")
Back to Journals » Journal of Inflammation Research » Volume 17
Novel Insights into Parkin–Mediated Mitochondrial Dysfunction and “Mito-Inflammation” in α-Synuclein Toxicity. The Role of the cGAS–STING Signalling Pathway
Authors Gąssowska-Dobrowolska M , Olech-Kochańczyk G, Culmsee C , Adamczyk A
Received 12 March 2024
Accepted for publication 22 June 2024
Published 11 July 2024 Volume 2024:17 Pages 4549—4574
DOI https://doi.org/10.2147/JIR.S468609
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Tara Strutt
Magdalena Gąssowska-Dobrowolska,1 Gabriela Olech-Kochańczyk,1 Carsten Culmsee,2,3 Agata Adamczyk1
1Department of Cellular Signalling, Mossakowski Medical Research Institute, Polish Academy of Sciences, Warsaw, Poland; 2Institute of Pharmacology and Clinical Pharmacy, University of Marburg, Marburg, Germany; 3Center for Mind Brain and Behavior – CMBB, University of Marburg, Marburg, Germany
Correspondence: Magdalena Gąssowska-Dobrowolska, Email [email protected]
Abstract: The prevalence of age-related neurodegenerative diseases, such as Parkinson’s disease (PD) and related disorders continues to grow worldwide. Increasing evidence links intracellular inclusions of misfolded alpha-synuclein (α-syn) aggregates, so-called Lewy bodies (LB) and Lewy neuritis, to the progressive pathology of PD and other synucleinopathies. Our previous findings established that α-syn oligomers induce S-nitrosylation and deregulation of the E3-ubiquitin ligase Parkin, leading to mitochondrial disturbances in neuronal cells. The accumulation of damaged mitochondria as a consequence, together with the release of mitochondrial-derived damage-associated molecular patterns (mtDAMPs) could activate the innate immune response and induce neuroinflammation (“mito-inflammation”), eventually accelerating neurodegeneration. However, the molecular pathways that transmit pro-inflammatory signals from damaged mitochondria are not well understood. One of the proposed pathways could be the cyclic GMP-AMP synthase (cGAS) – stimulator of interferon genes (STING) (cGAS–STING) pathway, which plays a pivotal role in modulating the innate immune response. It has recently been suggested that cGAS–STING deregulation may contribute to the development of various pathological conditions. Especially, its excessive engagement may lead to neuroinflammation and appear to be essential for the development of neurodegenerative brain diseases, including PD. However, the precise molecular mechanisms underlying cGAS–STING pathway activation in PD and other synucleinopathies are not fully understood. This review focuses on linking mitochondrial dysfunction to neuroinflammation in these disorders, particularly emphasizing the role of the cGAS–STING signaling. We propose the cGAS–STING pathway as a critical driver of inflammation in α-syn-dependent neurodegeneration and hypothesize that cGAS–STING–driven “mito-inflammation” may be one of the key mechanisms promoting the neurodegeneration in PD. Understanding the molecular mechanisms of α-syn–induced cGAS–STING–associated “mito-inflammation” in PD and related synucleinopathies may contribute to the identification of new targets for the treatment of these disorders.
Keywords: α-synuclein, Parkin, mtDAMPs, mito-inflammation, sterile inflammation, cGAS–STING pathway
Introduction
Parkinson’s disease (PD) is a widespread, age-related, fatal neurodegenerative disorder that impairs movement and is accompanied by dementia, affecting 3% of the global population over the age of 65.1 The most prominent neuropathological hallmarks of PD are the presence of intracellular inclusions containing misfolded α-synuclein (α-syn) aggregates, such as Lewy bodies (LB) and Lewy neuritis, and progressive dopaminergic neurodegeneration in the substantia nigra pars compacta (SNpc). Aggregated forms of α-syn have also been implicated in other neurodegenerative diseases such as dementia with Lewy bodies (DLB), multiple system atrophy (MSA), pure autonomic failure (PAF), and REM sleep behaviour disorder (RBD), collectively termed α-synucleinopathies.2 Although the discovery of the critical role of this protein in the pathogenesis of PD is almost thirty years old, many fundamental questions regarding successful strategies to target α-syn toxicity to prevent neurodegeneration remain unanswered. Proposed mechanisms of α-syn neurotoxicity include oxidative/nitrosative stress generation, mitochondrial and proteasomal dysfunction as well as neuroinflammation, but links between these detrimental processes are poorly understood.3,4 Our previous studies show that α-syn–induced nitrosative stress leads to S-nitrosylation and inactivation of another PD-associated protein, an E3 ubiquitin ligase, Parkin,5,6 and that α-syn–evoked Parkin down-regulation promotes mitochondrial dysfunction in neuronal cells.7 Parkin regulates protein breakdown by the ubiquitin–proteasome system and is essential for mitochondrial quality control (MQC) assurance.8 Upon loss of the mitochondrial membrane potential, Parkin is translocated to the mitochondrial surface and promotes degradation of the defective organelle by mitophagy, a selective type of autophagy.9 Therefore, Parkin could be a “multipurpose neuroprotective agent” that protects mitochondrial integrity and prevents mitochondrial stress–induced inflammation, oxidative stress, and cell death.10–18 In addition, Parkin can mitigate neuroinflammation through interaction with several key immune regulatory pathways, including those involving the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family pyrin domain containing protein 3 (NLRP3) inflammasome, nuclear factor κB (NF-κB) and tumour necrosis factor-α (TNF-α) receptor (TNFR) – associated factors.19–21 The effects of Parkin deficiency, combined with mitochondrial damage and release of mitochondrial-derived damage-associated molecular patterns (mtDAMPs), may induce the abnormal activation of NLRP3 and its signalling mediator, cyclic GMP-AMP synthase (cGAS) – stimulator of the interferon genes (STING) (cGAS–STING) immune pathway.22–24 This could render neurons extremely vulnerable to any additional mitochondria-targeting insults, effectively accelerating their degeneration. Overall, defects in Parkin–mediated mitophagy, resulting mitochondrial stress, and the accumulation of damaged mitochondria may contribute to PD. The involvement of Parkin in PD-associated inflammation has been confirmed in mouse models.11,25–27 Moreover, it was recently shown that mitochondrial damage linked to mutations in quality control proteins such as the PD-associated PTEN–induced putative kinase 1 (PINK1)/Parkin activated the DNA–sensing cGAS and its signaling effector– STING, thus leading to an inflammatory response.28,29 As an essential component of the innate immunity, the cGAS–STING pathway is pivotal in activating the inflammatory response.30 Beyond its involvement in host defence against infectious microbes (as cGAS–STING has emerged as a key player against pathogens by detecting cytosolic foreign DNA and prompting a robust innate immune response),31 cGAS is also the main sensor that detects self-DNA, including genomic and mitochondrial DNA (mtDNA), released from either damaged, dead, or transformed cells.32 Upon mitochondrial damage, mtDNA is released into the cytoplasmic matrix as a danger signal, triggering cGAS, which generates the 2′3′- cyclic GMP-AMP (2’3’-cGAMP or cGAMP) that then activates a STING–mediated innate immune response, culminating in the production of type I interferons (IFN-I) and several other inflammatory mediators such as pro-inflammatory interleukin-6 (IL-6) and TNF-α.33–35 After secretion, IFN-I acts through the type I interferon receptor/or interferon-α/β receptor (IFNAR) on target cells, stimulating the transcription of hundreds of interferon-stimulated genes (ISGs), including