")
Back to Journals » Journal of Inflammation Research » Volume 18
Neutrophils: From Inflammatory Bowel Disease to Colitis-Associated Colorectal Cancer
Authors Chen T , Liu J, Hang R, Chen Q, Wang D
Received 3 October 2024
Accepted for publication 6 January 2025
Published 22 January 2025 Volume 2025:18 Pages 925—947
DOI https://doi.org/10.2147/JIR.S497701
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Tianyi Chen,1 Jiachen Liu,2 Ruyi Hang,1 Qian Chen,1 Dong Wang1,3
1Cancer Center, Daping Hospital, Army Medical University, Chongqing, People’s Republic of China; 2Radiology Department, Daping Hospital, Army Medical University, Chongqing, People’s Republic of China; 3Oncology Department of Qianjiang Center Hospital, Chongqing University, Chongqing, People’s Republic of China
Correspondence: Dong Wang, Cancer Center, Daping Hospital, 10 Changjiang Zhi Road, Yuzhong Dist, Chongqing, 400042, People’s Republic of China, Tel +86-23-68757151, Fax +86-23-68894062, Email [email protected]
Abstract: Inflammatory bowel disease (IBD) is a non-specific inflammatory disease of digestive tract, primarily manifesting as ulcerative colitis (UC) and Crohn’s disease (CD). The precise etiology of IBD remains elusive. The interplay of genetic factors, environmental influences, and intestinal microbiota contributes to the establishment of an uncontrolled immune environment within the intestine, which can progressively lead to atypical hyperplasia and ultimately to malignancy over a long period. This colorectal malignant tumor that arises from chronic IBD is referred to as colitis-associated colorectal cancer (CAC). Dysregulation in the quantity and functionality of neutrophils plays a significant role in the onset, progression, and recurrence of IBD, as well as in the transition from IBD to CAC. Neutrophils affect the pathophysiology of IBD through various mechanisms, including the production of reactive oxygen species (ROS), degranulation, the release of inflammatory mediators and chemokines, and the formation of neutrophil extracellular traps (NETs). These processes can induce DNA mutations, thereby facilitating the development of colon cancer. Given the incomplete understanding of the disease mechanisms underlying IBD and CAC, effective treatment and prevention strategies remain challenging. Consequently, a comprehensive review of the functional roles of neutrophils in IBD and CAC is essential for advancing our understanding of IBD pathogenesis and identifying potential therapeutic targets.
Keywords: inflammatory bowel disease, colitis-associated colorectal cancer, neutrophil, neutrophil extracellular traps
Introduction
Inflammatory bowel disease (IBD) is a non-specific inflammatory disease of the digestive tract, primarily manifesting as ulcerative colitis (UC) and Crohn’s disease (CD). Despite significant advancements in medical science and clinical practice, the precise etiology of IBD remains elusive. The intricate interaction between genetic predispositions and environmental influences contributes to the challenges faced in elucidating the underlying causes of IBD. Additionally, dysregulation of immune responses, damage of the intestinal barrier, and alterations in gut microbiota further complicate the disease’s pathophysiology, presenting substantial obstacles to research on IBD’s pathological mechanisms. Although IBD is not typically a direct cause of death, it is recognized as a precancerous condition associated with colorectal cancer (CRC), particularly in the case of UC, thereby representing a significant contributor to the incidence of malignant tumors.1 Colitis-associated colorectal cancer (CAC) is a malignant tumor that arises from chronic IBD. The dysregulation of the immune system is a significant factor contributing to the onset, progression, and recurrence of IBD, and it may also play a role in the transformation of IBD into CAC. Neutrophil infiltration, a critical component of the innate immune response to pathogens, is a prominent pathological characteristic observed during active stage of IBD. Previous studies have indicated that neutrophils can influence the disease trajectory of IBD through mechanisms such as the production of reactive oxygen species (ROS),2 degranulation,3–5 the release of inflammatory mediators and chemokines,6 and the formation of neutrophil extracellular traps (NETs).7 These processes can induce DNA mutations, thereby facilitating the development of colon cancer. Given that the underlying mechanisms of IBD and CAC remain incompletely understood, effective treatment and prevention strategies are challenging to establish. Current therapeutic options often fail to adequately halt disease progression and may even exacerbate the condition due to adverse drug effects. Consequently, a comprehensive review of the functional roles of neutrophils in IBD and CAC is essential for advancing our understanding of the pathophysiology of IBD and for identifying novel therapeutic approaches.
From Inflammation to Cancer: IBD and CAC
IBD: An Increasingly Complex Interweave of Genetics, Environment, and Microorganisms
Since the initial documentation of IBD in 1859, its incidence has exhibited a consistent upward trend. Although epidemiological evidence shows that the incidence of IBD in developed countries has stabilized or even declined since the early 21st century, the prevalence remains notably high.8 Conversely, in a broader spectrum of developing countries, the incidence continues to rise at an alarming rate.8 As a global health concern, IBD imposes significant burdens on economies and healthcare systems worldwide.9 A contributing factor to this predicament is the ambiguity surrounding the precise etiology of IBD. Current understanding suggests that no singular cause-whether it be genetic predisposition, immune response, environmental factors, or gut microbiota-can comprehensively account for the disease.
First, genetic susceptibility is a critical factor in the pathophysiology of IBD. The risk of developing IBD is significantly elevated among first-degree relatives, particularly parents.10 And in more extreme instances, the concordance rate for identical twins with IBD ranges from 20% to 60%.10 In contrast, the concordance rate for fraternal twins is significantly lower, at approximately 3% to 5%, indicating that genetic factors may substantially influence the onset of IBD.10 Genome-wide association studies (GWAS) have identified over 250 loci associated with an increased risk of IBD; however, only 38 of these loci are thought to be directly involved in the disease’s mechanisms, predominantly linked to inflammatory processes and immune responses.11,12 The recent Activity-By-Contact (ABC) model has predicted that 43 genes located within enhancers may be implicated in IBD, and CRISPR interference experiments have validated that PPIF is closely associated with the disease’s pathogenesis, as it encodes cyclophilin D and is involved in mitochondrial activity in macrophages.13 NOD2/CARD15 is recognized as a significant susceptibility gene for IBD, with mutations in this gene heightening the risk of IBD among Caucasian populations.10 This pattern recognition receptor (PRR), which is expressed in Paneth cells, neutrophils, and macrophages, activates the NF-κB signaling pathway upon recognizing bacterial cell wall antigens.10,14 Additionally, interleukin-23/ interleukin-23 receptor (IL-23/IL-23R) have been identified as risk factors for IBD across both European and non-European populations.12 Various immune cells, including T cells, neutrophils, and macrophages, can produce IL-23, which interacts with receptors on intestinal epithelial cells (IECs), thereby influencing intestinal barrier integrity, immune homeostasis, and gut microbiota composition.15 The inflammatory gene profile associated with IBD overlaps with numerous complex autoimmune and immunodeficiency disorders.16 Furthermore, the extraintestinal manifestations of ulcerative colitis (UC) and Crohn’s disease (CD), such as erythema nodosum, pyoderma gangrenosum, ankylosing spondylitis, and peripheral arthritis with negative rheumatoid factor, suggest a distinct relationship between IBD and immune system dysregulation.17
Second, the onset and progression of IBD are significantly influenced by environmental factors. While above evidence underscores the importance of genetic predisposition in the manifestation and advancement of IBD, alterations in genetic composition alone do not adequately account for the pronounced geographical disparities and the rapidly escalating incidence of the disease. This observation suggests that environmental influences and epigenetic factors are also critical in the etiology of IBD. Notably, individuals migrating from regions with lower IBD incidence to those with higher rates experience an increased risk of developing the condition; specifically, the risk escalates by 14% for each decade of younger age at migration, indicating that earlier exposure to high-incidence environments correlates with a heightened risk of IBD.18 Furthermore, the scientific and technological advancements accompanying industrial transformation appear to have a nuanced relationship with IBD incidence, particularly as urbanization alters lifestyle habits. Epidemiological studies have identified several credible risk factors for IBD, including smoking, antibiotic use, and appendectomy, while factors such as physical activity, breastfeeding, tea consumption, and vitamin D intake are recognized as protective.19 Additionally, some analyses indicate that smoking exerts differential effects on the risk of colectomy in UC and CD; specifically, smoking is associated with an increased risk in CD patients, whereas cessation of smoking raises the risk in UC patients.20,21
Finally, the aforementioned risk factors associated with IBD are largely connected to the composition of the intestinal microbiota; however, establishing a definitive causal relationship between dysbiosis and IBD remains a challenge.22 Recent commentary posits that changes in gut microbiota may serve as both a contributing factor and a consequence to IBD.23 Current observational data, including the efficacy of fecal diversion in alleviating affected intestinal segments in patients with CD, the beneficial effects of antibiotics in treating certain forms of IBD, and the observed reduction in microbial diversity among IBD patients, underscore the significant role of intestinal microorganisms in the pathophysiology of IBD.22 During IBD, dysregulation of innate immunity, particularly involving neutrophils, leads to structural alterations and a loss of microbial homeostasis. Furthermore, microorganisms can modulate neutrophil expression and function, either directly or indirectly.24 Additionally, systematic screening associations has indicated that the risk of disease linked to specific genetic variations in IBD may be influenced by microorganisms.24 Numerous genetic markers associated with IBD, such as NOD2/CARD15,25 ATG16L1,26 and CARD9,27 exhibit intricate interactions with both the immune system and the microbiota.22,28,29 Intestinal microbes may play a role in the variability of IBD risk genes; for instance, Faecalibacterium prausnitzii and g_Roseburia have been identified as microorganisms capable of modulating NOD2 variations in recent observational study of feces from IBD patients.30 Table 1 presents a compilation of IBD susceptibility genes related to neutrophil function (refers to the comprehensive review conducted by Camille Danne et al24) and their interactions with gut microbiota.
 |
Table 1 IBD Susceptibility Genes Associated with Neutrophils |
The interplay among genetic factors, environmental influences, and intestinal microbiota is intricately linked to the dysfunction of the immune system, contributing to the complexity of IBD. The significant involvement of neutrophils in both the onset and progression of IBD is noteworthy (Figure 1). Recent research has indicated that the IBD-associated gene Card9 is expressed in neutrophils and influences colitis in DSS-induced experimental mice by modulating mitochondrial function.47 Additionally, Cxcr2, a receptor gene responsible for neutrophil chemotaxis, has been shown to alter the composition of the gut microbiota in mice when it is defective.48,49 The excessive infiltration of neutrophils, recognized as a hallmark of IBD, interacts with these multifaceted factors and plays a critical role in the disease’s progression. This review aims to elucidate the role of neutrophils in IBD, while the interactions between neutrophils and intestinal microbiota need to be further explored in the future.
