–327. doi:10.2478/jtim-2022-0068
11. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894–1905. doi:10.1056/NEJMoa1915745
12. de Duve C. The lysosome turns fifty. Nat Cell Biol. 2005;7(9):847–849. doi:10.1038/ncb0905-847
13. Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. 2020;21(2):101–118. doi:10.1038/s41580-019-0185-4
14. Chauhan N, Patro BS. Emerging roles of lysosome homeostasis (repair, lysophagy and biogenesis) in cancer progression and therapy. Cancer Lett. 2024;584:216599. doi:10.1016/j.canlet.2023.216599
15. Ma J, Ma R, Zeng X, et al. Lysosome blockade induces divergent metabolic programs in macrophages and tumours for cancer immunotherapy. J Exp Clin Cancer Res. 2023;42(1):192. doi:10.1186/s13046-023-02768-0
16. Zhang Z, Yue P, Lu T, Wang Y, Wei Y, Wei X. Role of lysosomes in physiological activities, diseases, and therapy. J Hematol Oncol. 2021;14(1):79.
17. Pechincha C, Groessl S, Kalis R, et al. Lysosomal enzyme trafficking factor LYSET enables nutritional usage of extracellular proteins. Science. 2022;378(6615):eabn5637. doi:10.1126/science.abn5637
18. Cao M, Luo X, Wu K, He X. Targeting lysosomes in human disease: from basic research to clinical applications. Signal Transduct Target Ther. 2021;6(1):379. doi:10.1038/s41392-021-00778-y
19. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi:10.1038/nm.3394
20. Rauner G, Kuperwasser C. Microenvironmental control of cell fate decisions in mammary gland development and cancer. Dev Cell. 2021;56(13):1875–1883. doi:10.1016/j.devcel.2021.06.016
21. Zou Y, Ye F, Kong Y, et al. The single-cell landscape of intratumoral heterogeneity and the immunosuppressive microenvironment in liver and brain metastases of breast cancer. Adv Sci. 2023;10(5):e2203699. doi:10.1002/advs.202203699
22. Tian B, Pang Y, Gao Y, et al. A pan-cancer analysis of the oncogenic role of Golgi transport 1B in human tumors. J Transl Int Med. 2023;11(4):433–448. doi:10.2478/jtim-2023-0002
23. Bejarano L, Jordao MJC, Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 2021;11(4):933–959. doi:10.1158/2159-8290.CD-20-1808
24. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2021;221:107753. doi:10.1016/j.pharmthera.2020.107753
25. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30(1):36–50. doi:10.1016/j.cmet.2019.06.001
26. Bader JE, Voss K, Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol Cell. 2020;78(6):1019–1033. doi:10.1016/j.molcel.2020.05.034
27. Ke M, Zhu H, Lin Y, et al. Actin-related protein 2/3 complex subunit 1B promotes ovarian cancer progression by regulating the AKT/PI3K/mTOR signaling pathway. J Transl Int Med. 2024;12(4):406–423. doi:10.2478/jtim-2024-0025
28. Chang CP, Su YC, Lee PH, Lei HY. Targeting NFKB by autophagy to polarize hepatoma-associated macrophage differentiation. Autophagy. 2013;9(4):619–621. doi:10.4161/auto.23546
29. Parekh VV, Pabbisetty SK, Wu L, et al. Autophagy-related protein Vps34 controls the homeostasis and function of antigen cross-presenting CD8alpha(+) dendritic cells. Proc Natl Acad Sci U S A. 2017;114(31):E6371–E6380. doi:10.1073/pnas.1706504114
30. Zhang W-Y, Chen Z-H, An -X-X, et al. Analysis and validation of diagnostic biomarkers and immune cell infiltration characteristics in pediatric sepsis by integrating bioinformatics and machine learning. World J Pediatr. 2023;19(11):1094–1103. doi:10.1007/s12519-023-00717-7