pro-inflammatory cytokine, that together assist in counteracting threats.36 Although innate immune activation and inflammatory response within the central nervous system (CNS) is a neuroprotective and desirable mechanism against a variety of pathogens, injuries and non-infectious stressors, prolonged and uncontrolled inflammation can result in tissue damage and disease.37,38 Growing evidence has indicated that excessive engagement of the cGAS–STING signaling pathway plays a role in various pathological processes.31 Inappropriate activation of the cGAS–STING triggered by the nuclear DNA, mtDNA, mitochondrial damage and failures in Parkin–mediated mitophagy has been linked to neurodegenerative diseases.31,39,40 Aberrant cGAS–STING response contributes to neuroinflammation and promotes degeneration of neuronal cells.41 In chronic neurodegenerative diseases states, exacerbated cGAS–STING–mediated IFN-I response shapes the glia phenotype and accelerates disease progression.42 Aberrant overstimulation of this highly versatile innate immune sensing system has recently been linked to the aetiology of PD.43–45 Inflammation may enhance progression of PD pathology, but the relationships between α-syn accumulation, Parkin deregulation, and detrimental inflammatory responses are not fully elucidated. Our main hypothesis discussed here is that α-syn–induced Parkin dysfunction disturbs the mitochondrial network homeostasis, leading to mitochondrial stress, cGAS–STING pathway activation and neuroinflammation (“mito-inflammation”), thereby promoting the progressive neurodegeneration in PD. We suggest that the cGAS–STING signalling pathway is a key trigger in α-syn–induced “mito-inflammation”, which could be one of the pivotal mechanisms for the initiation and spread of PD pathology.
Parkin as a Regulator of Mitochondrial Function
Mitochondria are pivotal organelles for many cellular functions and are the primary energy-generating system in most eukaryotic cells. The architecture of mitochondria is essential for their proper function and also for the confinement of mitochondria-derived immunogenic molecules.46 In addition to their role in cellular energy production in the form of adenosine triphosphate (ATP), mitochondria are implicated in the regulation of programmed cell death, calcium homeostasis and many other crucial physiological cellular processes.47,48
The disruption of mitochondrial homeostasis is a frequently reported key feature in ageing organisms and a major hallmark in neurodegenerative diseases, including Alzheimer’s, Huntington’s disease, and amyotrophic lateral sclerosis (ALS).49 These organelles are also important players in the pathophysiology of PD. Particularly, failure of MQC and the accumulation of damaged mitochondria in neurons along with the release of mtDAMPs are proposed to be important drivers of PD pathology.19,21,50 MQC includes a set of processes spanning from mitochondrial biogenesis to plasticity controlled by coordinated cycles of fusion and fission, autophagy, and quality control mechanism called the mitochondrial unfolded protein response (UPRmt) that are intended to ensure cellular and organismal homeostasis.51,52 A key player in orchestrating mitochondrial quality assurance across various tissues is Parkin, an E3 ubiquitin ligase, also known as Parkinson Disease Protein 2 (PARK2). Parkin is implicated in autophagic clearance of damaged mitochondria, regulation of mitochondrial fusion and fission dynamics, and in protecting mitochondrial biogenesis.
Mitophagy, the selective macroautophagy of mitochondria, can be triggered by various stimuli such as the loss of mitochondrial membrane potential,8 the presence of unfolded proteins in the mitochondrial matrix,53 or an altered protein composition of the mitochondrial outer membrane.54,55 Parkin plays a pivotal role in initiating mitophagy56–59 and this process is assisted by the PINK1 kinase, crucial for the recruitment of Parkin to the mitochondrial outer membrane of damaged mitochondria.58,60,61 Notably, mutations leading to the loss of PINK1 and Parkin function are the predominant causes of autosomal recessive early PD,62,63 characterised by a relatively benign course and positive responsiveness to dopamine replacement therapy.64
Under physiological conditions, by interacting with translocase complexes of the outer and inner mitochondrial membrane, PINK1 is constitutively translocated onto polarised mitochondria,65 where it is cleaved and then undergoes degradation in the proteasome.66 However, under pathological conditions associated with reactive oxygen species or the loss of mitochondrial membrane potential, PINK1 undergoes autophosphorylation, dimerisation, and rapid accumulation on the outer membrane of damaged or uncoupled mitochondria.59,67 Subsequently, PINK1 phosphorylates mitofusin 2 (Mfn2), enabling it to act as a mitochondrial outer membrane receptor for Parkin.68 PINK1’s multifaceted role includes the activation of Parkin not only by controlling its subcellular location, but also by phosphorylating its Ser65,69–73 thereby abolishing Parkin’s auto-inhibition.74 Furthermore, PINK1 phosphorylates ubiquitin,75–78 influencing the assembly of polyubiquitin chains and their susceptibility to hydrolysis.79 Critically, not only mitofusins but also phosphorylated polyubiquitin chains on the mitochondrial surface facilitate Parkin tethering,80–82 creating in this way a positive regulatory loop78 that amplifies the pro-mitophagic signaling initiated by PINK1.83
Activated Parkin primarily functions by ubiquitin-tagging proteins of damaged mitochondria, including mitofusin 1 (Mfn1), Mfn2, dynamin-related protein 1 (Drp1), mitochondrial fission protein 1 (Fis1) and voltage-dependent anion channel 1 (VDAC1).55,78,84 These proteins are directed to degradation in the ubiquitin–proteasome system, remodelling the composition of the mitochondrial outer membrane.54,55 Before degradation, these proteins may recruit two receptors possessing a ubiquitin-binding domain, nuclear dot protein 52 (NDP52) and optineurin (OPTN), that canonically initiate selective mitophagy.83 Notably, the affinity of OPTN towards ubiquitin chains increases upon its phosphorylation by TANK-binding kinase 1 (TBK1), which in turn has been shown to be activated by mitochondrial damage in a PINK1- and Parkin-dependent manner.83 Both NDP52 and OPTN initiate phagophore formation by recruiting a protein complex containing Unc-51-like kinase 1 (ULK1), the principal driver of autophagy. ULK1 is activated by AMP-activated protein kinase (AMPK) upon the drop of cellular ATP and inhibited by mammalian target of rapamycin (mTOR) complex 1 (mTORC1).85 AMPK-activated ULK1 rapidly phosphorylates conserved Ser108 of Parkin,86 which in turn can interact with and ubiquitinate mTOR kinase, the main component of mTORC1.87 Moreover, Parkin is able to interact with autophagy and beclin 1 regulator 1 (AMBRA1), another driver of autophagy,88 and is also involved in a non-canonical mechanism of mitochondrial quality assurance, in which vesicles budding off the oxidatively stressed mitochondria are ultimately directed to the lysosome.89,90 Deubiquitinases, which counteract the ubiquitin ligase activity of Parkin, remove Parkin-attached ubiquitin from damaged mitochondria, preventing induction of excessive mitophagy.91,92 Also, Parkin–mediated ubiquitination of Mfn2 eventually suppresses mitophagy,68 likely implementing a self-restraining mechanism, which prevents exaggerated mitochondrial elimination. Mfn1 also undergoes ubiquitination–mediated degradation.54,93–95