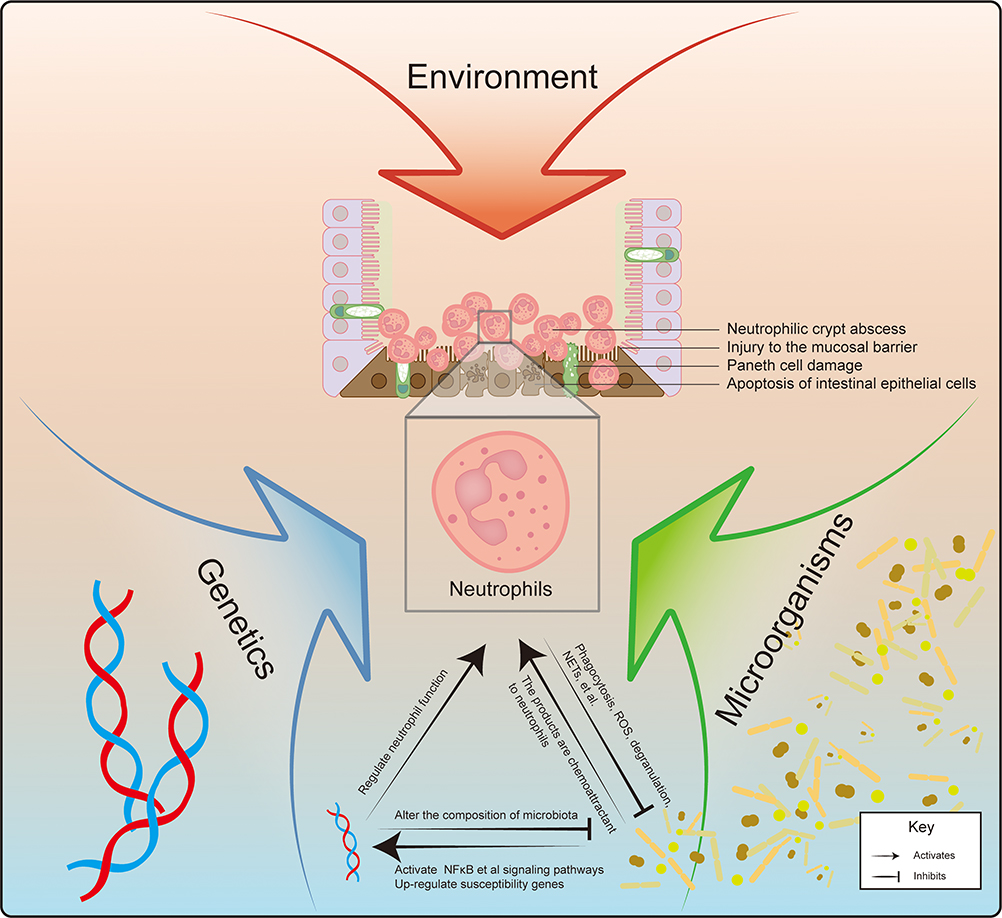 |
Figure 1 IBD is a multifaceted disorder resulting from a confluence of environmental factors, alterations in gut microbiota, and genetic predisposition. The interplay of these elements leads to the development of intestinal lesions in IBD patients, characterized by Paneth cell injury and apoptosis of IECs, ultimately resulting in mucosal damage and the formation of neutrophilic crypt abscesses. It is likely that the influence of IBD risk gene variants is mediated, at least in part, through their effects on the bacterial microbiota. Modifications in certain IBD susceptibility genes, such as Cxcr2, can directly impact the composition of gut flora. Neutrophils serve as critical mediators at the intersection of gut genetics and microbiota. Numerous genes associated with IBD susceptibility are involved in the neutrophil’s defense mechanisms against pathogens. Neutrophils play a vital role in limiting microbial dissemination and maintaining microbiota homeostasis. Their production and functional capabilities are regulated both directly and indirectly by microbial factors. Prolonged inflammation leads to significant and sustained neutrophilic infiltration, which alters the intestinal environment, conferring a selective advantage to facultative anaerobic bacteria and promoting the proliferation of Enterobacteriaceae within the context of inflammation.23 |
CAC: A Malignant Fruit That Grows in the Chronic Inflammation
Recent advancements in endoscopic technology, coupled with lifestyle modifications and smoking cessation efforts, have contributed to a decline in the incidence of CRC in recent years.50 However, individuals with a chronic history of IBD remain at an elevated risk for progression to cancer.51 Chronic inflammation is a significant factor in the initiation and progression of malignant tumors. In the context of CRC, long-standing and recurrent intestinal inflammation associated with IBD can lead to atypical hyperplasia, which may subsequently evolve into malignant tumors. The risk of developing CRC in patients with IBD is substantially higher than that observed in the general population.1,52,53 Furthermore, patients with colorectal cancer associated with IBD exhibit poorer prognoses and higher mortality rates compared to those with sporadic CRC.54 Notably, long-term UC is particularly prone to progress to CAC, representing a primary cause of mortality among UC patients,1,55 and effective management of UC can mitigate the risk of CAC.17 Additionally, individuals with CD face an increased risk of malignancy, particularly those with colonic involvement, who are at a heightened risk for developing CAC.52 The transition from colitis to colorectal cancer typically spans 16 to 21 years; however, CAC patients with a history of IBD tend to develop malignant tumors approximately 7.7 years earlier than those with sporadic CRC.1 Recent research has identified several risk factors for CAC, including extensive involvement of the gastrointestinal tract, low-grade atypical hyperplasia, colonic strictures, primary sclerosing cholangitis, polyps, and a family history of CRC.56 Conversely, protective factors against CAC include endoscopic surveillance, the use of 5-aminosalicylic acid (5-ASA), and smoking.56
In the context of IBD, the prolonged and recurrent nature of the condition can lead to damage to the mucosal membrane and the presence of aberrant white blood cells at the lesion sites, resulting in redox imbalances characterized by an excessive accumulation of ROS. This accumulation not only inflicts direct harm to intestinal microvilli, exacerbating the pathological inflammatory response, but also serves as terminal electron acceptors for anaerobic respiration, thereby facilitating the proliferation of facultative anaerobes.2,6,57 The abnormal infiltration of neutrophils in IBD appears to heighten the risk of progression to CAC,58,59 as the ROS released by these cells can induce DNA mutations that contribute to tumorigenesis.60,61 The transition from colorectal inflammation to cancer follows inflammation-atypical hyperplasia-adenocarcinoma sequence, during which markers associated with DNA double-strand breaks and damage repair progressively increase.62 In contrast to sporadic CRC, mutations in the p53 gene occur earlier and more readily in normal intestinal tissues affected by CAC,1 whereas alterations in the APC gene are less frequent and occur later in the progression from IBD to CAC.51 This distinction underscores the differing mechanisms underlying malignant tumorigenesis in CAC compared to sporadic CRC (Figure 2).
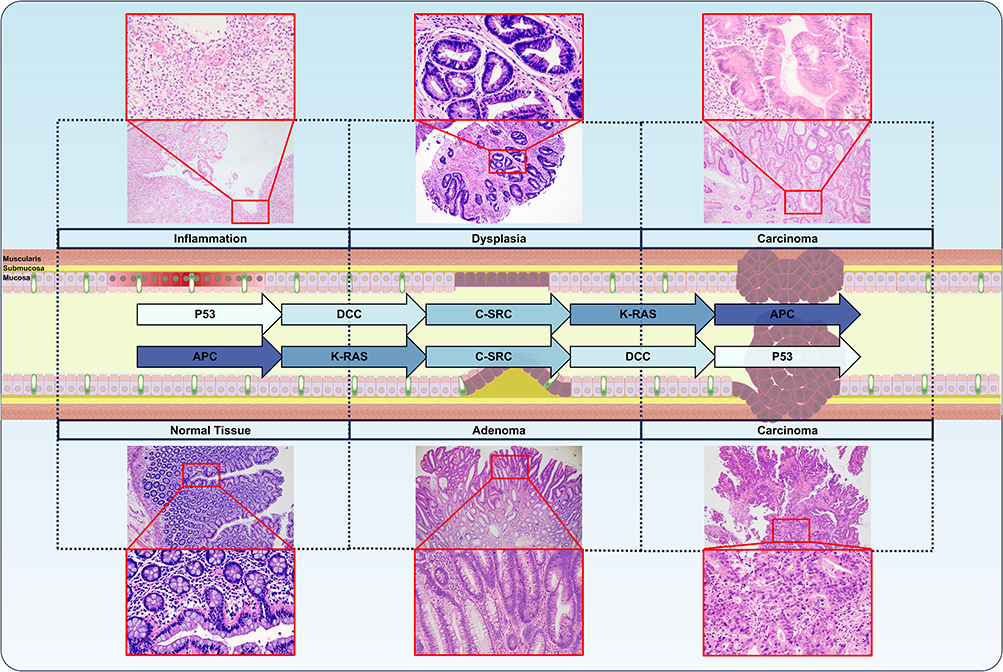 |
Figure 2 Overview of the variations in tumorigenicity between sporadic CRC and CAC based on IBD. In the inflammation-dysplasia-carcinoma sequence, there is a progressive increase in markers indicative of oxidative damage and DNA double-strand breaks. In the case of CAC, mutations in the p53 gene occur at an earlier stage, while dysfunction of the APC gene is observed at a later stage. Conversely, in sporadic CRC, mutations in the p53 gene arise later, whereas APC gene dysfunction is noted earlier, leading to the formation of a dysplasia-carcinoma sequence.51 All hematoxylin and eosin (HE) staining images of representative patients included in this article were sourced from the Daping Hospital, Chongqing, China and received approval from the Ethics Committee of the Daping Hospital [Ratification No.: 2024(127)]. |
Genetic and epigenetic abnormalities, including chromosome instability (CIN),63,64 microsatellite instability (MSI),65,66 DNA hypermethylation,67,68 DNA glycosylation defects,69 and aberrant non-coding RNA expression,70 are closely associated with the development of CAC. Furthermore, the extensive infiltration of dysfunctional immune cells, along with dysbiosis of intestinal microbiota, can activate nuclear factor κB (NF-κB), a critical factor that exacerbates the disease and facilitates the transition to CAC.1 Recent studies on pyroptosis have revealed that the stimulator of interferon genes (STING) can collaborate with spleen tyrosine kinase (SYK) to induce pyroptosis in CAC tumor cells within an azoxymethane/dextran sodium sulfate (AOM/DSS)-induced inflammation-cancer transformation model in experimental mice.71 In addition, pyroptosis results in the release of damage-associated molecular patterns (DAMPs), which promote CAC tumorigenesis through the ERK1/2 signaling pathway.72
Moreover, various ways that activate the Wnt/β-Catenin signaling pathway have been implicated in the development of CAC.73–75 Other molecular mechanisms contributing to the progression of CAC include Toll-like receptors (TLR), the JAK/STAT signaling pathway, the mammalian target of rapamycin complex (mTOR), autophagy, and oxidative stress.76 As a disease characterized by dysregulated intestinal immunity, IBD is marked by abnormal expression of inflammatory cytokines and the accumulation of immunosuppressive cells within the intestinal milieu, creating an immunosuppressive microenvironment conducive to tumor survival.67 A recent study identified five differential genes (AQP9, FCGR3B, GPR109B, PROK2, and S100A12) in neutrophils from individuals with IBD compared to healthy controls, proposing these genes as biomarkers for the transition from IBD to CAC,76 thereby establishing a close association between neutrophils and the inflammatory cancer transformation process in IBD.
Neutrophils Regulate IBD and CAC
The Function of Neutrophils
Neutrophils progressively develop bactericidal capabilities throughout their maturation in the bone marrow. Upon entering the bloodstream, these cells, which constitute approximately 60% of the circulating immune cell population, form a critical component of the innate immune system. They play a pivotal role in combating pathogen invasion and initiating inflammatory responses through various mechanisms, including phagocytosis, the production of ROS, degranulation, the formation of NETs, and the recruitment of other immune cells.6,77 (Figure 3A-C)
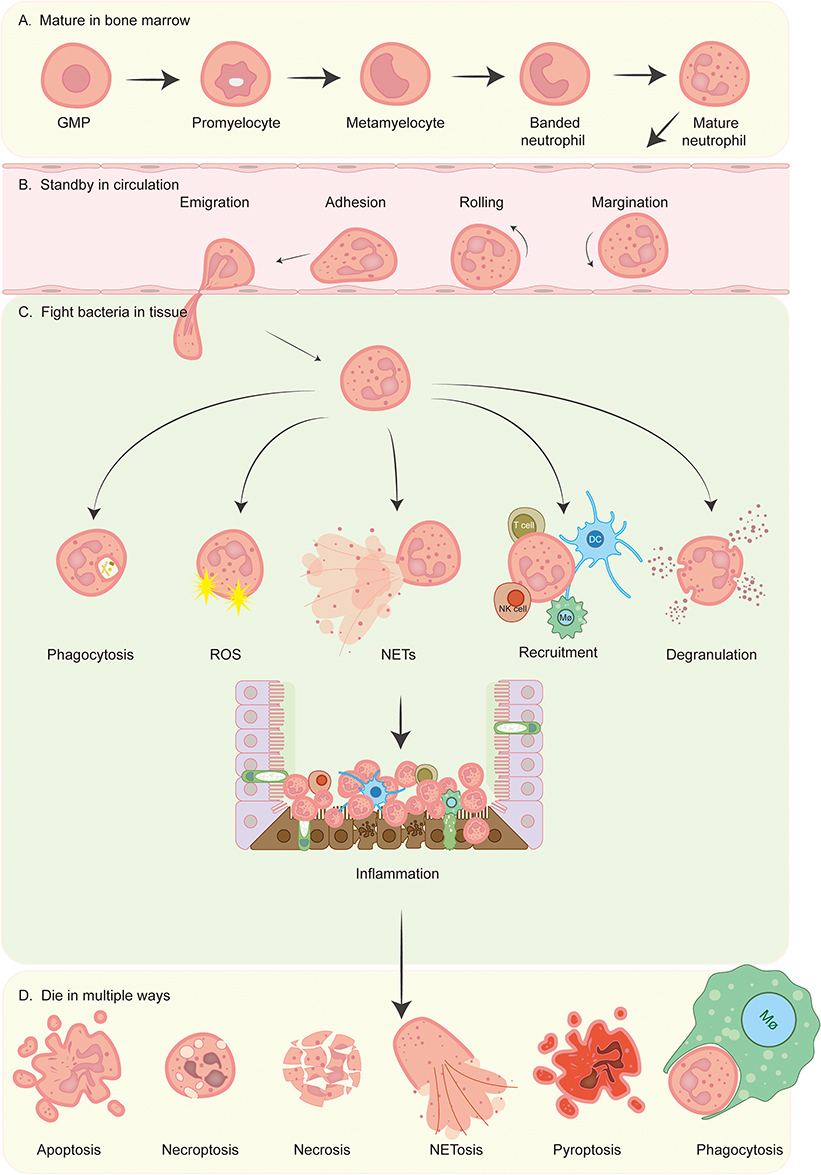 |
Figure 3 Overview of neutrophil’s entire life. (A) The differentiation of neutrophils occurs within the bone marrow, where granulocyte-monocyte progenitors (GMPs) are generated from common myeloid progenitors (CMPs), which themselves are derived from hematopoietic stem cells (HSCs). The process of neutrophil generation is regulated by granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage-colony stimulating factor (GM-CSF), which guide GMPs through a series of developmental stages, including promyelocyte, myelocyte, metamyelocyte, and band cell, ultimately resulting in the maturation of neutrophils from myeloblasts that then enter the circulatory system.78 (B) Neutrophils present in the bloodstream are activated by chemokines and undergo a series of processes including margination, rolling, adhesion, and migration, prior to exiting the circulatory system. This allows them to accumulate in sites of inflammation through the mechanism of chemotaxis. (C) Neutrophils employ various strategies to eliminate pathogenic bacteria at the site of inflammation, including phagocytosis, the production of ROS, the formation of NETs, the recruitment of a diverse array of immune cells (such as dendritic cells (DC), macrophages, T cells, natural killer (NK) cells, and additional neutrophils), and degranulation. (D) Neutrophils can undergo cell death through multiple pathways, including apoptosis, necroptosis, necrosis, NETosis, pyroptosis, phagocytosis by macrophages, etc.79 |
Neutrophils possess the ability to identify and engulf pathogens through a diverse array of membrane and intracellular receptors.79 Following this recognition, they generate superoxide free radicals via respiratory bursts that are facilitated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.80 These superoxide radicals are subsequently converted into hydrogen peroxide and other cytotoxic reactive oxygen species (ROS), which are released within intracellular phagosomes or onto extracellular plasma membranes.80 Neutrophils also contain a distinct pool of granules that can release various antimicrobial proteins to eliminate pathogens. This release occurs in a specific sequence, beginning with tertiary granules that contain matrix metalloproteinase-9 (MMP9), followed by secondary granules (or special granules) that house lactoferrin, and concluding with primary granules (or azurophilic granules) that contain myeloperoxidase (MPO).81,82 Furthermore, neutrophils employ NETs as a highly conserved antimicrobial strategy, which involves the formation of an extracellular network composed of DNA and proteins that effectively traps and neutralizes pathogens, thereby preventing their further spread.7 Upon migration to the site of infection, neutrophils are capable of recruiting various immune cells, including additional neutrophils, to engage in the inflammatory response through the release of a range of cytokines and chemokines.6 The regulated death of neutrophils is essential for the resolution of inflammation. In addition to apoptosis, neutrophils can modulate the number in the lesion through mechanisms such as necroptosis, necrosis, NETosis, pyroptosis, and phagocytosis, thereby maintaining the homeostasis of the inflammatory response (Figure 3D).79
Neutrophils that are isolated via density gradient centrifugation can be categorized into several distinct groups: normal-density neutrophils (NDNs), low-density neutrophils (LDNs), which exhibit either pro-inflammatory or anti-inflammatory characteristics, and tumor-associated neutrophils (TANs), which can further be classified into those with anti-tumor effects (N1 subtype) and those with pro-tumor effects (N2 subtype).24 Furthermore, polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) represent a specialized subset of “neutrophils” found in both tumor and non-tumor conditions, the latter of which encompasses diseases such as systemic lupus erythematosus, multiple sclerosis, rheumatoid arthritis, and IBD. These PMN-MDSCs are often indistinguishable from conventional neutrophils and possess the ability to inhibit T cells and natural killer (NK) cells through various mechanisms.83
The chemotaxis of neutrophils from the bloodstream to localized tissues marks the onset of the inflammatory response. However, inflammation characterized by a predominance of neutrophils may also signify the initiation of chronic inflammatory processes and the potential development of malignant tumors. As research in this area progresses, the role of neutrophils in IBD has become increasingly complex. On one hand, the protective function of neutrophils against intestinal pathogens is significant and cannot be overlooked. Conversely, the abnormal infiltration of neutrophils into intestinal tissues can exacerbate inflammation and contribute to the formation of crypt abscesses. In the context of IBD, the delicate equilibrium that neutrophils strive to maintain is crucial; any disruption of this balance may result in the recurrence or worsening of the disease.