31. Zhang X, Tan L, Zhu C, et al. Key genes and immune infiltration patterns and the clinical implications in psoriasis patients. Skin Res Technol. 2024;30(8):e13889. doi:10.1111/srt.13889
32. Li K, Li S, Zhang H, Lei D, Lo WLA, Ding M. Computational analysis of the immune infiltration pattern and candidate diagnostic biomarkers in lumbar disc herniation. Front Mol Neurosci. 2022;15:846554. doi:10.3389/fnmol.2022.846554
33. Wu LD, Li F, Chen JY, Zhang J, Qian LL, Wang RX. Analysis of potential genetic biomarkers using machine learning methods and immune infiltration regulatory mechanisms underlying atrial fibrillation. BMC Med Genomics. 2022;15(1):64. doi:10.1186/s12920-022-01212-0
34. Gilles H, Garbutt T, Landrum J. Hepatocellular carcinoma. Crit Care Nurs Clin North Am. 2022;34(3):289–301. doi:10.1016/j.cnc.2022.04.004
35. Tay BWR, Huang DQ, Mark M, et al. Comparable outcomes in early hepatocellular carcinomas treated with trans-arterial chemoembolization and radiofrequency ablation. Biomedicines. 2022;10(10):2361. doi:10.3390/biomedicines10102361
36. Wallace MC, Preen D, Jeffrey GP, Adams LA. The evolving epidemiology of hepatocellular carcinoma: a global perspective. Expert Rev Gastroenterol Hepatol. 2015;9(6):765–779. doi:10.1586/17474124.2015.1028363
37. Parikh ND, Pillai A. Recent advances in hepatocellular carcinoma treatment. Clin Gastroenterol Hepatol. 2021;19(10):2020–2024. doi:10.1016/j.cgh.2021.05.045
38. Saoudi Gonzalez N, Castet F, Elez E, Macarulla T, Tabernero J. Current and emerging anti-angiogenic therapies in gastrointestinal and hepatobiliary cancers. Front Oncol. 2022;12:1021772. doi:10.3389/fonc.2022.1021772
39. Shen W, Chen Y, Lei P, et al. Immunotherapeutic approaches for treating hepatocellular carcinoma. Cancers. 2022;14(20):5013. doi:10.3390/cancers14205013
40. Sim HW, Knox J. Hepatocellular carcinoma in the era of immunotherapy. Curr Probl Cancer. 2018;42(1):40–48. doi:10.1016/j.currproblcancer.2017.10.007
41. Torimura T, Iwamoto H. Treatment and the prognosis of hepatocellular carcinoma in Asia. Liver Int. 2022;42(9):2042–2054. doi:10.1111/liv.15130
42. Galle PR, Dufour JF, Peck-Radosavljevic M, Trojan J, Vogel A. Systemic therapy of advanced hepatocellular carcinoma. Future Oncol. 2021;17(10):1237–1251. doi:10.2217/fon-2020-0758
43. Chaisaingmongkol J, Budhu A, Dang H, et al. Common molecular subtypes among Asian hepatocellular carcinoma and cholangiocarcinoma. Cancer Cell. 2017;32(1):57–70e53. doi:10.1016/j.ccell.2017.05.009
44. Li B, Li Y, Zhou H, et al. Multiomics identifies metabolic subtypes based on fatty acid degradation allocating personalized treatment in hepatocellular carcinoma. Hepatology. 2024;79(2):289–306. doi:10.1097/HEP.0000000000000553
45. Song F, Wang CG, Mao JZ, et al. PANoptosis-based molecular subtyping and HPAN-index predicts therapeutic response and survival in hepatocellular carcinoma. Front Immunol. 2023;14:1197152. doi:10.3389/fimmu.2023.1197152
46. Lee SH, Yim SY, Shim JJ, Lee JS. Molecular subtypes and genomic signatures of hepatocellular carcinoma for prognostication and therapeutic decision-making. In: Hoshida Y, editor. Hepatocellular Carcinoma: Translational Precision Medicine Approaches. Cham (CH); 2019:109–123.