Moreover, the PINK1/Parkin pathway influences mitochondrial function through the control of their dynamics. Proper mitochondrial functioning relies on a balance between fusion and fission processes, and disruption of which impairs the entire mitochondrial network. Respiratory active cells exhibit mitochondrial fusion, while resting cells often have fragmented mitochondria.96 Oxidative stress alters mitochondrial dynamics, with moderate stress promoting fusion,97 whereas severe stress induces excessive fragmentation98 leading to decreased membrane potential, decreased ATP levels, increased mitochondrial free radicals, and cell death. In rat hippocampal and dopaminergic neurons, PINK1 or Parkin overexpression tilts the fusion–fission balance toward fission, whereas inactivation of PINK1 shifts it in the reverse direction.99 In mammals, mitochondrial fusion involves proteins anchored in the outer mitochondrial membrane, specifically, Mfn1 and Mfn2 from the mitofusin family, and the dynamin-like GTPase localised to mitochondrial inner membrane, optical atrophy 1 (Opa-1) protein. Under severe stress, Parkin is involved in ubiquitinating mitofusin family proteins, inhibiting the fusion of damaged mitochondria.93,95,100–102 This ubiquitination leads to mitochondrial network fragmentation, facilitating the removal of malfunctioning mitochondria. Drp1 is a cytoplasmic protein crucially involved in the mitochondrial fission process. Some adapter proteins, including mitochondrial fission factor (Mff), facilitate Drp1’s interaction with Fis1 on the outer mitochondrial membrane. Parkin interferes with this interaction by ubiquitinating Drp1, eventually preventing the fission of functional mitochondria.103
The generation of new mitochondria is as crucial as the removal of damaged ones. Mitochondrial biogenesis is tightly regulated by several members of the peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1) family of transcription factors, including PGC-1α, PGC-1β, and PGC-1-related coactivator (PRC).104 Parkin plays a pivotal role in regulating mitochondrial biogenesis by ubiquitinating and targeting to degradation the transcription factor Parkin-interacting substrate (PARIS) that inhibits the expression of PGC-1α.105 The control of PARIS and its clearance by Parkin is orchestrated by PINK1, which phosphorylates PARIS on two residues, Ser322 and Ser613.106 The knockout of Parkin in the adult mouse ventral midbrain leads to defective mitochondrial biogenesis.107 In line with this regulatory mechanism, Parkin silencing significantly reduces the level of PGC-1α in PC12 cells.7 The abrogation of Parkin function in a mouse model of age-related sporadic PD coincides with increased PARIS levels and reduced PGC-1α signalling.108 Recently, PARIS has been implicated in the suppression of nuclear factor-erythroid 2 related factor 2 (NRF2)–driven transcription,109 suggesting that Parkin may also be essential for appropriate cellular responses to oxidative stress.
Relationship Between Mitochondrial Dysfunction and Neuroinflammation – the “Mito-Inflammation” Concept
Both mitochondrial dysfunction and chronic neuroinflammation are strictly involved in neurodegeneration and pathogenesis of neurodegenerative diseases, although the underlying mechanisms engaged in neuronal death have not been fully explained.39,110,111 New data indicate a relationship between mitochondrial pathology and neuroinflammation. In particular, defective MQC together with mtDAMPs generation are proposed to be major contributors to the pathogenic mechanisms of neuronal degeneration.112 The plethora of evidence strongly suggests that mitochondrial integrity and innate immunity are closely interlinked.113–115 Mitochondrial dysfunction may precede neuroinflammation and act as an inflammation-promoting agent during neurodegenerative disease progression.116 In turn, chronic inflammation may promote and exacerbate mitochondrial damage, thus generating a vicious cycle of neurotoxic events. This vicious cycle leads to the release of mtDAMPs and activates specific inflammatory cascades.111 Therefore, an in-depth understanding of the potential molecular mechanisms of the interaction between mitochondrial dysfunction and neuroinflammation as well as signalling pathways leading to mitochondria–induced inflammation (“mito-inflammation”) in neurodegenerative disorders may prove helpful in identifying new therapeutic targets and approaches for the treatment of neurodegenerative diseases, especially PD.
mtDAMPs - Key Mediators Linking Mitochondrial Dysfunction to Neuroinflammation
Upon deleterious stimulus, compromising the integrity of the mitochondrial membrane results in the release of some molecules that normally reside inside the mitochondria.39,110 These released molecules, often referred to as mtDAMPs (also known as mitochondrial alarmins),117,118 act as a stress signal for the body, activating innate immune system receptors and downstream signalling engaged in “sterile inflammation” and associated pathology, according to a theory developed by Matzinger in 2002.119,120 The endosymbiotic origin of mitochondria121 causes the immune system to erroneously recognise mtDAMPs as bacteria and triggers an innate immune response.122 Therefore, in light of the endosymbiotic theory, mitochondria are the main regulators of innate immunity.123
The innate immune system, as a first line of defence against various pathogens, injury, and stress, deploys germline-encoded receptors named pattern-recognition receptors (PRRs) to detect pathogens and danger signals through the recognition of conserved molecular motifs, called pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs).117 PRRs that recognise and detect both pathogenic PAMPs and mitochondrial DAMPs are NLRs, cGAS, and toll-like receptors (TLRs) at the plasma membrane or endosomes.34 Most of the PRRs are expressed not only in specialised innate immune cells, such as macrophages, microglia, dendritic cells or neutrophils, but also in non-immune cells, including neurons.34 Although an inflammatory response within CNS, activated in response to infection, toxic accumulation and other pathological injuries and non-infectious stressors, is a protective mechanism, excessive, prolonged and uncontrolled overactivation of glial cells can result in tissue damage and disease.37,38,124 Under pathological conditions, when mitochondrial malformations are uncontrolled and/or damaged mitochondria cannot be properly removed by mitophagy, mtDAMPs are released into the cytoplasm or out of the cell leading to abnormal activation of the PRRs and the innate immune system. This, in turn, enhances neuroinflammatory processes.112,125,126 Long-term overactivated glial cells cooperate, overproduce and release various harmful pro-inflammatory, stress-inducing factors, which aggravate mitochondrial damage, cause a gradual loss of their function and form a vicious cycle of mitochondrial dysfunction and neuroinflammation. The interactions between mitochondrial dysfunction and neuroinflammation ultimately result in neurodegenerative diseases. Among the mitochondrial DAMPs, considered as danger signals, the pro-inflammatory molecules that induce and exacerbate the inflammatory response within CNS include inter alia: mtDNA, mitochondrial-derived reactive oxygen species (mtROS), ATP, mitochondrial transcription factor A (TFAM), cardiolipin, and cytochrome C.39,127–133
mtDNA
In response to certain cellular stress or environmental insults, mtDNA, a key signalling molecule that regulates energy production and cell metabolism, can be released from damaged organelles into the cytoplasm or even outside the cell, where it engages various PRRs to trigger an immune response and activate pro-inflammatory pathways.39,134 There are several potential routes by which mtDNA is released into the cytosol to mediate pro-inflammatory responses.42 However, the exact mechanisms of mtDNA entry into the cytosol are not well elucidated. Likewise, little is known about the mechanism responsible for the loss of mitochondrial inner membrane integrity.