Pathological Mechanism of Neutrophils in IBD
Numerous susceptibility genes associated with IBD are intricately linked to neutrophil functionality, encompassing processes such as pathogen recognition and phagocytosis (NOD2), ROS production (CYBA, CYBB, NCF4, IFNGR2), mitochondrial-dependent apoptosis (CARD9), and autophagy (ATG16L1).24,47 A hallmark of IBD is the excessive infiltration of neutrophils within the intestinal mucosa,21 which may play a significant role in the disease’s pathogenesis.77,84 Specifically, these aberrantly infiltrated neutrophils contribute to the formation of crypt abscesses, leading to heightened apoptosis of IECs and subsequent impairment of the intestinal barrier2. Furthermore, the hyperactivation of neutrophils results in the generation of substantial quantities of ROS, which can rapidly deplete oxygen and nutrients in the surrounding microenvironment, as well as induce lipid peroxidation, thereby compromising the integrity of cellular membranes and organelles.2,85
Neutrophil Transepithelial Migration
The onset of intestinal inflammation in IBD prompts IECs to secrete a variety of recruitment signals that facilitate the chemotaxis of neutrophils from the bloodstream to the lamina propria. These signals include cytokines such as IL-8, IL-6, and IL-33, chemokines such as CXCL5, CXCL7, CXCL10, and CCL20, arachidonic acid metabolites including leukotriene B4 (LTB4) and hepoxilin A3 (HXA3), as well as MMPs such as MMP3 and MMP7.6,86,87 Following this recruitment, neutrophils increase the permeability of the intestinal epithelium by releasing proteolytic enzymes, including neutrophil elastase (NE) and MMPs, thereby facilitating transepithelial migration.86 This migration serves a dual purpose: it enhances the ability of neutrophils to recognize pathogen antigens and stimulates the immune response; however, it also exacerbates mucosal injury, leading to heightened apoptosis of IECs, which ultimately compromises the integrity of the epithelial barrier.86 (Figure 4A)
 |
Figure 4 Overview of the pathogenesis of neutrophils in IBD. (A) IECs are activated by pathogenic bacteria to release various signaling molecules, including chemokines, cytokines, lipid mediators, and MMPs, which serve to recruit neutrophils to the site of inflammation. This recruitment leads to mucosal damage, apoptosis of IECs, and dysfunction of the intestinal barrier, as neutrophils secrete NE and MMPs that penetrate the intestinal epithelium86,87 (B) The delayed apoptosis of neutrophils may be influenced by several factors, including cytokines, inflammatory mediators, CSF, hypoxic conditions, extracellular acidosis, and LPS, LP, PG, and LTA produced by pathogens.88,89 Additionally, the ChemR2390 and PD-L1-PI3K-AKT91 signaling pathways in neutrophils have been associated with delayed regression. (C) The accumulation of neutrophils at the lesion site is a consequence of abnormal aggregation and delayed apoptosis, which ultimately resulting in the formation of a neutrophilic crypt abscess. (D and E). Neutrophils exhibit dual functionality; they release ROS and NETs that effectively eliminate pathogenic microorganisms. However, excessive inflammation can also result in tissue damage due to the overactivity of these immune responses. |
Delayed Apoptosis of Neutrophils
Neutrophils are characterized by a brief half-life of approximately 8 to 12 hours in circulation, which can extend to 1 to 4 days following their migration into tissues.79 In IBD, the process of neutrophil apoptosis is notably delayed.92,93 While this prolongation can enhance bactericidal activity, the accumulation of neutrophils poses a heightened risk of damage to normal tissues. The recruitment of neutrophils within the intestines of patients with Crohn’s disease (CD) correlates with the severity of the condition,94 whereas the complete resolution of neutrophils in the intestinal mucosa is associated with improved long-term clinical outcomes in patients with UC.95 The regulation of neutrophil apoptosis is primarily mediated by integrin receptors.88 Various cytokines and inflammatory mediators, including IL-1β, IL-6, IL-8, tumor necrosis factor (TNF), interferons (IFN-α, IFN-β, IFN-γ), chemokines such as CXCL1, complement components like C5a, leukotriene B4 (LTB4), granulocyte-macrophage colony-stimulating factor (GM-CSF), and granulocyte colony-stimulating factor (G-CSF) can prolong neutrophil survival by inhibiting apoptosis.88,89,96 Additionally, factors such as hypoxia, extracellular acidosis, and bacterial product including lipopolysaccharides (LPS), lipoproteins (LP), peptidoglycans (PG), and lipoteichoic acid (LTA) have also been implicated in the delay of neutrophil apoptosis.88,89 ChemR23 has been linked to drug resistance in IBD immunotherapy; the administration of anti-ChemR23 antibody has been shown to facilitate neutrophil clearance in experimental models of IBD by promoting the transition of pro-inflammatory macrophages (M1) to an anti-inflammatory phenotype (M2).90 Furthermore, the expression of programmed death-ligand 1 (PD-L1) on neutrophils is upregulated under inflammatory conditions.97 Recent investigations indicate that the overexpression of PD-L1 in neutrophils, both in humans and mice, can delay apoptosis through the activation of the PI3K-AKT signaling pathway.91 The extensive infiltration of neutrophils in intestinal lesions, coupled with their delayed apoptosis, contributes to increased neutrophil accumulation and the formation of crypt abscesses, which not only induces local hypoxia but also compromises the integrity of the intestinal barrier, thereby elevating the risk of treatment failure in IBD.6,21 (Figure 4B)
Neutrophilic Crypt Abscess
Acute inflammation in individuals with IBD is marked by a significant influx of neutrophils into the intestinal mucosa.21 Delayed apoptosis contributes to the accumulation of neutrophils. Furthermore, neutrophils have the capacity to recruit various immune cells, including monocytes, macrophages, NK cells, DCs, T cells, and additional neutrophils, to the site of inflammation through the secretion of diverse cytokines and chemokines, such as IL-1β, IL-8, IL-17, TNF, CXCL1, CXCL2, and CXCL5, among others.24 The aggregation of neutrophils within the intestinal environment can damage the integrity of the intestinal epithelial barrier, leading to the formation of crypt abscesses. Proteases released by neutrophils can disrupt cellular junctions, resulting in crypt deformation and the development of abscesses.24 In contrast to non-IBD intestinal inflammatory conditions, which predominantly feature apoptotic crypt abscesses, the majority of cases of IBD are characterized by neutrophilic crypt abscesses98 (Figure 4C). Cryptitis and crypt abscesses have become established criteria for the pathological diagnosis and scoring of IBD.99 Moreover, excessive infiltration of neutrophils is recognized as a contributing factor to the ineffectiveness of IBD treatments.90
Neutrophils Release ROS
Neutrophils generate ROS through the action of NADPH oxidase.100 Furthermore, these immune cells directly influence mitochondrial function via the glycerol 3-phosphate pathway, thereby maintaining mitochondrial polarization and facilitating ROS production.101 Neutrophils employ respiratory bursts, characterized by the release of ROS, to eliminate pathogens. In the context of IBD, aberrantly activated neutrophils can release excessive amounts of ROS, leading to rapid depletion of oxygen and nutrients in the surrounding environment, as well as inducing lipid peroxidation that compromises cell membranes and organelles.2 Additionally, extracellular ROS can activate redox-sensitive inflammatory pathways and function as signaling molecules to modulate the inflammatory response.100 The ROS generated by neutrophils can damage the telomeres of adjacent cells, resulting in increased cellular senescence both in vitro and ex vivo.102 While ROS serves as a crucial mechanism for neutrophils to combat pathogens, chronic inflammation-associated ROS production is linked to an increase in electron receptors (such as nitrates, sulfur oxides, and nitrogen oxides) within the intestines of humans and mice, which may promote the proliferation of facultative anaerobic bacteria and other pathogens.24 Typically, the levels of intestinal ROS in patients with IBD are elevated compared to those in healthy individuals.103 However, it has been reported that a reduction in ROS levels due to mutations in the NADPH oxidase gene is associated with refractory Crohn’s disease,104 indicating a need for further investigation into the underlying mechanisms to resolve this apparent contradiction. (Figure 4D)
Neutrophils Release NETs
NETs represent a highly conserved antimicrobial defense mechanism employed by neutrophils, characterized by an extracellular network composed of DNA and proteins that effectively capture and eliminate pathogens, thereby preventing their dissemination7 (Figure 4E). The proteins found within NETs, such as histones, granule proteins, and various proteases—including the prominent calprotectin S100A8/9—are recognized as significant biomarkers in IBD, constituting approximately 60% of neutrophil cytoplasmic proteins and playing a crucial role in resistance to fungal infections.7 Under normal physiological conditions, NE and MPO are sequestered within the azurophilic granules of neutrophils.105 Upon activation, NE translocates to the nucleus from these granules, leading to histone cleavage and subsequent chromatin de-condensation.105 MPO then binds to chromatin, further facilitating de-condensation, which culminates in cellular rupture and the release of NETs.105
The formation of NETs is notably elevated in the intestinal mucosa,106 feces,7 and blood107 of patients with IBD, which cause a positive correlation with disease activity.108 Furthermore, the stimulation of neutrophils from healthy individuals with serum derived from patients experiencing active IBD can significantly induce NETs release.109 CD177+ neutrophils exhibit protective properties in IBD due to their enhanced bactericidal activity, and concurrently producing lower levels of inflammatory mediators such as IL-6, IL-17A, and IFN-γ.24 In patients with active IBD, there is a marked increase in CD177+ neutrophils within peripheral blood and inflamed mucosa, which are associated with heightened NETs formation.110 In contrast, CD177-deficient colitis mouse models exhibit more severe colitis, attributed to compromised intestinal barrier function and immune mechanisms.110 In patients with UC, IL-1β has been shown to modulate NETs formation through an autophagy-dependent mechanism, thereby facilitating NETs generation.111 Additionally, NETs can induce apoptosis in epithelial cells, compromising the integrity of the intestinal barrier and exacerbating intestinal inflammation in IBD mouse models.112 In the colon of IBD patients and DSS-induced mice, there is a synchronous increase in the levels of DNase and NETs within the affected areas, alongside the release of NET-associated proteins that damage vascular endothelium.113 Notably, the concurrent inhibition of high mobility group protein 1 (HMGB1) and DNase I has been shown to significantly reduce the release of NETs-related markers in experimental mouse models.114 Moreover, hydrogen sulfide may exert anti-inflammatory effects by inhibiting NETs formation and the expression of NF-κB and HMGB1, as demonstrated in rat model of IBD induced by 2,4,6-trinitrobenzenesulfonic acid (TNBS).115
NETs have been identified as a factor that elevates the risk of thrombosis in patients with IBD.107 While NETs serve the purpose of inhibiting the dissemination of pathogens, they also facilitate thrombosis by acting as adhesive structures for platelets, erythrocytes, and various adhesion molecules.116 Numerous constituents of these structures, such as histones, DNA, NE, and cathepsin G, have the potential to activate platelet function and promote coagulation through the stimulation of both intrinsic and extrinsic coagulation pathways.116 Patients diagnosed with UC and CD exhibit a threefold increased risk of thromboembolic events compared to healthy individuals.117 Recent in vitro investigations have demonstrated that LINC00668, present in exosomes, enhances the translocation of NE from cytoplasmic compartments to the nucleus via its interaction with NE, thereby promoting the release of NETs and subsequent thrombosis.116 Furthermore, the application of DNase I has been shown to diminish disease activity indices and thrombotic events in mouse models of colitis.107 In conclusion, the release of NETs by neutrophils in the context of IBD contributes to the impairment of the intestinal barrier by inducing apoptosis in IECs, thereby heightening the risk of thrombosis.