47. Peng X, Zhu J, Liu S, et al. Signature construction and molecular subtype identification based on cuproptosis-related genes to predict the prognosis and immune activity of patients with hepatocellular carcinoma. Front Immunol. 2022;13:990790. doi:10.3389/fimmu.2022.990790
48. Ye J, Lin Y, Gao X, et al. Prognosis-related molecular subtypes and immune features associated with hepatocellular carcinoma. Cancers. 2022;14(22):5721. doi:10.3390/cancers14225721
49. Xu Z, Peng B, Liang Q, et al. Construction of a ferroptosis-related nine-lncRNA signature for predicting prognosis and immune response in hepatocellular carcinoma. Front Immunol. 2021;12:719175. doi:10.3389/fimmu.2021.719175
50. Liu X, Sun B, Yao Y, et al. Identification of copper metabolism and cuproptosis-related subtypes for predicting prognosis tumor microenvironment and drug candidates in hepatocellular carcinoma. Front Immunol. 2022;13:996308. doi:10.3389/fimmu.2022.996308
51. Zeng C, Zhong L, Liu W, et al. Targeting the lysosomal degradation of Rab22a-NeoF1 fusion protein for osteosarcoma lung metastasis. Adv Sci. 2023;10(5):e2205483. doi:10.1002/advs.202205483
52. Belmonte-Fernandez A, Herrero-Ruiz J, Galindo-Moreno M, et al. Cisplatin-induced cell death increases the degradation of the MRE11-RAD50-NBS1 complex through the autophagy/lysosomal pathway. Cell Death Differ. 2023;30(2):488–499. doi:10.1038/s41418-022-01100-1
53. Ediriweera MK, Tennekoon KH, Samarakoon SR. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: biological and therapeutic significance. Semin Cancer Biol. 2019;59:147–160. doi:10.1016/j.semcancer.2019.05.012
54. Hu W, Shi Y, Han T, et al. A panel of E2F target gene signature predicting the prognosis of hepatocellular carcinoma. Front Genet. 2022;13:879299. doi:10.3389/fgene.2022.879299
55. Kim J, Guan KL. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol. 2019;21(1):63–71. doi:10.1038/s41556-018-0205-1
56. Yin L, Chang C, Xu C. G2/M checkpoint plays a vital role at the early stage of HCC by analysis of key pathways and genes. Oncotarget. 2017;8(44):76305–76317. doi:10.18632/oncotarget.19351
57. Cai K, Chen S, Zhu C, et al. FOXD1 facilitates pancreatic cancer cell proliferation, invasion, and metastasis by regulating GLUT1-mediated aerobic glycolysis. Cell Death Dis. 2022;13(9):765. doi:10.1038/s41419-022-05213-w
58. Wang B, Zhang H, Chen YF, et al. Acyl-CoA thioesterase 9 promotes tumour growth and metastasis through reprogramming of fatty acid metabolism in hepatocellular carcinoma. Liver Int. 2022;42(11):2548–2561. doi:10.1111/liv.15409
59. Wu J, Guo L, Qiu X, et al. Genkwadaphnin inhibits growth and invasion in hepatocellular carcinoma by blocking DHCR24-mediated cholesterol biosynthesis and lipid rafts formation. Br J Cancer. 2020;123(11):1673–1685. doi:10.1038/s41416-020-01085-z
60. Li L, Yu R, Cai T, et al. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int Immunopharmacol. 2020;88:106939. doi:10.1016/j.intimp.2020.106939
61. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019;79(18):4557–4566. doi:10.1158/0008-5472.CAN-18-3962
62. Anderson NM, Simon MC. The tumor microenvironment. Curr Biol. 2020;30(16):R921–R925. doi:10.1016/j.cub.2020.06.081
63. Baharom F, Ramirez-Valdez RA, Khalilnezhad A, et al. Systemic vaccination induces CD8(+) T cells and remodels the tumor microenvironment. Cell. 2022;185(23):4317–4332e4315. doi:10.1016/j.cell.2022.10.006
64. Dolina JS, Lee J, Brightman SE, et al. Linked CD4+/CD8+ T cell neoantigen vaccination overcomes immune checkpoint blockade resistance and enables tumor regression. J Clin Invest. 2023;133(17). doi:10.1172/JCI164258