The protein that organises mtDNA into nucleoids (discrete protein–DNA complexes protecting mtDNA against oxidative damage) and regulates its segregation and number is the mitochondrial DNA binding protein, TFAM.135 A haplodeficiency of TFAM via disorganisation of the mitochondrial genome and mitochondrial stress leads to the release of mtDNA into the cytosol, which elicits the expression of several ISGs in a cGAS–STING pathway activation-dependent manner.136,137 Under physiological conditions, an mtDNA-binding TFAM is localised in the mitochondrial inner membrane. However, when mitochondria are damaged due to conditions of stress, the combination of caspase and B-cell lymphoma-2 (Bcl-2) inhibitors induces the translocation of this protein to the cytosol, where it assists cGAS in sensing cytosolic DNA.42,136 Studies show that loss of TFAM causes a three-to-four-fold rise in mtDNA in the cytosol associated with activation and an increase in the expression of more than 39 ISGs.42 Additionally, knocking down cGAS, STING, TBK1 or interferon regulatory factor 3 (IRF3) in the TFAM± cells leads to the suppression of the ISGs, overall confirming that TFAM depletion–induced mtDNA stress involves the cGAS–STING pathway.42,135
The proposed mechanism of mtDNA leakage involves the participation of apoptosis regulator, Bcl-2-associated X (BAX) and Bcl-2 antagonist/killer 1 (BAK1) protein.136,138 Under conditions of extreme stress, activation of BAX/BAK induces the formation of extremely large macropores in the outer mitochondrial membrane. These progressively widen, allowing the extrusion/herniation of the inner mitochondrial membrane into the cytosol, driven by the increase in osmotic pressure in the mitochondrial matrix. Formation of these macropores in the outer membrane leads to permeabilisation of the inner mitochondrial membrane and a leak of mitochondrial macromolecules into the cytosol, including mtDNA.139 Another theory assumes the escape of mtDNA to the cytoplasm via the VDAC, where VDAC oligomers can form large pores on the outer mitochondrial membrane under mild-stress conditions.140 Other possible routes include the transit of mtDNA through the mitochondrial permeability transition pore (mPTP)141 or the formation of mitochondria-derived vesicles (MDVs), which transport mtDNA to the endosomal-lysosomal pathway or to the plasma membrane, as alternative routes for inner mitochondrial membrane traversal.34 The opening of mPTP allows the release of comparatively small mtDNA fragments. Thus, it has been proposed that mtDNA released through this pore is limited to fragments of sizes smaller than 700 bp.142,143
mtDNA derived from damaged mitochondria acts as a mtDAMP to regulate the inflammatory response. mtDNA potently triggers the innate immune response and its high immunogenic potential is due to the presence of unmethylated (or hypomethylated) CpG motifs within its structure. The motifs are similar to those of bacterial DNA and are recognised by PRRs.34,144–146
mtDNA modulates the inflammatory response by triggering Toll-like receptor 9 (TLR9),39 cytosolic inflammasomes,114,147,148 and stimulation of ISGs transcription.117 mtDNA, containing an unmethylated CpG-DNA sequence, is a ligand for endosomal TLR9 and can mediate pro-inflammatory response via the NF-κB pathway. A downstream signalling cascade includes myeloid differentiation primary response 88 (MyD88), which activates the p38 mitogen-activated protein kinase (MAPK) and finally triggers the nuclear transcription factor NF-κB signalling to promote pro-inflammatory immune response.149–151
Released from damaged mitochondria into the cytoplasm, mtDNA can also activate the NLRP3 inflammasome, promoting the cleavage of pro-interleukins into mature interleukin-1β (IL-1β), interleukin-18 (IL-18) by activating caspase-1 subunit of the NLRP3 complex, thereby inducing inflammation.147,152 NLRP3 is the best-characterised inflammasome and one of the most involved in mtDNA sensing.117 In particular, oxidised mtDNA preferentially activates NLRP3.147 Interestingly, Nakahira and Zhou discovered that activated NLRP3 can also increase cytosolic mtDNA release, and this was mediated through altered Parkin function and impaired mitophagy,153 which means that NLRP3 stimulation could play a role as a positive feedback loop, and further exacerbate the pro-inflammatory response.114,154 It is suggested that NLRP3 is one of the key factors responsible for the initiation and progression of chronic inflammation.