Neutrophils are Potential Therapeutic Targets for IBD
In the management of IBD, no definitive pharmacological cure has been identified. The primary objectives of current therapeutic strategies are to alleviate symptoms and prevent disease recurrence.17 5-ASA, commonly referred to as mesalamine, is a traditional medication utilized in the treatment of IBD. It functions as a non-steroidal anti-inflammatory agent, mitigating intestinal mucosal inflammation through the inhibition of prostaglandin and leukotriene synthesis. Nevertheless, 5-ASA is not devoid of risks, as patients may experience adverse effects such as abdominal pain, fever, joint pain, and allergic reactions. Clinically, the efficacy of 5-ASA is limited, with an effective response rate of only 65%, and approximately 50% of patients are able to sustain remission, particularly those with severe manifestations of the disease.118 Furthermore, the effectiveness of 5-ASA in conjunction with biological agents or immunomodulators for maintaining remission in Crohn’s disease has yet to be substantiated.17,52 The use of corticosteroids in patients unresponsive to 5-ASA carries a significant risk of side effects, providing symptomatic relief in only 46% of cases, without contributing to sustained remission or recurrence prevention.17,119 Endogenous antioxidants present in the gastrointestinal tract have the potential to mitigate mucosal injury, facilitate healing, and reduce ulceration;2 however, their clinical application in IBD remains limited. Other natural antioxidants, such as resveratrol and curcumin, face challenges including high renal clearance rates, non-specific distribution, and low therapeutic efficacy.57 For patients exhibiting resistance to 5-ASA or those with severe disease, immunosuppressive or corticosteroid therapies are recommended, with azathioprine being a viable option for preventing IBD recurrence and maintaining remission in corticosteroid-dependent individuals.17 Nonetheless, these treatments necessitate careful consideration due to the associated risks of infection, long-term medication side effects, and potential serious complications.
Conventional therapies for IBD do not specifically address neutrophils, which play a critical role in the pathogenesis of the condition. Neutrophils are integral to the regulation of intestinal inflammation in IBD.120 Notably, excessive infiltration of neutrophils has been linked to the development of tolerance to anti-TNF therapies and other forms of immunotherapy.90 The progression of IBD may be mitigated by inhibiting leukocyte migration to the intestine or by utilizing antibodies to antagonize neutrophils in experimental murine models.6,16 Anti-TNF-α therapy, specifically infliximab, has been employed in the management of IBD. Clinical evidence indicates that this treatment can significantly reduce neutrophil infiltration in the inflamed mucosa of IBD patients and suppress the production of pro-inflammatory mediators, including ROS, calprotectin, IL-8, IL-6, and TNF-α.121 Neutrophil adsorption therapy has received approval for the treatment of IBD in several countries and has been shown to decrease inflammatory levels and alleviate parenteral symptoms in affected individuals.122 Table 2 provides a summary of key research focused on targeting neutrophils for IBD treatment, and the need for further clinical trials to validate these findings and assess their therapeutic efficacy.
 |
Table 2 Some Exploring Neutrophil Targets for IBD Therapy |
In the investigation of therapeutic approaches aimed at neutrophils, it is important to note that mere inhibition of these cells may not yield the intended therapeutic outcomes.23 This is attributable to the essential function of neutrophils in controlling pathogens within the immune system. Consequently, treatments that induce neutrophil deficiency could potentially counteract the intended treatment objectives. Therefore, a more promising avenue for developing treatment strategies for IBD may involve the regulation of hyperactivated neutrophils to facilitate their appropriate resolution, rather than merely diminishing their population.
Neutrophils in the Transformation from IBD to CAC
Chronic inflammation is known to facilitate the renewal of epithelial cells, thereby exerting selective pressure on mutant clones. This phenomenon is associated with an increased propensity for cancer, as it elevates the rate of DNA mutations while simultaneously fostering epigenetic alterations.131 Inflammatory processes involving neutrophils can disrupt the balance of intestinal microbiota, leading to ecological disturbances that contribute to CAC through the production of reactive metabolites and carcinogens, as well as damage to the epithelial barrier.132 Neutrophils, as pivotal cells in the inflammatory response, play a significant role in the progression from IBD to CAC.133 The neutrophil/lymphocyte ratio has emerged as a prognostic marker indicative of poor overall survival in cancer patients.81 Furthermore, excessive neutrophil infiltration in IBD not only exacerbates inflammation and promotes tumorigenesis through aberrant lipid metabolism77 but also facilitates cancer development by inducing cellular stagnation and replication errors.59 Research indicates that patients with UC exhibiting heightened neutrophil infiltration are at an increased risk of developing CAC.76 The multifaceted roles of neutrophils, including the production of reactive oxygen species (ROS) and neutrophil extracellular traps (NETs), have been implicated in the pathogenesis of colorectal malignancies.
ROS have been shown to facilitate tumor growth and regulate various processes associated with tumor proliferation, including anti-apoptotic mechanisms, epithelial-mesenchymal transition (EMT), and angiogenesis.134 This regulation occurs through interactions with several key signaling pathways, such as PI3K/Akt/mTOR, RAS/ERK, JNK/p38, NF-κB, HIF, and Src kinase.134 In mice model of CAC, the silencing of Glutathione peroxidase 4 (GPX4), an enzyme responsible for the detoxification of ROS, results in increased tumor invasiveness.61 Additionally, MMP9, a zinc-dependent endopeptidase that targets extracellular matrix proteins, is associated with reduced levels of ROS, diminished DNA damage, and enhanced activation of the mismatch repair pathway in CAC mouse models, thereby inhibiting tumorigenesis via the p53 signaling axis.135 Furthermore, ROS generated by neutrophils can contribute to cancer metastasis, angiogenesis, and immunosuppression within the tumor microenvironment.134,136 In chronic inflammatory conditions, neutrophils can induce DNA mutations through the release of ROS, thereby promoting tumor initiation, progression, and metastasis.60,61,137,138
NETs are present at elevated levels in tissue and blood samples from patients with CRC,139 and this elevation is associated with a poor prognosis for individuals undergoing radical resection of CRC.140 In murine models, the presence of NETs increases in response to LPS induction,140 and these structures facilitate the capture and transfer of platelet-tumor cell aggregates, thereby contributing to the metastasis of CRC.141 Additionally, NETs promote tumor growth and metastasis indirectly through mechanisms such as angiogenesis, which is characterized by epithelial-mesenchymal transition (EMT)-related cell migration, degradation of basement membrane proteins mediated by MMPs, and the entrapment of CRC cells.139
MPO, a heme peroxidase, is one of the most prevalent proteins found in neutrophils. It is primarily located within the primary granules of these cells and is released following neutrophil activation.76 Elevated levels of MPO in NETs contribute to oxidative stress in epithelial cells, which in turn aggravates DNA damage and increases the likelihood of mutations.76 Patients exhibiting high levels of MPO-DNA demonstrate a heightened risk of postoperative infections and a reduced overall survival rate when compared to those with lower MPO-DNA levels.140 Furthermore, the expression of MPO is significantly greater in the AOM/DSS-induced CAC animal model than in the DSS-induced IBD model.76
MDSCs represent a dysfunctional subset of neutrophils in the context of cancer, which poses challenges in differentiating them from conventional neutrophils. MDSCs are widely recognized as significant contributors to the ineffectiveness of immunotherapy across various malignancies.142 The presence of chronic inflammation leads to the recruitment of numerous immunosuppressive cells, including MDSCs and N2 subtype TANs, thereby establishing an immunosuppressive microenvironment that facilitates tumor survival.143,144 MDSCs exert their immunosuppressive effects on T cells through the production of ROS-dependent peroxynitrite (ONOO−), which further supports tumor viability.134 Research has demonstrated an increase in MDSC populations in IBD, correlating strongly with the severity of intestinal inflammation.145,146 In mouse models, the progression from inflammation to cancer in the colon is associated with an accumulation of MDSCs regulated by GM-CSF within the lesions.147 Notably, the depletion of MDSCs has been shown to mitigate the transition from IBD to CAC in experimental mice.148 Research indicates that a deficiency in STAT1 results in diminished recruitment of neutrophils and MDSCs during the initial phase of AOM/DSS-induced CAC in murine models, consequently delaying tumorigenesis.149 Furthermore, the inhibition of neutrophil chemokines has been shown to impede disease progression in CAC model mice. The APE1-NF-κB-CXCL1 signaling pathway in intestinal epithelial cells plays a significant role in modulating the chemotaxis and functionality of neutrophils, thereby exacerbating the progression of IBD.150 The chemokine CXCL1, which is associated with neutrophil recruitment, has been implicated in promoting CAC by recruiting MDSCs and inducing immunosuppressive mechanisms in mice subjected to AOM/DSS treatment.151,152 CXCL5, similar to CXCL1, functions as a ligand for CXCR2 and is capable of recruiting neutrophils via its interaction with the CXCR2 receptor, thereby playing a role in the progression of CAC.133 Furthermore, the inhibition of IL-17 in CAC mouse models results in a reduction of neutrophil recruitment to intestinal tissues, as well as a decrease in the expression of inflammatory mediators and the intestinal signal transducer and activator of transcription (STAT3).149 This inhibition also leads to diminished expression of arginase-1 (ARG1) and inducible nitric oxide synthase (iNOS) in the colon, both of which are associated with the immunosuppressive functions of MDSCs.149 Additionally, the intestinal microbiota has been shown to enhance the immunosuppressive capabilities of MDSCs, thereby contributing to the development of CAC.153
Recent investigations into the role of neutrophils in CAC have yielded promising findings. The administration of anti-ChemR23 antibody has been shown to inhibit the transformation of IBD to CAC in AOM/DSS-induced mouse models.90 Furthermore, CD177+ neutrophils have been identified as playing a crucial role in suppressing the tumorigenesis of IECs in mice with IBD, serving as an independent prognostic factor for patients diagnosed with colorectal cancer.154 Additionally, α-1 antitrypsin (α-1-AT) has demonstrated efficacy in reducing neutrophil populations in CAC mouse model,155 as well as preventing the progression from IBD to CAC and inhibiting the advancement of established CAC through the inhibition of neutrophil-activated serine protease.156 Moreover, calcitonin S100a9 has emerged as a significant pro-inflammatory mediator in both acute and chronic inflammatory processes, contributing to persistent inflammation that may lead to carcinogenesis. Targeting S100a9 could potentially mitigate the risk of tumorigenesis in CAC within AOM/DSS-induced mouse models.54 (Figure 5)
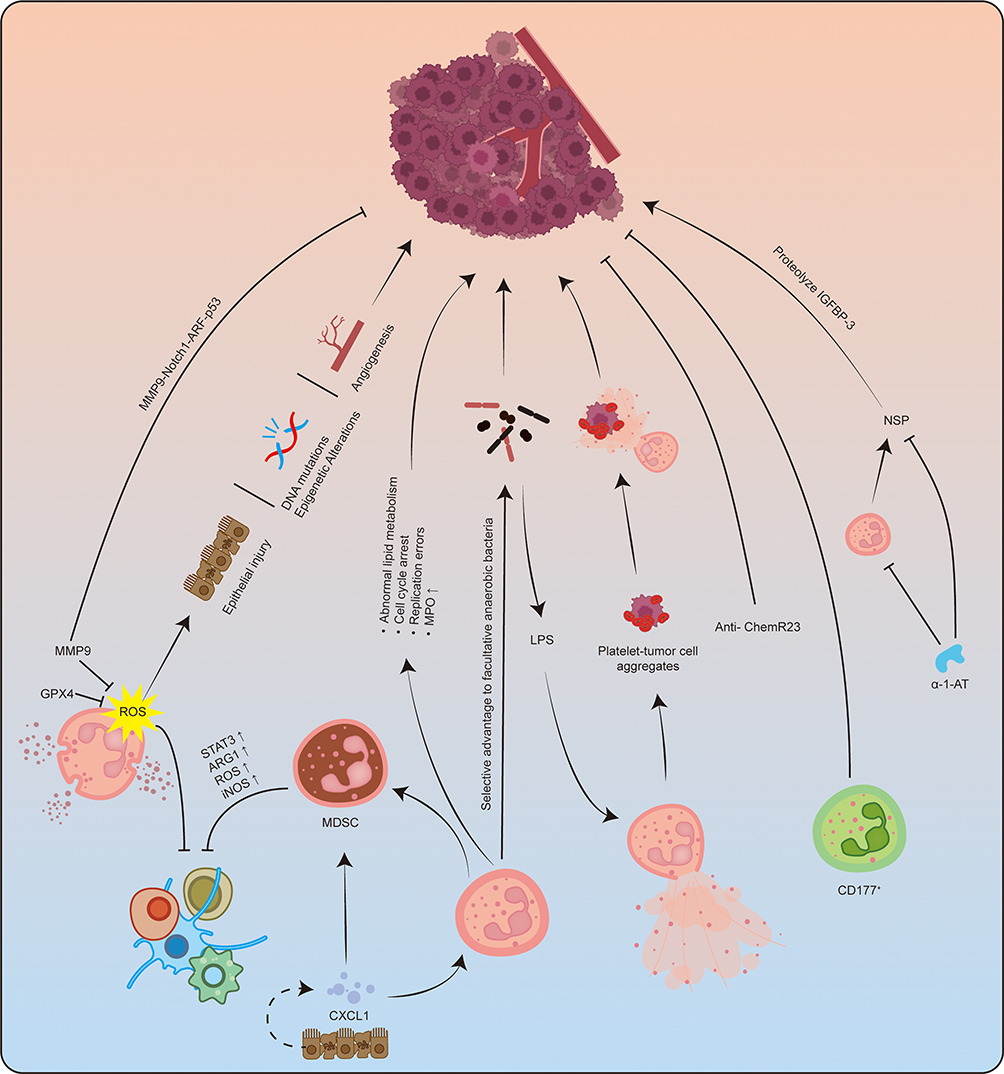 |