65. Xu Y, Mou J, Wang Y, et al. Regulatory T cells promote the stemness of leukemia stem cells through IL10 cytokine-related signaling pathway. Leukemia. 2022;36(2):403–415. doi:10.1038/s41375-021-01375-2
66. Zhang L, Yi H, Chen J, et al. Neutrophil extracellular traps facilitate A549 cell invasion and migration in a macrophage-maintained inflammatory microenvironment. Biomed Res Int. 2022;2022(1):8316525. doi:10.1155/2022/8316525
67. Assy L, Khalil SM, Attia M, Salem ML. IL-12 conditioning of peripheral blood mononuclear cells from breast cancer patients promotes the zoledronate-induced expansion of gammadelta T cells in vitro and enhances their cytotoxic activity and cytokine production. Int Immunopharmacol. 2023;114:109402. doi:10.1016/j.intimp.2022.109402
68. Lukhele S, Rabbo DA, Guo M, et al. The transcription factor IRF2 drives interferon-mediated CD8(+) T cell exhaustion to restrict anti-tumor immunity. Immunity. 2022;55(12):2369–2385e2310. doi:10.1016/j.immuni.2022.10.020
69. Wang F, Wu H, Zhang S, et al. LAPTM4B facilitates tumor growth and induces autophagy in hepatocellular carcinoma. Cancer Manag Res. 2019;11:2485–2497. doi:10.2147/CMAR.S201092
70. Wang H, Zhou Q, Xie DF, Xu Q, Yang T, Wang W. LAPTM4B-mediated hepatocellular carcinoma stem cell proliferation and MDSC migration: implications for HCC progression and sensitivity to PD-L1 monoclonal antibody therapy. Cell Death Dis. 2024;15(2):165. doi:10.1038/s41419-024-06542-8
71. Meng Y, Wang L, Xu J, Zhang Q. AP4 positively regulates LAPTM4B to promote hepatocellular carcinoma growth and metastasis, while reducing chemotherapy sensitivity. Mol Oncol. 2018;12(3):373–390. doi:10.1002/1878-0261.12171
72. Zhai G, Yang H, Ji X, et al. Correlation of LAPTM4B polymorphisms with hepatocellular carcinoma in Chinese patients. Med Oncol. 2012;29(4):2744–2749. doi:10.1007/s12032-011-0139-y
73. Li X, Zhao K, Lu Y, Wang J, Yao W. Genetic analysis of platelet-related genes in hepatocellular carcinoma reveals a novel prognostic signature and determines PRKCD as the potential molecular bridge. Biol Proced Online. 2022;24(1):22. doi:10.1186/s12575-022-00185-9
74. Xu D, Wang Y, Wu J, et al. Systematic characterization of novel immune gene signatures predicts prognostic factors in hepatocellular carcinoma. Front Cell Dev Biol. 2021;9:686664. doi:10.3389/fcell.2021.686664
75. Sun Q, Fu C, Liu J, Li S, Zheng J. Knockdown of LPCAT1 repressed hepatocellular carcinoma growth and invasion by targeting S100A11. Ann Clin Lab Sci. 2023;53(2):212–221.
76. Zhang H, Xu K, Xiang Q, et al. LPCAT1 functions as a novel prognostic molecular marker in hepatocellular carcinoma. Genes Dis. 2022;9(1):151–164. doi:10.1016/j.gendis.2020.07.007
77. He RQ, Li JD, Du XF, et al. LPCAT1 overexpression promotes the progression of hepatocellular carcinoma. Cancer Cell Int. 2021;21(1):442. doi:10.1186/s12935-021-02130-4
78. Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378(2):158–168. doi:10.1056/NEJMra1703481
79. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275–287. doi:10.1038/nrc.2016.36
80. Funes SC, Manrique de Lara A, Altamirano-Lagos MJ, Mackern-Oberti JP, Escobar-Vera J, Kalergis AM. Immune checkpoints and the regulation of tolerogenicity in dendritic cells: implications for autoimmunity and immunotherapy. Autoimmun Rev. 2019;18(4):359–368. doi:10.1016/j.autrev.2019.02.006
81. Ning Z, Liu K, Xiong H. Roles of BTLA in immunity and immune disorders. Front Immunol. 2021;12:654960. doi:10.3389/fimmu.2021.654960