Another well-described cytoplasmic mtDNA sensor, and innate immune receptor, is the AIM2 inflammasome, also known as absent in melanoma-2. It belongs to the IFI20X-IFI16 (PYHIN) protein family and binds to DNA via hematopoietic interferon-inducible nuclear proteins. AIM2 responds preferentially to double-stranded DNA (dsDNA) and non-oxidised DNA released from damaged host cells, causing the gasdermin D–mediated secretion of the bioactive effectors IL-1β and IL-18, as well as triggering pyroptotic cell death, thereby driving the progression of “sterile inflammatory” diseases.155,156
mtDNA released into the cytoplasm by defective and damaged mitochondria also causes the activation of the DNA–induced cGAS–STING pathway and the promotion of neuroinflammation.135 mtDNA damage and high levels of mtDNA fragments circulating in plasma positively correlate with chronic inflammation and oxidative stress, suggesting an important role of free mtDNA as a mtDAMP and stimulators of the cGAS–STING–TBK1 signalling cascade.157 Thus, circulating mtDNA is increasingly recognised as a key mediator linking mitochondrial dysfunction to inflammation.39
Other mtDAMPs
Besides mtDNA, mitochondria also contain other pro-inflammatory molecules classified as mtDAMPs, which are released from damaged mitochondria and amplify the pro-inflammatory response. Among them, we distinguish mitochondrial-derived mtROS, an upstream mediator required for NLRP3 inflammasome activation.130 Mainly produced by the mitochondrial electron transport chain (ETC) and mitochondrial NADPH oxidase (NOX), mtROS are the major source of oxygen radicals, including superoxide (O2•−), nitric oxide (NO), hydrogen peroxide (H2O2), and singlet oxygen.116 The imbalance between mtROS generation and their removal, due to overproduction of ROS and/or decreased activity of the antioxidant defence system, results in oxidative stress, which leads to oxidative damage and mitochondrial destruction.158 Mitochondrial dysfunction and exacerbation of mtROS levels have been proposed to contribute to “mito-inflammation”. Excessive mtROS levels sustain and intensify inflammation causing tissue damage and becoming a chronic phenomenon in pathological conditions.116 mtROS may trigger the stimulation of pro-inflammatory signalling through activation of the redox-sensitive transcription factors NF-κB,159,160 hypoxia-inducible factor 1 (HIF-1)161 and activator protein 1 (AP-1).162 As well as inducing the expression of inflammasome genes, such as Nlrp3, Nlrc4, and Il-1b genes. mtROS accumulation acts destructively on the mtDNA particularly susceptible to the effect of mtROS due to the lack of protective histones and effective complex mechanism to repair DNA.39 Synergistic stimulation between the inflammasome and redox-vulnerable inflammation reinforces the inflammatory response.163 NLRP3 facilitates mPTP opening, thereby contributing to the release of mtDNA.114 In this manner, NLRP3 activation could act as an upstream checkpoint of the innate immune system during the deployment of “sterile inflammation”, involving the DNA-sensing cGAS–STING pathway. Hence, a self-sustaining circle of events involving loss of mitochondrial function, ROS bursts, and mtDNA damage caused by NLRP3 activators has been proposed.147 Oxidised mtDNA may serve as the ultimate NLRP3 ligand,147 thus confirming the crucial role played by ROS in inflammasome activation.154
Similarly, extracellular ATP, released by the Pannexin 1 (Panx1) channel from injured or dying mitochondria under stress conditions,164 serves as a mtDAMP and may activate the caspase-1 signalling cascade mediated by NLRP3 through the purinergic P2X7 receptor of glial cells, leading to neuroinflammation.118,165 P2X7 receptors are preferentially located on microglia, and their activation is associated with inflammation.166 The binding of extracellular ATP to the P2X7 receptor leads to the opening of this cation channel that allows K+ efflux from and Ca2+ and Na+ influx into the cells. Calcium influx into the cell triggers several intracellular signalling cascades, resulting in the activatory cleavage of caspase-1, which in turn catalyses the formation of mature IL-1β and IL-18 from their biologically inert propeptides.117 Moreover, ATP–induced activation of the P2X7 receptor leads to the stimulation of the nuclear factor of activated T-cells (NFAT) and MAPK cascades, also known as protein kinase C/mitogen-activated protein kinase (PKC/MAPK) pathways, leading to the production and release of C-X-C Motif Chemokine Ligand 2 (CXCL2) in microglia and exacerbating inflammation.167
It has been reported that cardiolipin externalisation and relocation to the surface of dysregulated mitochondria also mediates the activation of NLRP3 to initiate a caspase-1-dependent neuroinflammatory response and release bioactive IL-18 and IL-1β.130 Cardiolipin, tetra-acylated diphosphatidylglycerol lipid, primarily exists in the mitochondria as a component of the mitochondrial inner membrane and is indispensable in maintaining the electron transport chain.118,168 However, under stress conditions, mitochondrial damage and depolarisation result in the translocation of cardiolipin to the outer mitochondrial membrane. Attachment of cardiolipin to NLRP3 activates the inflammasome, resulting in a neuroinflammatory response.130 Depending on the amount, and level of cardiolipin saturation and oxidation it can exert both pro-mitochondrial or pro-apoptotic signals affecting mitochondrial function and inflammatory response.169 Exposed cardiolipin mediates the degradation of damaged mitochondria and their phagocytosis from the extracellular milieu, thus serving as an “eat-me” signal for eliminating defective mitochondria.170 Cardiolipin in the inner mitochondrial membrane acts as an anchor for cytochrome C. Oxidation of cardiolipin and its exposure on the outer mitochondrial membrane induce cytochrome C release and apoptosis.171
Cytochrome C is a 15-kDa water-soluble mitochondrial haemoprotein that can act also as mtDAMP when released into the extracellular space from damaged cells to activate neuroinflammation.172 Released from damaged mitochondria to the outside cytochrome C activates the mitogen-activated protein kinase/c-Jun N-terminal kinase (MAPK/JNK) signalling cascade by interacting with glial TLR4 receptors to regulate the function of immune cells in the brain.132,133 Damaged glial cells are a potential source of extracellular cytochrome C in the CNS.39 Cytochrome C causes immune activation of both microglia and astrocytes in a TLR4-dependent manner.132,133 In addition, extracellular cytochrome c may display direct pro-inflammatory properties mediated by the activation of NF-κB and causing neutrophil and monocyte-triggered inflammation.173
TFAM is a member of the high mobility group box (HMGB) family of proteins and acts as a guardian of the mitochondrial genome, playing important roles in mtDNA replication, maintenance and transcription.118 The correct expression of TFAM is crucial for the functioning of mitochondria and, consequently, cellular homeostasis. The presence of extracellular TFAM is described as inducing an inflammatory response, similar to the action of another important member of mtDAMPs, including HMGB1.165 When released externally from neurons or glia, TFAM can activate the receptor for advanced glycation endproducts (RAGE) or TLR4 on the plasma membrane surface of the glia, thus initiating the NF-κB–mediated nuclear signalling cascade to activate a pro-inflammatory response.39,129 TFAM also exacerbates the immunogenicity of mtDNA.46 TFAM, bound to mtDNA, can interact with the plasma membrane receptor RAGE and induce the internalization of mtDNA, thereby promoting its recognition by TLR9 and amplifying TNF-α release from TLR9-expressing plasmacytoid dendritic cells (pDCs).174,175
Despite the existence of many safeguards that prevent mitochondria from triggering harmful DAMPs-associated inflammatory reactions, when the homeostatic capacity of such systems is exceeded or when these systems become damaged, inflammatory reactions induced by mitochondria may become pathogenic and contribute to the aetiology of neurodegenerative diseases, including PD.156