Figure 5 Overview of neutrophils in CAC. Neutrophils play a crucial role in the regulation of the development, progression, and metastasis of CAC through a series of intricate signaling pathways. ROS generated by neutrophils have been implicated in the promotion of CAC through various mechanisms, including the induction of epithelial cell damage, DNA mutations, epigenetic alterations, modulation of angiogenesis and immune suppression. The proteins GPX4 and MMP9 are correlated with diminished levels of ROS. Furthermore, MMP9 has the capacity to inhibit CAC via the MMP9-Notch1-ARF-P53 signaling pathway. Intestinal epithelial and tumor cells secrete neutrophil chemokines, such as CXCL1, which facilitate the increased infiltration of neutrophils and MDSCs. MDSCs contribute to the establishment of an immunosuppressive tumor microenvironment through multiple pathways, thereby enhancing the survival of CAC. Additionally, neutrophils may promote CAC through dysregulated lipid metabolism, which can lead to cell cycle arrest and replication errors, as well as the upregulation of MPO. The bactericidal functions of neutrophils confer a selective advantage to facultative anaerobes. Moreover, bacterial LPS can stimulate neutrophils to release NETs, which have the potential to capture aggregates of platelets and tumor cells, thereby facilitating CAC metastasis. The administration of anti-ChemR23 antibodies has been shown to impede the progression from chronic colitis to colorectal cancer. In murine models of IBD, CD177+ neutrophils have been observed to suppress tumorigenesis in intestinal epithelial cells. Additionally, alpha-1 antitrypsin has been demonstrated to decrease neutrophil populations in CAC model mice and to inhibit the transition from IBD to CAC by obstructing the activation of neutrophil serine proteases, thereby preventing the further progression of established CAC. |
The intricate mechanisms underlying the development of CAC remain inadequately understood, complicating both prevention and treatment strategies. Meta-analysis indicates that 5-ASA may reduce the risk of malignant tumor progression in individuals with IBD.157–159 Conversely, another analysis suggests that 5-ASA does not effectively halt the progression of CAC.160 Furthermore, natural antioxidants, including water-soluble vitamins, selenium, and β-carotene, exhibit minimal preventive effects in individuals at high risk for CRC.1 Although the use of anti-TNF-α monoclonal antibodies in the management of IBD has been shown to mitigate the severity of inflammation,161 there is a lack of substantial evidence supporting their efficacy in preventing CAC in humans.1,17 Patients diagnosed with severe IBD accompanied by atypical hyperplasia or malignant tumors require surgical intervention. The associated risks and potential complications of such procedures underscore the critical importance of early intervention in the management of IBD.
Discussion and Prospect
IBD is a multifactorial condition characterized by a dysregulated intestinal immune environment, which arises from the interplay of genetic, environmental, and microbiotic factors. Over time, this dysregulation can lead to atypical hyperplasia and ultimately cancer. While neutrophils are not the sole contributors to IBD, their significance in the pathophysiology of the disease is substantial. Neutrophils function as a double-edged sword in the context of IBD; they play a crucial role in controlling bacterial proliferation, yet an excessive immune response can result in detrimental outcomes. Abnormalities in neutrophil numbers and functions lead to their infiltration into the intestinal mucosa, where they recruit various immune cells, including additional neutrophils, through the release of diverse mediators. This accumulation is exacerbated by resistance to apoptosis, culminating in the formation of crypt abscesses within intestinal lesions. During this process, the integrity of the intestinal epithelial barrier is compromised by ROS, NETs, and proteases released by neutrophils. IBD is characterized by a chronic course punctuated by periods of remission and acute exacerbation.162 The persistent assault by neutrophils on the intestinal tissue over the disease’s progression can ultimately result in the development of irreversible malignancies.
CAC is a multi-stage disease influenced by various cells within the tumor microenvironment. The role of chronic inflammation resulting from IBD is a critical aspect of CAC research that cannot be ignored. A pertinent question arises: at what stage can a disease characterized by persistent intestinal inflammation be classified as malignant? Identifying specific temporal markers to accurately predict and differentiate these stages is a complex challenge. Throughout the progression of CAC, there exists a dynamic interplay between neutrophils (or their various subtypes) and intestinal epithelial or tumor cells. For instance, during acute inflammation, neutrophils migrate from the intestinal mucosa, and in chronic inflammation, neutrophils exhibit resistance to apoptosis, which damage the intestinal epithelial cells while killing the pathogen. Additionally, TANs are prominent during tumorigenesis, NETs are involved in tumor metastasis, and MDSCs are present within the tumor microenvironment. Furthermore, neutrophils engage in intricate interactions with other immune cells and gut microbiota. Consequently, the investigation of neutrophils at specific temporal intervals is insufficient for comprehensively understanding the cell-to-cell interactions that occur during the progression of CAC, and the precise determination of such temporal markers remains unclear.
As previously noted, the transition from IBD to CAC typically spans approximately 20 years, with each episode of relapse and remission in IBD contributing to the dynamics of neutrophil accumulation and regression. Numerous studies have indicated a gradual increase in the risk of CAC correlating with the duration of IBD;163 however, it is important to recognize that not all individuals with IBD will ultimately progress to CAC. Distinguishing between patients with chronic IBD and those who develop CAC presents a significant challenge, as it remains uncertain whether a certain patients with chronic IBD will inevitably progress to CAC. Variations in genetic predisposition may play a crucial role in the development of CAC, while recurrent episodes of acute inflammation, primarily involving neutrophils, may facilitate tumorigenesis to some extent. A retrospective study may be feasible by analyzing samples of IBD phase from patients with established CAC, but several uncertainties persist. These include the regularity of endoscopic sampling intervals. In instances where samples are collected only during episodes of inflammation recurrence, significant uncertainties arise regarding the potential impact of acute inflammation on chronic inflammatory processes. Specifically, fluctuations in gene expression, immune cell infiltration, and alterations in gut microbiota associated with acute inflammation may confound the underlying chronic inflammation. Moreover, it is noteworthy that a limited number of IBD patients undergo colonoscopy during remission periods between flare-ups, which does not preclude the possibility of DNA damage occurring in the intestinal tissue during remission, potentially leading to dysplasia or malignancy. The dynamics of neutrophil changes within the intestinal tissue during this timeframe remain poorly understood. Additionally, challenges in the clinical study of the progression from IBD to CAC include determining whether samples were consistently obtained from the same anatomical site and whether these samples were adequately preserved over a nearly two-decade span.
Furthermore, the recognized methodology for developing a mouse model of CAC typically involves the administration of the carcinogenic agent AOM in conjunction with the inflammatory agent DSS.164 Alternative models include those induced by various chemical agents or through genetic modifications.165 It is important to acknowledge that, while these mouse models are widely regarded as representative of CAC, they still exist significant differences from human IBD and CAC. Additionally, the duration required for the establishment of these models ranges from 10 to 40 weeks.166,167 The determination of specific time points for research purposes remains a topic of ongoing debate. Inflammatory agents such as DSS can induce acute intestinal inflammation, and the repeated induction of such acute inflammatory responses may lead to sustained chronic inflammation within the murine intestine. During DSS-induced acute inflammation, there is a substantial accumulation of neutrophils, which subsequently diminish following the cessation of the treatment.150 However, with repeated DSS exposure, neutrophils that exhibit resistance to apoptosis infiltrate the intestinal tissue and differentiate into immunosuppressive subsets. Given that IBD serves as a precursor to CAC, it appears that the role of chronic inflammation in tumorigenesis is more pronounced, as chronic inflammation is associated with the accumulation of DNA damage. Consequently, it is imperative to consider whether and how the influence of acute inflammation on the overall disease should be excluded when collecting samples and analyzing genetic alterations during tumorigenesis. Another critical consideration is animal welfare; the development of the aforementioned mouse model is time-intensive and can result in adverse health effects, including weight loss, diarrhea, and hematochezia, which are detrimental to the well-being of the animals involved.166
It seems difficult to answer the question perfectly enough: how neutrophils influence the progression of IBD to CAC. Initially, the relationship between intestinal epithelial or tumor cells and immune cells, such as neutrophils, can be analogously described using the metaphor of seeds and soil. In this analogy, the seed represents the potential for growth, while the soil influences the rate and timing of that growth. Historically, research has predominantly concentrated on the genetic alterations occurring in intestinal epithelial cells as they transition into tumor cells, with comparatively limited investigation into the interactions between these “seeds” and the “soil”. Furthermore, CAC is recognized as a multi-stage disease. Chronic inflammation is both a characteristic of IBD and the initiating stage of CAC. Consequently, IBD can be viewed as an integral component of CAC, complicating the ability to distinctly differentiate between the two conditions. The progression of CAC is characterized by a dynamic continuum that includes chronic inflammation, atypical hyperplasia, and ultimately, malignant tumor formation. Within this continuum, it is challenging to pinpoint specific demarcations that separate these stages. Traditional research methodologies often involve the establishment of fixed time intervals for sampling from CAC patients or animal models, which fails to account for the dynamic nature of the disease, particularly the fluctuations associated with the recurrence and resolution of inflammation. This represents a critical gap that necessitates further exploration in contemporary studies.
Lastly, returning to the seed versus soil metaphor, advancements in methodological approaches have significantly enhanced the understanding of the interplay between tumors and immune cells. The evolution from traditional techniques such as flow cytometry and immunohistochemical staining to more sophisticated methods, including single-cell sequencing, multiplex immunohistochemical staining, and organoid cultures, has the potential to elucidate the intricate relationship between neutrophils, IBD, and CAC. Future research endeavors are anticipated to provide a comprehensive understanding of this complex landscape.
In summary, numerous studies have investigated the impact of neutrophils on IBD; however, their role in CAC remains inadequately understood. The complexity of the disease mechanisms contributes to the incomplete elucidation of the etiology of IBD and its associated malignancies. Nevertheless, neutrophils may offer a novel avenue for clarifying these mechanisms and could emerge as potential targets for the prevention and treatment of both IBD and CAC.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82073170).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. Tianyi Chen and Jiachen Liu contributed equally to this manuscript.