82. Li X, Xu Z, Cui G, Yu L, Zhang X. BTLA expression in stage I-III non-small-cell lung cancer and its correlation with PD-1/PD-L1 and clinical outcomes. Onco Targets Ther. 2020;13:215–224. doi:10.2147/OTT.S232234
83. Long J, Wang A, Bai Y, et al. Development and validation of a TP53-associated immune prognostic model for hepatocellular carcinoma. EBioMedicine. 2019;42:363–374. doi:10.1016/j.ebiom.2019.03.022
84. Gao X, Xu N, Li Z, et al. Safety and antitumour activity of cadonilimab, an anti-PD-1/CTLA-4 bispecific antibody, for patients with advanced solid tumours (COMPASSION-03): a multicentre, open-label, phase 1b/2 trial. Lancet Oncol. 2023;24(10):1134–1146. doi:10.1016/S1470-2045(23)00411-4
85. Hou K, Xu X, Ge X, Jiang J, Ouyang F. Blockade of PD-1 and CTLA-4: a potent immunotherapeutic approach for hepatocellular carcinoma. Biofactors. 2024;50(2):250–265. doi:10.1002/biof.2012
86. Huang PY, Guo SS, Zhang Y, et al. Tumor CTLA-4 overexpression predicts poor survival in patients with nasopharyngeal carcinoma. Oncotarget. 2016;7(11):13060–13068. doi:10.18632/oncotarget.7421
87. Kern R, Panis C. CTLA-4 expression and its clinical significance in breast cancer. Arch Immunol Ther Exp. 2021;69(1):16. doi:10.1007/s00005-021-00618-5
88. Wang SD, Li HY, Li BH, et al. The role of CTLA-4 and PD-1 in anti-tumor immune response and their potential efficacy against osteosarcoma. Int Immunopharmacol. 2016;38:81–89. doi:10.1016/j.intimp.2016.05.016
89. Qin S, Bai Y, Lim HY, et al. Randomized, multicenter, open-label study of oxaliplatin plus fluorouracil/leucovorin versus doxorubicin as palliative chemotherapy in patients with advanced hepatocellular carcinoma from Asia. J Clin Oncol. 2013;31(28):3501–3508. doi:10.1200/JCO.2012.44.5643
90. Qin S, Cheng Y, Liang J, et al. Efficacy and safety of the FOLFOX4 regimen versus doxorubicin in Chinese patients with advanced hepatocellular carcinoma: a subgroup analysis of the EACH study. Oncologist. 2014;19(11):1169–1178. doi:10.1634/theoncologist.2014-0190
91. Wang Y, Deng B. Hepatocellular carcinoma: molecular mechanism, targeted therapy, and biomarkers. Cancer Metastasis Rev. 2023;42(3):629–652. doi:10.1007/s10555-023-10084-4
92. Huang A, Yang XR, Chung WY, Dennison AR, Zhou J. Targeted therapy for hepatocellular carcinoma. Signal Transduct Target Ther. 2020;5(1):146. doi:10.1038/s41392-020-00264-x
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Decaprenyl Diphosphate Synthase Subunit 1 (PDSS1): A Potential Prognostic Biomarker and Immunotherapy-Target for Hepatocellular Carcinoma
Yang Y, Li J, Tang M, Nie B, Huang W
Cancer Management and Research 2022, 14:1627-1639
Published Date: 3 May 2022
Identification of KRBA1 as a Potential Prognostic Biomarker Associated with Immune Infiltration and m6A Modification in Hepatocellular Carcinoma
Liu Y, Fu B, Yu Z, Song G, Zeng H, Gong Y, Ding Y, Huang D
Journal of Hepatocellular Carcinoma 2022, 9:497-516
Published Date: 31 May 2022
Histological Severity of Cirrhosis Influences Surgical Outcomes of Hepatocellular Carcinoma After Curative Hepatectomy
Liang BY, Gu J, Xiong M, Zhang EL, Zhang ZY, Lau WY, Wang SF, Guan Y, Chen XP, Huang ZY
Journal of Hepatocellular Carcinoma 2022, 9:633-647
Published Date: 23 July 2022
Development and Validation of a Propionate Metabolism-Related Gene Signature for Prognostic Prediction of Hepatocellular Carcinoma
Xiao J, Wang J, Zhou C, Luo J
Journal of Hepatocellular Carcinoma 2023, 10:1673-1687
Published Date: 2 October 2023
Construction of a Prognostic Model for Hepatocellular Carcinoma Based on Macrophage Polarization-Related Genes
Chen H, Li J, Cao D, Tang H
Journal of Hepatocellular Carcinoma 2024, 11:857-878
Published Date: 11 May 2024