mtDAMPs-Dependent Activation of cGAS–STING Signalling Pathway
cGAS, a major sensor of cytoplasmic viral, retroviral, parasitic and bacterial DNA, as well as any dsDNA, including self-DNA (both genomic and mitochondrial DNA), is an innate immune system receptor, that undergoes a conformational change to an active state and generates the production of the second messenger molecule, cGAMP using ATP and guanosine triphosphate (GTP) as substrates.176–178 cGAS is a cytosolic nucleotidyltransferase, which is activated by DNA-inducing conformational changes around the catalytic active site of cGAS (encompassing its dimerisation, oligomerisation, and ultimately phase separation).179 The immune-stimulatory DNA-binding capacity of cGAS is contained in the less conserved, disordered, and containing a high density of positively charged residues in the N-terminal structural domain (1–160) of cGAS, while the highly conserved C-terminal catalytic domain (161–522) (composed of three DNA-binding sites, named “site A”, “site B”, and “site C”) is critical for the enzyme activity of cGAS and the synthesis of cGAMP.180,181 Recent studies have provided evidence that the Ser (13, 37, 64, 129 and 143) residues in the cGAS N-terminus are crucial for sensing genomic/chromatin DNA.182 In turn, three structures/sites in the catalytic part of cGAS suggest a sequential reaction mechanism in the cGAS-catalysed production of 2′3′- cGAMP.183 Site A is the primary site that mediates DNA binding–induced conformational changes of the activation loop in cGAS (rearranging the catalytic pocket of the enzyme to allow for optimal interaction with the substrates). DNA binding to the juxtaposed secondary B-site leads to the formation of the core 2:2 cGAS-DNA complex - the minimal active enzymatic unit. Site C provides an additional interaction between cGAS and DNA, contributing to the phase separation of the cGAS-DNA complex.35,183 Interestingly, structural studies uncovered mechanistic details of DNA binding to cGAS, providing explanations for the preferential response of human cGAS to DNA fragments longer than 45 bp and the enhanced activation of cGAS by TFAM-associated mtDNA.135,184 To ensure the activation of cGAS and its sufficient catalytic activity to lead to a robust induction of IFN-I transcription in vitro, an amount of a>50 mer of DNA is necessary.185,186 Thus, mechanistically, any dsDNA is bound by cGAS in a sequence-independent but length-dependent manner.187 The interactions are mostly mediated by positively charged residues in cGAS with a sugar-phosphate backbone of DNA, explaining the lack of sequence specificity of the interaction.183 Moreover, additional evidence for a DNA-length-dependent mechanism of cGAS activation was provided by the recent discovery of DNA–induced liquid-liquid phase separation (LLPS) of cGAS.188 The DNA–induced phase transition of cGAS, critically dependent on the concentration of both cGAS and DNA in the cell, promotes cGAMP production and innate immune signalling while providing a mechanism to avoid autoimmune reactions to low concentrations of self-DNA.183
cGAMP formed by cGAS subsequently binds to the adaptor protein STING. STING, also known as transmembrane protein 173 or MPYS, MITA, and ERIS, is a small (~40kDa) transmembrane protein located in the endoplasmic reticulum (ER) membrane.180 The binding of cGAMP to STING causes its profound and extensive conformational rearrangement that trigger a half-turn rotation, oligomerisation of STING dimers, and its liberation from anchoring factors (such as stromal interaction molecule 1 (STIM1)). The resulting active STING unit is capable of initiating effector functions. In the following steps, a prerequisite for the initiation of downstream signalling is the translocation of oligomerised STING from the ER through the ER–Golgi intermediate compartment (ERGIC) to the Golgi apparatus.35,42,189–191 So far, very little is known about the precise molecular events and the relationship between putative players involved in this trafficking process. It is only known that the canonical ER-to-Golgi transport machinery, composed of coatomer protein complex II (COPII) vesicles, relying on the GTPase SAR1A (secretion associated, Ras related GTPase 1A) and the COPII complex components, including SEC24-related protein C (SEC24C) as well as the ARF-GTPase ARF1, facilitates the STING translocation to the Golgi.192 Upon reaching the ERGIC and Golgi compartments, the C-terminal tail of STING recruits the downstream TBK1 through a conserved PLPLRT/SD amino acid binding motif, which promotes dimerisation–mediated TBK1 autophosphorylation of (Thr172) in the activation loop and thus TBK1 activation.193,194 In turn, activated TBK1 phosphorylates STING (at Ser366 in humans and Ser365 in mouse) in the C-terminal tail, providing a binding site for IRF3, thereby recruiting IRF3 for phosphorylation by nearby TBK1.34,195 A master regulator of IFN-I, IRF3, phosphorylated by TBK1 forms a dimer and translocates to the nucleus, where it turns on the expression of IFN-I.35,196
The STING oligomer translocated to the Golgi apparatus can also recruit and activate IκB kinase (IKK). This event results in the phosphorylation of IκBα (inhibitor of the NF-κB). Subsequent ubiquitination directs IκBα for its proteasomal degradation, resulting in the release of NF-κB heterodimers (comprising the p65 and p50 subunits) from inhibitory binding, and their subsequent translocation to the nucleus to up-regulate the production of pro-inflammatory cytokines and chemokines.34,197–199 Thus, induction of both downstream signalling pathways such as NF-κB signalling together with IRF3 activation, as a result of cGAS–STING stimulation, turns on robust transcription of IFN-I and a number of pro-inflammatory factors to enhance the innate immune response.200
After secretion, the IFN-I (interferon-alpha (IFN-α) and interferon-beta (IFN-β)) act on target cells through IFNAR, comprising interferon-α/β receptor alpha chain (IFNAR1) and interferon-α/β receptor beta chain (IFNAR2) subunits, associated with the Tyrosine kinase 2 (TYK2) and Janus kinase 1 (JAK1), respectively, and located on the target cells. Stimulation of TYK2 and JAK1 promotes activation of signal transducer and activator of transcription 1 and 2 (STAT1 and STAT2), which bind to the interferon regulatory factor 9 (IRF9), forming a heterotrimeric transcription factor complex termed IFN-stimulated gene factor-3 (ISGF3).42 By binding genes harbouring the interferon-sensitive response element (ISRE), ISGF3 complex leads to the stimulation of the transcription of hundreds of ISGs, including pro-inflammatory IL-1β and IL-6.36,201,202
Interferons are a group of cytokines, which are classified as type I (especially IFN-α and IFN-β), type II (INF-γ) and type III (INF-λ1–λ4 in humans).203 The brain is more vulnerable to the effects of IFNs than the peripheral tissues. IFN-I can directly change both the structural and functional integrity of neurons.42 Not only neurons but also microglia and astrocytes do express IFNAR and respond to IFN-I.204,205 The predominant type of cell which activates the cGAS–STING signalling pathway are the immunocompetent cells of the CNS, microglia.204,205 These tissue-resident macrophages, activated by danger signals, secrete IFN-I, which act on the IFNAR receptor on neurons, thereby eliciting defence mechanisms.42 In parallel, the astrocytes are also stimulated to alleviate inflammation.42 Activated microglia release a wide spectrum of pro-inflammatory cytokines, which act on astrocytes leading to their activation and the release of pro-inflammatory molecules. The activated glial cells cooperate and produce pro-inflammatory molecules (cytokines and chemokines), as well as ROS and reactive nitrogen species (RNS) (IL-6, IL-1β, TNF-α, iNOS, ROS, NO, ONOO-) to trigger an immune response.38 However, prolonged overactivation of glial cells together with the excessive engagement of this signalling system can cause chronic neuroinflammation contributing to neurodegeneration.37,39 Long-term overstimulation of the glia leads to the overproduction and release of various harmful stress-inducing factors that affect neurons and cause a gradual loss of their function.124,206 Moreover, the pro-inflammatory factors released by activated glia may aggravate mitochondrial damage, forming a vicious circle of toxic events. In chronic neurodegenerative states, aberrant cGAS–STING–induced generation of IFN-I shapes the microglia phenotype and accelerates disease progression.207