References
1. Keller DS, Windsor A, Cohen R, Chand M. Colorectal cancer in inflammatory bowel disease: review of the evidence. Tech Coloproctol. 2019;23(1):3–13. doi:10.1007/s10151-019-1926-2
2. Campbell EL, Colgan SP. Control and dysregulation of redox signalling in the gastrointestinal tract. Nat Rev Gastroenterol Hepatol. 2019;16(2):106–120. doi:10.1038/s41575-018-0079-5
3. Slater TW, Finkielsztein A, Mascarenhas LA, Mehl LC, Butin-Israeli V, Sumagin R. Neutrophil microparticles deliver active myeloperoxidase to injured mucosa to inhibit epithelial wound healing. J Immunol. 2017;198(7):2886–2897. doi:10.4049/jimmunol.1601810
4. Kuno Y, Ina K, Nishiwaki T, et al. Possible involvement of neutrophil elastase in impaired mucosal repair in patients with ulcerative colitis. J Gastroenterol. 2002;37(Suppl 14):22–32. doi:10.1007/BF03326409
5. Butin-Israeli V, Houser MC, Feng M, et al. Deposition of microparticles by neutrophils onto inflamed epithelium: a new mechanism to disrupt epithelial intercellular adhesions and promote transepithelial migration. FASEB J. 2016;30(12):4007–4020. doi:10.1096/fj.201600734R
6. Chen H, Wu X, Xu C, Lin J, Liu Z. Dichotomous roles of neutrophils in modulating pathogenic and repair processes of inflammatory bowel diseases. Precis Clin Med. 2021;4(4):246–257. doi:10.1093/pcmedi/pbab025
7. Drury B, Hardisty G, Gray RD, Ho GT. Neutrophil extracellular traps in inflammatory bowel disease: pathogenic mechanisms and clinical translation. Cell Mol Gastroenterol Hepatol. 2021;12(1):321–333. doi:10.1016/j.jcmgh.2021.03.002
8. Kaplan GG, Windsor JW. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2021;18(1):56–66. doi:10.1038/s41575-020-00360-x
9. Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol. 2015;12(12):720–727. doi:10.1038/nrgastro.2015.150
10. Borowitz SM. The epidemiology of inflammatory bowel disease: clues to pathogenesis? Front Pediatr. 2022;10:1103713. doi:10.3389/fped.2022.1103713
11. Agrawal M, Allin KH, Petralia F, Colombel JF, Jess T. Multiomics to elucidate inflammatory bowel disease risk factors and pathways. Nat Rev Gastroenterol Hepatol. 2022;19(6):399–409. doi:10.1038/s41575-022-00593-y
12. Liu JZ, van Sommeren S, Huang H, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47(9):979–986. doi:10.1038/ng.3359
13. Nasser J, Bergman DT, Fulco CP, et al. Genome-wide enhancer maps link risk variants to disease genes. Nature. 2021;593(7858):238–243. doi:10.1038/s41586-021-03446-x
14. Trindade BC, Chen GY. NOD1 and NOD2 in inflammatory and infectious diseases. Immunol Rev. 2020;297(1):139–161. doi:10.1111/imr.12902
15. Neurath MF. IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev. 2019;45:1–8. doi:10.1016/j.cytogfr.2018.12.002
16. Ramos GP, Papadakis KA. Mechanisms of disease: inflammatory bowel diseases. Mayo Clin Proc. 2019;94(1):155–165. doi:10.1016/j.mayocp.2018.09.013
17. Nakase H, Uchino M, Shinzaki S, et al. Evidence-based clinical practice guidelines for inflammatory bowel disease 2020. J Gastroenterol. 2021;56(6):489–526. doi:10.1007/s00535-021-01784-1
18. Dhaliwal J, Tuna M, Shah BR, et al. Incidence of inflammatory bowel disease in South Asian and Chinese people: a population-based cohort study from Ontario, Canada. Clin Epidemiol. 2021;13:1109–1118. doi:10.2147/CLEP.S336517
19. Piovani D, Danese S, Peyrin-Biroulet L, Nikolopoulos GK, Lytras T, Bonovas S. Environmental risk factors for inflammatory bowel diseases: an umbrella review of meta-analyses. Gastroenterology. 2019;157:647–659e644.
20. Kuenzig ME, Lee SM, Eksteen B, et al. Smoking influences the need for surgery in patients with the inflammatory bowel diseases: a systematic review and meta-analysis incorporating disease duration. BMC Gastroenterol. 2016;16(1):143. doi:10.1186/s12876-016-0555-8
21. Guan Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res. 2019;2019:7247238. doi:10.1155/2019/7247238
22. Glassner KL, Abraham BP, Quigley EMM. The microbiome and inflammatory bowel disease. J Allergy Clin Immunol. 2020;145(1):16–27. doi:10.1016/j.jaci.2019.11.003
23. Danne C. Neutrophils: old cells in IBD, new actors in interactions with the gut microbiota. Clin Transl Med. 2024;14(6):e1739. doi:10.1002/ctm2.1739
24. Danne C, Skerniskyte J, Marteyn B, Sokol H. Neutrophils: from IBD to the gut microbiota. Nat Rev Gastroenterol Hepatol. 2024;21(3):184–197. doi:10.1038/s41575-023-00871-3
25. Liu Z, Zhang Y, Jin T, Yi C, Ocansey DKW, Mao F. The role of NOD2 in intestinal immune response and microbiota modulation: a therapeutic target in inflammatory bowel disease. Int Immunopharmacol. 2022;113:109466. doi:10.1016/j.intimp.2022.109466
26. Liu H, Gao P, Jia B, Lu N, Zhu B, Zhang F. IBD-associated Atg16L1T300A polymorphism regulates commensal microbiota of the intestine. Front Immunol. 2021;12:772189. doi:10.3389/fimmu.2021.772189
27. Hartjes L, Ruland J. CARD9 signaling in intestinal immune homeostasis and oncogenesis. Front Immunol. 2019;10:419. doi:10.3389/fimmu.2019.00419
28. Cohen LJ, Cho JH, Gevers D, Chu H. Genetic factors and the intestinal microbiome guide development of microbe-based therapies for inflammatory bowel diseases. Gastroenterology. 2019;156:2174–2189. doi:10.1053/j.gastro.2019.03.017
29. Lavoie S, Conway KL, Lassen KG, et al. The Crohn’s disease polymorphism, ATG16L1 T300A, alters the gut microbiota and enhances the local Th1/Th17 response. Elife. 2019;8. doi:10.7554/eLife.39982
30. Aschard H, Laville V, Tchetgen ET, et al. Genetic effects on the commensal microbiota in inflammatory bowel disease patients. PLoS Genet. 2019;15(3):e1008018. doi:10.1371/journal.pgen.1008018
31. Li Y, Kundu P, Seow SW, et al. Gut microbiota accelerate tumor growth via c-jun and STAT3 phosphorylation in APCMin/+ mice. Carcinogenesis. 2012;33(6):1231–1238. doi:10.1093/carcin/bgs137
32. Loh JT, Lee KG, Lee AP, et al. DOK3 maintains intestinal homeostasis by suppressing JAK2/STAT3 signaling and S100a8/9 production in neutrophils. Cell Death Dis. 2021;12(11):1054. doi:10.1038/s41419-021-04357-5
33. Mei J, Liu Y, Dai N, et al. Cxcr2 and Cxcl5 regulate the IL-17/G-CSF axis and neutrophil homeostasis in mice. J Clin Invest. 2012;122(3):974–986. doi:10.1172/JCI60588
34. Yang Y, Li L, Xu C, et al. Cross-talk between the gut microbiota and monocyte-like macrophages mediates an inflammatory response to promote colitis-associated tumourigenesis. Gut. 2020;70(8):1495–1506. doi:10.1136/gutjnl-2020-320777
35. Chen K, Fu Y, Wang Y, et al. Therapeutic effects of the in vitro cultured human gut microbiota as transplants on altering gut microbiota and improving symptoms associated with autism spectrum disorder. Microb Ecol. 2020;80(2):475–486. doi:10.1007/s00248-020-01494-w
36. Wang Q, Ye J, Fang D, et al. Multi-omic profiling reveals associations between the gut mucosal microbiome, the metabolome, and host DNA methylation associated gene expression in patients with colorectal cancer. BMC Microbiol. 2020;20(S1):83. doi:10.1186/s12866-020-01762-2
37. Yan Q, Jia L, Wen B, Wu Y, Zeng Y, Wang Q. Clostridium butyricum protects against pancreatic and intestinal injury after severe acute pancreatitis via downregulation of MMP9. Front Pharmacol. 2022;13:919010. doi:10.3389/fphar.2022.919010
38. de Bruyn M, Sabino J, Vandeputte D, Vermeire S, Raes J, Opdenakker G. Comparisons of gut microbiota profiles in wild-type and gelatinase B/matrix metalloproteinase-9-deficient mice in acute DSS-induced colitis. NPJ Biofilms Microbiomes. 2018;4(1):18. doi:10.1038/s41522-018-0059-0
39. Lejeune J, Brachet G, Watier H. Evolutionary story of the low/medium-affinity igg fc receptor gene cluster. Front Immunol. 2019;10:1297. doi:10.3389/fimmu.2019.01297
40. Yoshida K, Murayama MA, Shimizu K, et al. IL-1R2 deficiency suppresses dextran sodium sulfate-induced colitis in mice via regulation of microbiota. Biochem Biophys Res Commun. 2018;496(3):934–940. doi:10.1016/j.bbrc.2018.01.116
41. Hu G, Liu L, Miao X, et al. The response of cecal microbiota to inflammatory state induced by Salmonella enterica serovar Enteritidis. Front Microbiol. 2022;13:963678. doi:10.3389/fmicb.2022.963678
42. Zhang Q, Pan Y, Yan R, et al. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat Immunol. 2015;16(9):918–926. doi:10.1038/ni.3233
43. Yan J, Yu W, Wang G, et al. LRRK2 deficiency mitigates colitis progression by favoring resolution of inflammation and restoring homeostasis of gut microbiota. Genomics. 2022;114(6):110527. doi:10.1016/j.ygeno.2022.110527
44. Wang H, Latorre JD, Bansal M, et al. Microbial metabolite deoxycholic acid controls Clostridium perfringens-induced chicken necrotic enteritis through attenuating inflammatory cyclooxygenase signaling. Sci Rep. 2019;9(1):14541. doi:10.1038/s41598-019-51104-0
45. Pircalabioru G, Aviello G, Kubica M, et al. Defensive mutualism rescues NADPH oxidase inactivation in gut infection. Cell Host Microbe. 2016;19(5):651–663. doi:10.1016/j.chom.2016.04.007
46. Sadaghian Sadabad M, Regeling A, de Goffau MC, et al. The ATG16L1–T300A allele impairs clearance of pathosymbionts in the inflamed ileal mucosa of Crohn’s disease patients. Gut. 2015;64(10):1546–1552. doi:10.1136/gutjnl-2014-307289
47. Danne C, Michaudel C, Skerniskyte J, et al. CARD9 in neutrophils protects from colitis and controls mitochondrial metabolism and cell survival. Gut. 2023;72(6):1081–1092. doi:10.1136/gutjnl-2022-326917
48. Jee J, Mourya R, Shivakumar P, Fei L, Wagner M, Bezerra JA. Cxcr2 signaling and the microbiome suppress inflammation, bile duct injury, and the phenotype of experimental biliary atresia. PLoS One. 2017;12(8):e0182089. doi:10.1371/journal.pone.0182089
49. Dong D, Su T, Chen W, et al. Clostridioides difficile aggravates dextran sulfate solution (DSS)-induced colitis by shaping the gut microbiota and promoting neutrophil recruitment. Gut Microbes. 2023;15(1):2192478. doi:10.1080/19490976.2023.2192478
50. Morgan E, Arnold M, Gini A, et al. Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN. Gut. 2023;72(2):338–344. doi:10.1136/gutjnl-2022-327736
51. Shah SC, Itzkowitz SH. Colorectal cancer in inflammatory bowel disease: mechanisms and management. Gastroenterology. 2022;162(3):715–730e713. doi:10.1053/j.gastro.2021.10.035
52. Lichtenstein GR, Loftus EV, Isaacs KL, Regueiro MD, Gerson LB, Sands BE. ACG clinical guideline: management of crohn’s disease in adults. Am J Gastroenterol. 2018;113(4):481–517. doi:10.1038/ajg.2018.27
53. Kobayashi T, Siegmund B, Le Berre C, et al. Ulcerative colitis. Nat Rev Dis Primers. 2020.
54. Fantini MC, Guadagni I. From inflammation to colitis-associated colorectal cancer in inflammatory bowel disease: pathogenesis and impact of current therapies. Dig Liver Dis. 2021;53(5):558–565. doi:10.1016/j.dld.2021.01.012
55. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48(4):526–535. doi:10.1136/gut.48.4.526
56. Wijnands AM, de Jong ME, Lutgens M, et al. Prognostic factors for advanced colorectal neoplasia in inflammatory bowel disease: systematic review and meta-analysis. Gastroenterology. 2021;160(5):1584–1598. doi:10.1053/j.gastro.2020.12.036
57. Huang Q, Yang Y, Zhu Y, et al. Oral metal-free melanin nanozymes for natural and durable targeted treatment of inflammatory bowel disease (IBD). Small. 2023;19(19):e2207350. doi:10.1002/smll.202207350
58. Mazaki J, Katsumata K, Kasahara K, et al. Neutrophil-to-lymphocyte ratio is a prognostic factor for colon cancer: a propensity score analysis. BMC Cancer. 2020;20(1):922. doi:10.1186/s12885-020-07429-5
59. Wang Y, Wang K, Han GC, et al. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunol. 2014;7(5):1106–1115. doi:10.1038/mi.2013.126
60. Bui TM, Butin-Israeli V, Wiesolek HL, et al. Neutrophils alter DNA Repair landscape to impact survival and shape distinct therapeutic phenotypes of colorectal cancer. Gastroenterology. 2021;161(1):225–238e215. doi:10.1053/j.gastro.2021.03.027
61. Canli O, Nicolas AM, Gupta J, et al. Myeloid cell-derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell. 2017;32(6):869–883e865. doi:10.1016/j.ccell.2017.11.004
62. Frick A, Khare V, Paul G, et al. Overt increase of oxidative stress and DNA Damage in murine and human colitis and colitis-associated neoplasia. Mol Cancer Res. 2018;16(4):634–642. doi:10.1158/1541-7786.MCR-17-0451
63. Meyer R, Freitag-Wolf S, Blindow S, Buning J, Habermann JK. Combining aneuploidy and dysplasia for colitis’ cancer risk assessment outperforms current surveillance efficiency: a meta-analysis. Int J Colorectal Dis. 2017;32(2):171–182. doi:10.1007/s00384-016-2684-5
64. Wanders LK, Cordes M, Voorham Q, et al. IBD-associated dysplastic lesions show more chromosomal instability than sporadic adenomas. Inflamm Bowel Dis. 2020;26(2):167–180. doi:10.1093/ibd/izz171
65. Gene M, Cuatrecasas M, Amat I, et al. Alterations in p53, microsatellite stability and lack of MUC5AC expression as molecular features of colorectal carcinoma associated with inflammatory bowel disease. Int J Mol Sci. 2023;24(10):8655. doi:10.3390/ijms24108655
66. Granofszky N, Lang M, Khare V, et al. Identification of PMN-released mutagenic factors in a co-culture model for colitis-associated cancer. Carcinogenesis. 2018;39(2):146–157. doi:10.1093/carcin/bgx118
67. Yin Y, Wan J, Yu J, Wu K. Molecular pathogenesis of colitis-associated colorectal cancer: immunity, genetics, and intestinal microecology. Inflamm Bowel Dis. 2023;29(10):1648–1657. doi:10.1093/ibd/izad081
68. Emmett RA, Davidson KL, Gould NJ, Arasaradnam RP. DNA methylation patterns in ulcerative colitis-associated cancer: a systematic review. Epigenomics. 2017;9(7):1029–1042. doi:10.2217/epi-2017-0025
69. Bergstrom K, Liu X, Zhao Y, et al. Defective intestinal mucin-type o-glycosylation causes spontaneous colitis-associated cancer in mice. Gastroenterology. 2016;151(1):152–164e111. doi:10.1053/j.gastro.2016.03.039
70. Gu Y, Zhao H, Zheng L, et al. Non-coding RNAs and colitis-associated cancer: mechanisms and clinical applications. Clin Transl Med. 2023;13(5):e1253. doi:10.1002/ctm2.1253
71. Gong W, Liu P, Zhao F, et al. STING-mediated Syk Signaling Attenuates Tumorigenesis Of Colitis‑Associated Colorectal Cancer Through Enhancing Intestinal Epithelium Pyroptosis. Inflamm Bowel Dis. 2022;28(4):572–585. doi:10.1093/ibd/izab217
72. Tan G, Huang C, Chen J, Zhi F. HMGB1 released from GSDME-mediated pyroptotic epithelial cells participates in the tumorigenesis of colitis-associated colorectal cancer through the ERK1/2 pathway. J Hematol Oncol. 2020;13(1):149. doi:10.1186/s13045-020-00985-0
73. Xu H, Li J, Chen H, Ghishan FK. NHE8 deficiency promotes colitis-associated cancer in mice via expansion of Lgr5-expressing cells. Cell Mol Gastroenterol Hepatol. 2019;7(1):19–31. doi:10.1016/j.jcmgh.2018.08.005
74. Swafford D, Shanmugam A, Ranganathan P, et al. The Wnt-beta-Catenin-IL-10 signaling axis in intestinal APCs protects mice from colitis-associated colon cancer in response to gut microbiota. J Immunol. 2020;205(8):2265–2275. doi:10.4049/jimmunol.1901376
75. Zhao Q, Bi Y, Zhong J, et al. Pristimerin suppresses colorectal cancer through inhibiting inflammatory responses and Wnt/beta-catenin signaling. Toxicol Appl Pharmacol. 2020;386:114813. doi:10.1016/j.taap.2019.114813
76. Zhang C, Zhang J, Zhang Y, et al. Identifying neutrophil-associated subtypes in ulcerative colitis and confirming neutrophils promote colitis-associated colorectal cancer. Front Immunol. 2023;14:1095098. doi:10.3389/fimmu.2023.1095098
77. Ghimire J, Iftikhar R, Penrose HM, Snarski P, Ruiz E, Savkovic SD. FOXO3 deficiency in neutrophils drives colonic inflammation and tumorigenesis. Int J Mol Sci. 2023;24(11):9730. doi:10.3390/ijms24119730
78. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. 2020;20(9):485–503. doi:10.1038/s41568-020-0281-y
79. Perez-Figueroa E, Alvarez-Carrasco P, Ortega E, Maldonado-Bernal C. Neutrophils: many ways to die. Front Immunol. 2021;12:631821. doi:10.3389/fimmu.2021.631821
80. Ellson CD, Rica IG, Kim JS, et al. An integrated pharmacological, structural, and genetic analysis of extracellular versus intracellular ROS production in neutrophils. J Mol Biol. 2022;434(9):167533. doi:10.1016/j.jmb.2022.167533
81. Mollinedo F. Neutrophil degranulation, plasticity, and cancer metastasis. Trends Immunol. 2019;40(3):228–242. doi:10.1016/j.it.2019.01.006
82. Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev. 2016;273(1):11–28. doi:10.1111/imr.12440
83. Antuamwine BB, Bosnjakovic R, Hofmann-Vega F, et al. N1 versus N2 and PMN-MDSC: a critical appraisal of current concepts on tumor-associated neutrophils and new directions for human oncology. Immunol Rev. 2023;314(1):250–279. doi:10.1111/imr.13176
84. Alsultan A, Sokol RJ, Lovell MA, Thurman G, Ambruso DR. Long term G-CSF-induced remission of ulcerative colitis-like inflammatory bowel disease in a patient with glycogen storage disease Ib and evaluation of associated neutrophil function. Pediatr Blood Cancer. 2010;55(7):1410–1413. doi:10.1002/pbc.22706
85. Nalle SC, Turner JR. Intestinal barrier loss as a critical pathogenic link between inflammatory bowel disease and graft-versus-host disease. Mucosal Immunol. 2015;8(4):720–730. doi:10.1038/mi.2015.40
86. Kang L, Fang X, Song YH, et al. Neutrophil-epithelial crosstalk during intestinal inflammation. Cell Mol Gastroenterol Hepatol. 2022;14(6):1257–1267. doi:10.1016/j.jcmgh.2022.09.002
87. Allocca M, Jovani M, Fiorino G, Schreiber S, Danese S. Anti-IL-6 treatment for inflammatory bowel diseases: next cytokine, next target. Curr Drug Targets. 2013;14(12):1508–1521. doi:10.2174/13894501113146660224
88. Noseykina EM, Schepetkin IA, Atochin DN. Molecular mechanisms for regulation of neutrophil apoptosis under normal and pathological conditions. J Evol Biochem Physiol. 2021;57(3):429–450. doi:10.1134/S0022093021030017
89. Kobayashi SD, DeLeo FR, Quinn MT. Microbes and the fate of neutrophils. Immunol Rev. 2023;314(1):210–228. doi:10.1111/imr.13163
90. Trilleaud C, Gauttier V, Biteau K, et al. Agonist anti-ChemR23 mAb reduces tissue neutrophil accumulation and triggers chronic inflammation resolution. Sci Adv. 2021;7(14). doi:10.1126/sciadv.abd1453
91. Wang JF, Wang YP, Xie J, et al. Upregulated PD-L1 delays human neutrophil apoptosis and promotes lung injury in an experimental mouse model of sepsis. Blood. 2021;138(9):806–810. doi:10.1182/blood.2020009417
92. Ina K, Kusugami K, Hosokawa T, et al. Increased mucosal production of granulocyte colony-stimulating factor is related to a delay in neutrophil apoptosis in Inflammatory Bowel disease. J Gastroenterol Hepatol. 1999;14(1):46–53. doi:10.1046/j.1440-1746.1999.01807.x
93. Brannigan AE, O’Connell PR, Hurley H, et al. Neutrophil apoptosis is delayed in patients with inflammatory bowel disease. Shock. 2000;13(5):361–366. doi:10.1097/00024382-200005000-00003
94. Therrien A, Chapuy L, Bsat M, et al. Recruitment of activated neutrophils correlates with disease severity in adult Crohn’s disease. Clin Exp Immunol. 2019;195(2):251–264. doi:10.1111/cei.13226
95. Pai RK, Hartman DJ, Rivers CR, et al. Complete resolution of mucosal neutrophils associates with improved long-term clinical outcomes of patients with ulcerative colitis. Clin Gastroenterol Hepatol. 2020;18(11):2510–2517e2515. doi:10.1016/j.cgh.2019.12.011
96. Glennon-Alty L, Moots RJ, Edwards SW, Wright HL. Type I interferon regulates cytokine-delayed neutrophil apoptosis, reactive oxygen species production and chemokine expression. Clin Exp Immunol. 2021;203(2):151–159. doi:10.1111/cei.13525
97. Fallon EA, Biron-Girard BM, Chung CS, et al. A novel role for coinhibitory receptors/checkpoint proteins in the immunopathology of sepsis. J Leukoc Biol. 2018;103(6):1151–1164. doi:10.1002/JLB.2MIR0917-377R
98. Talmon G, Manasek T, Miller R, Muirhead D, Lazenby A. The apoptotic crypt abscess: an underappreciated histologic finding in gastrointestinal pathology. Am J Clin Pathol. 2017;148(6):538–544. doi:10.1093/ajcp/aqx100
99. Villanacci V, Del Sordo R, Parigi TL, Leoncini G, Bassotti G. Inflammatory bowel diseases: does one histological score fit all? Diagnostics. 2023;13(12). doi:10.3390/diagnostics13122112
100. Zhou J, Li M, Chen Q, et al. Programmable probiotics modulate inflammation and gut microbiota for inflammatory bowel disease treatment after effective oral delivery. Nat Commun. 2022;13(1):3432. doi:10.1038/s41467-022-31171-0
101. Willson JA, Arienti S, Sadiku P, et al. Neutrophil HIF-1alpha stabilization is augmented by mitochondrial ROS produced via the glycerol 3-phosphate shuttle. Blood. 2022;139(2):281–286. doi:10.1182/blood.2021011010
102. Lagnado A, Leslie J, Ruchaud-Sparagano MH, et al. Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. EMBO J. 2021;40(9):e106048. doi:10.15252/embj.2020106048
103. Zhang T, Jiang J, Liu J, et al. MK2 is required for neutrophil-derived ros production and inflammatory bowel disease. Front Med Lausanne. 2020;7:207. doi:10.3389/fmed.2020.00207
104. Denson LA, Jurickova I, Karns R, et al. Clinical and genomic correlates of neutrophil reactive oxygen species production in pediatric patients with Crohn’s disease. Gastroenterology. 2018;154(8):2097–2110. doi:10.1053/j.gastro.2018.02.016
105. Mozzini C, Pagani M. Cardiovascular Diseases: consider Netosis. Curr Probl Cardiol. 2022;47(10):100929. doi:10.1016/j.cpcardiol.2021.100929
106. Dinallo V, Marafini I, Di Fusco D, et al. Neutrophil extracellular traps sustain inflammatory signals in ulcerative colitis. J Crohns Colitis. 2019;13(6):772–784. doi:10.1093/ecco-jcc/jjy215
107. Li T, Wang C, Liu Y, et al. Neutrophil extracellular traps induce intestinal damage and thrombotic tendency in inflammatory bowel disease. J Crohns Colitis. 2020;14(2):240–253. doi:10.1093/ecco-jcc/jjz132
108. Schroder AL, Chami B, Liu Y, et al. Neutrophil extracellular trap density increases with increasing histopathological severity of Crohn’s disease. Inflamm Bowel Dis. 2022;28(4):586–598. doi:10.1093/ibd/izab239
109. He Z, Si Y, Jiang T, et al. Phosphotidylserine exposure and neutrophil extracellular traps enhance procoagulant activity in patients with inflammatory bowel disease. Thromb Haemost. 2016;115(04):738–751. doi:10.1160/TH15-09-0710
110. Zhou G, Yu L, Fang L, et al. CD177+ neutrophils as functionally activated neutrophils negatively regulate IBD. Gut. 2018;67(6):1052–1063. doi:10.1136/gutjnl-2016-313535
111. Angelidou I, Chrysanthopoulou A, Mitsios A, et al. REDD1/autophagy pathway is associated with neutrophil-driven IL-1beta inflammatory response in active ulcerative colitis. J Immunol. 2018;200(12):3950–3961. doi:10.4049/jimmunol.1701643