In addition to facilitating the inflammatory response, increasing evidence suggest that the cGAS–STING signalling pathway can induce and regulate various cell death pathways. In response to various stresses in dynamic microenvironments, it can stimulate the process of apoptosis,208 autophagy/mitophagy, necroptosis, pyroptosis, and ferroptosis.209–212
From Mitochondrial Dysfunction Through cGAS–STING to “Mito-Inflammation” in PD
Among the pathogenic mechanisms of neurodegeneration in PD, the co-occurrence of mitochondrial dysfunction and neuroinflammation may be crucial to induce neuronal degeneration.52,213,214 In particular, defective mitochondrial quality control, mtDAMPs generation and activation of innate immune pathways, including cGAS–STING are proposed to be major contributing factors.215
The majority of PD-associated genes play a role in mitochondrial homeostasis.216,217 Mutations in the genes encoding PINK1 and/or PARKIN are linked to familial, autosomal recessive inherited forms of PD, providing evidence that the accumulation of damaged mitochondria may contribute to PD.34,62,63,218,219 Both the E3 ubiquitin ligase Parkin and PINK1 work together in the same cellular process, called mitophagy, thus playing pivotal roles in the clearance of damaged mitochondria.42,220 Disturbances of the mitophagy machinery may result in the accrual of damaged mitochondrial components, including mtDAMPs, which can be released into the cytosol or in the extracellular space and activate the innate immune receptors, which leads to stimulation of NF-κB, cGAS–STING pathway, NLRP3 inflammasome, and/or MAPK/JNK signalling cascade to promote inflammation, an important driver of PD pathology.19,21,50,114,183,221–223 The presence of mtDAMPs in circulating extracellular vesicles (EVs) has been described in older adults with PD and is characterised by a specific inflammatory signature.224 Interestingly, mitophagy was identified as a protective mechanism to limit the release of both mtROS and mtDNA from defective mitochondria.34,114,154 Mitophagy prevents the initiation of extensive inflammatory responses and favours the inhibition of inflammation driven by a small number of permeabilised, dysfunctional mitochondria (limited signalling via cGAS–STING and the NLRP3 promotes mitophagy, associated with the recruitment of Parkin to dysfunctional mitochondria, and TBK1-dependent phosphorylation of OPTN and consequent engulfment of mitochondria by formed autophagosomes).156
However, unrecoverable and uncontrolled massive dysfunction of the mitochondrion is associated with robust cGAS signalling and strong NLRP3 stimulation. This is accompanied by inhibition of mitophagy – as a consequence of caspase 1-dependent cleavage of Parkin elicited by robust NLRP3 activity, despite OPTN phosphorylation, which maximises fully-developed inflammatory responses.153 Thus, aberrant or inactivated mitophagy can lead to the accumulation of potent triggers of the innate immunity system response.42,135 Trinh et al have established that mutations in PINK1/Parkin increased the heteroplasmic mtDNA variants burden in blood, which was correlated with serum pro-inflammatory IL-6 levels.225
By linking impaired mitophagy and insufficient clearance of damaged mitochondria and inflammation, the release of mtDNA into the cytosol may trigger cGAS–STING–mediated activation of innate immunity.226 The cGAS–STING–driven inflammation has been implicated in neurodegeneration following mitophagy impairment. Increasing evidence suggests that mtDNA is a vector of neuroinflammation via the cGAS–STING axis in Parkin loss–mediated pathology.227 Midbrain neurons from patients with PD with biallelic Parkin mutations exhibited impaired mtDNA dynamics and increased cytosolic mtDNA levels. Loss of Parkin in the presence of mtDNA stress mirrored these phenotypes together with up-regulation of the cytosolic DNA sensor, cGAS protein.228 Furthermore, a recent study has also found that the depletion of PINK1, PARK9 and/or glucocerebrosidase beta 1 (GBA1) increased cytosolic dsDNA of mitochondrial origin and induced IFN-I responses and cell death in cellular models. In addition, interferon-inducible protein 16 (IFI16) (an innate immune sensor of intracellular DNA) and cytosolic dsDNA puncta of mitochondrial origin accumulate in the brains of patients with PD.229 Notably, IFI16 has the ability to the recruitment of STING and the production of IFN-I, or else forms the inflammasome with apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain (ASC) to induce the production of IL-1β and IL-18. Additionally, IFI16 activation stimulates the production of other cytokines and chemokines, such as IL-6, C-X-C Motif Chemokine Ligand 10 (CXCL10) and C-C Motif Chemokine Ligand 20 (CCL20).230 In a study conducted by Jimenez-Loygorri, pharmacological induction of PINK1/Parkin–mediated mitophagy curtailed cytosolic mtDNA-dependent activation of cGAS–STING inflammation and ameliorated deterioration of neurological function in old mito-QC reporter mice.29 Additionally, Parkin-deficient Drosophila melanogaster mutant’s exhibit disturbed mitochondria morphology together with their dysfunctional dynamics, which can be suppressed by loss of STING. This suggests feedback of STING signalling on mitochondria integrity.28 In a model of chronic mitochondrial stress, cross-breeding Parkin−/− mice with the “Mutator” mice (mice that express a proofreading-deficient mtDNA polymerase (PolG) and accumulate mtDNA mutations) caused dopaminergic neuron degeneration and motor defects.231 Similarly, constitutively active STING was associated with DA neuron degeneration and promotion of neuroinflammation in mice,45 suggesting that the accrual of dysfunctional mitochondria may be necessary for the release of mtDNA to drive cGAS–STING-associated inflammation in PD. According to these observations, Borsche et al demonstrated elevated levels of both serum IL-6 and circulating cell-free mtDNA in patients biallelic for either Parkin or PINK1 gene mutations, implicating inflammation in Parkinson’s disease.232 This may suggest an interplay between mitochondrial stress and STING signalling in the pathology of PD.190 This is an attractive hypothesis, if true, the inhibition of the cGAS–STING pathway or finding ways to limit mitochondria pathology and release of mtDAMPs, especially mtDNA into the cytoplasm or circulation, may have therapeutic value in PD.218
Other mutations which were linked to PD occur in leucine-rich repeat kinase 2 (LRRK2, PARK8) as a cause of hereditary PD. Mutation of LRRK2 accompanied by impaired basal mitophagy achieves the accumulation of damaged mitochondria in the brain of the PD mouse model.233 In macrophages with LRRK2 knockout, aberrant increase in Drp1-associated mitochondrial fission as well as excessive oxidative stress result in chronic activation of the cGAS–STING signalling along with increased expression of ISGs.43 Overexpression of Drp1, in the preclinical models of optic atrophy 1 ablation, causes hyper-fragmentation of mitochondria and giant mtDNA nucleoids outside the mitochondria that are capable of eliciting the immune response.234 Thus, mtDNA–driven inflammation was also associated with altered regulation of mitochondrial dynamics.235,236 Circulating mtDNA is increasingly acknowledged as a pivotal mediator linking mitochondrial dysfunction to inflammation.112,144,235,237 When released from damaged mitochondria, cytosolic mtDNA engages the cGAS–STING pathway. Accordingly, stimulation of autophagy can also decrease the load of cytoplasmic DNA and may provide therapeutic benefits.238,239 Similarly, subtle regulation of mitochondrial dynamics could be a possible therapeutic strategy to relieve inflammatory stress and thus alleviate PD pathology.