112. Lin EY, Lai HJ, Cheng YK, et al. Neutrophil extracellular traps impair intestinal barrier function during experimental colitis. Biomedicines. 2020;8(8):275. doi:10.3390/biomedicines8080275
113. Shao Y, Li L, Yang Y, et al. DNase aggravates intestinal microvascular injury in IBD patients by releasing NET-related proteins. FASEB J. 2024;38(1):e23395. doi:10.1096/fj.202301780R
114. Chen X, Bao S, Liu M, et al. Inhibition of HMGB1 improves experimental mice colitis by mediating NETs and macrophage polarization. Cytokine. 2024;176:156537. doi:10.1016/j.cyto.2024.156537
115. Torok S, Almasi N, Valkusz Z, Posa A, Varga C, Kupai K. Investigation of H(2)S donor treatment on neutrophil extracellular traps in experimental colitis. Int J Mol Sci. 2021;22(23):12729. doi:10.3390/ijms222312729
116. Zhang L, Zheng B, Bai Y, et al. Exosomes-transferred LINC00668 contributes to thrombosis by promoting NETs formation in inflammatory bowel disease. Adv Sci. 2023;10(28):e2300560. doi:10.1002/advs.202300560
117. Giannotta M, Tapete G, Emmi G, Silvestri E, Milla M. Thrombosis in inflammatory bowel diseases: what’s the link? Thromb J. 2015;13(1):14. doi:10.1186/s12959-015-0044-2
118. Lie MR, Kanis SL, Hansen BE, van der Woude CJ. Drug therapies for ulcerative proctitis: systematic review and meta-analysis. Inflamm Bowel Dis. 2014;20(11):2157–2178. doi:10.1097/MIB.0000000000000141
119. Marshall JK, Irvine EJ. Rectal corticosteroids versus alternative treatments in ulcerative colitis: a meta-analysis. Gut. 1997;40(6):775–781. doi:10.1136/gut.40.6.775
120. Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012;5(4):354–366. doi:10.1038/mi.2012.24
121. Zhang C, Shu W, Zhou G, et al. Anti-TNF-alpha therapy suppresses proinflammatory activities of mucosal neutrophils in inflammatory bowel disease. Mediators Inflamm. 2018;2018:3021863. doi:10.1155/2018/3021863
122. Bamias G, Zampeli E, Domenech E. Targeting neutrophils in inflammatory bowel disease: revisiting the role of adsorptive granulocyte and monocyte apheresis. Expert Rev Gastroenterol Hepatol. 2022;16(8):721–735. doi:10.1080/17474124.2022.2100759
123. Camba-Gomez M, Gualillo O, Conde-Aranda J. New perspectives in the study of intestinal inflammation: focus on the resolution of inflammation. Int J Mol Sci. 2021;22(5):2605. doi:10.3390/ijms22052605
124. Dos Santos Ramos A, Viana GCS, de Macedo Brigido M, Almeida JF. Neutrophil extracellular traps in inflammatory bowel diseases: implications in pathogenesis and therapeutic targets. Pharmacol Res. 2021;171:105779. doi:10.1016/j.phrs.2021.105779
125. Li G, Lin J, Zhang C, et al. Microbiota metabolite butyrate constrains neutrophil functions and ameliorates mucosal inflammation in inflammatory bowel disease. Gut Microbes. 2021;13(1):1968257. doi:10.1080/19490976.2021.1968257
126. Yan YX, Shao MJ, Qi Q, et al. Artemisinin analogue SM934 ameliorates DSS-induced mouse ulcerative colitis via suppressing neutrophils and macrophages. Acta Pharmacol Sin. 2018;39(10):1633–1644. doi:10.1038/aps.2017.185
127. Lu H, Lin J, Xu C, et al. Cyclosporine modulates neutrophil functions via the SIRT6-HIF-1alpha-glycolysis axis to alleviate severe ulcerative colitis. Clin Transl Med. 2021;11(2):e334. doi:10.1002/ctm2.334
128. Lin CM, Huang TH, Chi MC, et al. N-acetylcysteine alleviates fine particulate matter (PM2.5)-induced lung injury by attenuation of ROS-mediated recruitment of neutrophils and Ly6C(high) monocytes and lung inflammation. Ecotoxicol Environ Saf. 2022;239:113632. doi:10.1016/j.ecoenv.2022.113632
129. Couto D, Ribeiro D, Freitas M, Gomes A, Lima JL, Fernandes E. Scavenging of reactive oxygen and nitrogen species by the prodrug sulfasalazine and its metabolites 5-aminosalicylic acid and sulfapyridine. Redox Rep. 2010;15(6):259–267. doi:10.1179/135100010X12826446921707
130. Chami B, Martin NJJ, Dennis JM, Witting PK. Myeloperoxidase in the inflamed colon: a novel target for treating inflammatory bowel disease. Arch Biochem Biophys. 2018;645:61–71. doi:10.1016/j.abb.2018.03.012
131. Baker KT, Salk JJ, Brentnall TA, Risques RA. Precancer in ulcerative colitis: the role of the field effect and its clinical implications. Carcinogenesis. 2018;39(1):11–20. doi:10.1093/carcin/bgx117
132. Nagao-Kitamoto H, Kitamoto S, Kamada N. Inflammatory bowel disease and carcinogenesis. Cancer Metastasis Rev. 2022;41(2):301–316. doi:10.1007/s10555-022-10028-4
133. Lin Y, Cheng L, Liu Y, et al. Intestinal epithelium-derived BATF3 promotes colitis-associated colon cancer through facilitating CXCL5-mediated neutrophils recruitment. Mucosal Immunol. 2021;14(1):187–198. doi:10.1038/s41385-020-0297-3
134. Kennel KB, Greten FR. Immune cell - produced ROS and their impact on tumor growth and metastasis. Redox Biol. 2021;42:101891. doi:10.1016/j.redox.2021.101891
135. Walter L, Canup B, Pujada A, et al. Matrix metalloproteinase 9 (MMP9) limits reactive oxygen species (ROS) accumulation and DNA damage in colitis-associated cancer. Cell Death Dis. 2020;11(9):767. doi:10.1038/s41419-020-02959-z
136. Zhong J, Li Q, Luo H, Holmdahl R. Neutrophil-derived reactive oxygen species promote tumor colonization. Commun Biol. 2021;4(1):865. doi:10.1038/s42003-021-02376-8
137. He L, Kang Q, Chan KI, Zhang Y, Zhong Z, Tan W. The immunomodulatory role of matrix metalloproteinases in colitis-associated cancer. Front Immunol. 2022;13:1093990.
138. Short SP, Pilat JM, Barrett CW, et al. Colonic epithelial-derived selenoprotein P is the source for antioxidant-mediated protection in colitis-associated cancer. Gastroenterology. 2021;160:1694–1708e1693.
139. Kong X, Zhang Y, Xiang L, et al. Fusobacterium nucleatum-triggered neutrophil extracellular traps facilitate colorectal carcinoma progression. J Exp Clin Cancer Res. 2023;42:236.
140. Wang WW, Wu L, Lu W, et al. Lipopolysaccharides increase the risk of colorectal cancer recurrence and metastasis due to the induction of neutrophil extracellular traps after curative resection. J Cancer Res Clin Oncol. 2021;147(9):2609–2619. doi:10.1007/s00432-021-03682-8
141. Ren J, He J, Zhang H, et al. Platelet TLR4-ERK5 Axis Facilitates NET-mediated capturing of circulating tumor cells and distant metastasis after surgical stress. Cancer Res. 2021;81(9):2373–2385. doi:10.1158/0008-5472.CAN-20-3222
142. Wang C, Zheng X, Zhang J, et al. CD300ld on neutrophils is required for tumour-driven immune suppression. Nature. 2023;621(7980):830–839. doi:10.1038/s41586-023-06511-9
143. Zhao H, Wu L, Yan G, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. 2021;6(1):263. doi:10.1038/s41392-021-00658-5
144. Wang Y, Ding Y, Deng Y, Zheng Y, Wang S. Role of myeloid-derived suppressor cells in the promotion and immunotherapy of colitis-associated cancer. J Immunother Cancer. 2020;8(2):e000609. doi:10.1136/jitc-2020-000609
145. Guan Q, Moreno S, Qing G, et al. The role and potential therapeutic application of myeloid-derived suppressor cells in TNBS-induced colitis. J Leukoc Biol. 2013;94(4):803–811. doi:10.1189/jlb.0113050
146. Oh SY, Cho KA, Kang JL, Kim KH, Woo SY. Comparison of experimental mouse models of inflammatory bowel disease. Int J Mol Med. 2014;33(2):333–340. doi:10.3892/ijmm.2013.1569
147. Ma N, Liu Q, Hou L, Wang Y, Liu Z. MDSCs are involved in the protumorigenic potentials of GM-CSF in colitis-associated cancer. Int J Immunopathol Pharmacol. 2017;30(2):152–162. doi:10.1177/0394632017711055
148. Poh TW, Madsen CS, Gorman JE, et al. Downregulation of hematopoietic MUC1 during experimental colitis increases tumor-promoting myeloid-derived suppressor cells. Clin Cancer Res. 2013;19(18):5039–5052. doi:10.1158/1078-0432.CCR-13-0278
149. Delgado-Ramirez Y, Baltazar-Perez I, Martinez Y, et al. STAT1 is required for decreasing accumulation of granulocytic Cells via IL-17 during initial steps of colitis-associated cancer. Int J Mol Sci. 2021;22.
150. Chen Q, Chen T, Xiao H, et al. APEX1 in intestinal epithelium triggers neutrophil infiltration and intestinal barrier damage in Ulcerative colitis. Free Radic Biol Med. 2024;225:359–373. doi:10.1016/j.freeradbiomed.2024.10.260
151. Liu ZY, Zheng M, Li YM, et al. RIP3 promotes colitis-associated colorectal cancer by controlling tumor cell proliferation and CXCL1-induced immune suppression. Theranostics. 2019;9(12):3659–3673. doi:10.7150/thno.32126
152. Tianyi C, Chaofan L, Lingbo B, et al. APE1 mediates the occurrence and development of colitis-associated colorectal cancer through immunosuppressive tumor microenvironment. J Army Med University. 2024;46:1825–1837.
153. Zhang Z, Zheng Y, Chen Y, et al. Gut fungi enhances immunosuppressive function of myeloid-derived suppressor cells by activating PKM2-dependent glycolysis to promote colorectal tumorigenesis. Exp Hematol Oncol. 2022;11(1):88. doi:10.1186/s40164-022-00334-6
154. Zhou G, Peng K, Song Y, et al. CD177+ neutrophils suppress epithelial cell tumourigenesis in colitis-associated cancer and predict good prognosis in colorectal cancer. Carcinogenesis. 2018;39(2):272–282. doi:10.1093/carcin/bgx142
155. Al-Omari M, Al-Omari T, Batainah N, Al-Qauod K, Olejnicka B, Janciauskiene S. Beneficial effects of alpha-1 antitrypsin therapy in a mouse model of colitis-associated colon cancer. BMC Cancer. 2023;23(1):722. doi:10.1186/s12885-023-11195-5
156. Cai Q, Kim M, Harada A, et al. Alpha-1 antitrypsin inhibits tumorigenesis and progression of colitis-associated colon cancer through suppression of inflammatory neutrophil-activated serine proteases and IGFBP-3 proteolysis. Int J Mol Sci. 2022;23(22):13737. doi:10.3390/ijms232213737
157. O’Connor A, Packey CD, Akbari M, Moss AC. Mesalamine, but not sulfasalazine, reduces the risk of colorectal neoplasia in patients with inflammatory bowel disease: an agent-specific systematic review and meta-analysis. Inflamm Bowel Dis. 2015;21(11):2562–2569. doi:10.1097/MIB.0000000000000540
158. Zhao LN, Li JY, Yu T, Chen GC, Yuan YH, Chen QK. 5-Aminosalicylates reduce the risk of colorectal neoplasia in patients with ulcerative colitis: an updated meta-analysis. PLoS One. 2014;9(4):e94208. doi:10.1371/journal.pone.0094208
159. Bonovas S, Fiorino G, Lytras T, Nikolopoulos G, Peyrin-Biroulet L, Danese S. Systematic review with meta-analysis: use of 5-aminosalicylates and risk of colorectal neoplasia in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2017;45(9):1179–1192. doi:10.1111/apt.14023
160. Nguyen GC, Gulamhusein A, Bernstein CN. 5-aminosalicylic acid is not protective against colorectal cancer in inflammatory bowel disease: a meta-analysis of non-referral populations. Am J Gastroenterol. 2012;107(9):1298–1304;quiz1297,1305. doi:10.1038/ajg.2012.198
161. M’Koma AE. Inflammatory bowel disease: clinical diagnosis and surgical treatment-overview. Medicina. 2022;58(5). doi:10.3390/medicina58050567
162. Sahakian L, Robinson AM, Sahakian L, Stavely R, Kelley MR, Nurgali K. APE1/Ref-1 as a therapeutic target for inflammatory bowel disease. Biomolecules. 2023;13(11):1569. doi:10.3390/biom13111569
163. Bopanna S, Ananthakrishnan AN, Kedia S, Yajnik V, Ahuja V. Risk of colorectal cancer in Asian patients with ulcerative colitis: a systematic review and meta-analysis. Lancet Gastroenterol. 2017;2:269–276.
164. Dzhalilova D, Zolotova N, Fokichev N, Makarova O. Murine models of colorectal cancer: the azoxymethane (AOM)/dextran sulfate sodium (DSS) model of colitis-associated cancer. PeerJ. 2023;11:e16159. doi:10.7717/peerj.16159
165. Sussman DA, Santaolalla R, Strobel S, Dheer R, Abreu MT. Cancer in inflammatory bowel disease: lessons from animal models. Curr Opin Gastroenterol. 2012;28(4):327–333. doi:10.1097/MOG.0b013e328354cc36
166. Burtin F, Mullins CS, Linnebacher M. Mouse models of colorectal cancer: past, present and future perspectives. World J Gastroenterol. 2020;26(13):1394–1426. doi:10.3748/wjg.v26.i13.1394
167. De Robertis M, Massi E, Poeta ML, et al. The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J Carcinog. 2011;10(1):9. doi:10.4103/1477-3163.78279
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.