α-syn – a New Player in cGAS–STING Activation
Clearance of dysfunctional mitochondria may also be affected by α-syn – a central component to the pathogenesis of PD.216 α-syn aggregates engage both innate and adaptive inflammatory responses in CNS (gliosis, increased microglia antigen presentation, T cell infiltration, increased pro-inflammatory cytokines and chemokines), similar to classic DAMPs.240–245 Many studies implicate PRR-based mechanisms, including TLRs and NLRs signalling, as mediators of neuroinflammation in α-synucleinopathies.241–243,246,247 A relatively new issue is the α-syn–induced stimulation of the cGAS–STING signalling pathway. Pathologic α-syn aggregates evoke oxidative and nitrosative stress, which may entail genomic and mitochondrial DNA damage.44 The cGAS–STING pathway axis can be aberrantly stimulated by both mtDNA, as well as by genomic DNA in the context of damage to the cell nucleus, particularly when micronucleation and DNA breaks are present.44,248,249 Increasing evidence suggests that DNA damage and repair deficiency may be the direct mechanism involved in the pathophysiology of PD.250–252 Numerous studies revealed a significant up-regulation of the phosphorylated form of H2A Histone Family Member X (H2AX), γ-H2AX, a sensitive molecular marker of DNA double-strand breaks (DSBs), in PD, which type of damage was included not only DA neurons but also microglia.253,254 The study of Milanese demonstrated accumulation of DNA damage together with activation of the DNA damage response (DDR) in two different α-synucleinopathy PD mouse models. Interestingly, the observed accumulation of DSBs has been the initiating lesion of neurotoxicity, which preceded the onset of the motor phenotype and DA neuron degeneration.253 Pathogenic α-syn can damage DNA indirectly through ROS generation (α-syn aggregates localize to mitochondria and compromise respiratory chain components, resulting in mitochondrial dysfunction together with massive production of harmful ROS255) or directly cleave DNA, leading to DNA strand breaks.256,257 In neurons, α-syn induces DSBs in genomic DNA due to increased nitrosative stress.258 Recent research provides evidence that α-syn aggregates (intrastriatal α-syn performed fibril, α-syn-PFF) induce DSBs in microglial, genomic DNA, which in turn stimulates the STING-dependent IFN-I response resulting ultimately in inflammation that precedes neurodegeneration.44 Furthermore, the same authors provided evidence that genetic knockout of STING was neuroprotective in this α-syn-PFF model of PD. Mice without functional STING showed reduced inflammation, motor deficits, α-syn accumulation and DA neuron loss.44 Interestingly, there is also biochemical evidence that STING protein expression is up-regulated in the SNpc of PD patients, and this stimulation is correlated with the deposition of pathological α-syn.44 From these data, the authors suggest that stimulation of cGAS–STING pathway may exacerbate pathologic neuroinflammation and mediate neurodegeneration arising from nigrostriatal α-synucleinopathies, including idiopathic PD.44,259 Additionally, a study by Szego et al demonstrated that the chronic activation of STING was sufficient to cause neurodegeneration. In a mouse model that expresses the constitutively active STING variant N153S, researchers observed degeneration of DA neurons, a lower density of dopaminergic axon terminals and the concentration of dopamine in the striatum, α-syn pathology and a lower density of synaptic puncta, and neuroinflammation.45 Moreover, the histological evidence shows that the cGAS–STING pathway appears to be activated in endothelial and neural cells in multiple sclerosis, Alzheimer’s disease, Parkinson’s disease, and ALS. Together with the in vitro data, this suggests that the cGAS–STING pathway might be activated via α-syn–induced generation and accumulation of genomic DNA damage, as well as by mitochondrial stress and DNA leakage, resulting in downstream neuroinflammation.260
Moreover, α-syn–evoked Parkin deficiency may also lead to increased levels of oxidative genomic and mtDNA damage.261 There is evidence that Parkin protects nuclear DNA and regulates nucleotide excision repair (NER) mechanism.262 Nuclear translocation of Parkin seems to be required to promote DNA repair.263 DNA damage is able to induce inflammatory responses in microglia, astrocytes, and neurons,264,265 and the innate immune response induced by the cGAS–STING pathway appears to play a major role.256 Thus, beside genomic and mitochondrial DNA damage generation, overexpression or pathogenic aggregation of α-syn can also cause DNA repair defects by affecting the expression of DNA repair proteins, including the above-mentioned Parkin as well MRE11 and APE1.256,266,267 APE1 (DNA (apurinic/apyrimidinic site) endonuclease 1) is a pivotal enzyme of one of the major DNA repair routes - base excision repair (BER) pathway, which mainly repairs oxidative DNA damage and the alkylation of bases and is the dominant DNA repair pathway in neurons.268 The loss of APE1 expression was observed in the α-syn-PFF mouse model compared to controls,269 which exacerbates the accumulation of oxidised DNA damage.270 It has been also demonstrated that α-syn overexpression can lead to reduced expression of double-strand break repair protein MRE11 (MRE11) in the SH-SY5Y cells, thus promoting DNA DSBs.267 Considering the role of MRE11 in recognizing DSBs, and initiating the downstream repair process via forming an MRN complex with RAD50 double strand break repair protein (RAD50) and nibrin 1 (NBS1), down-regulation of MRE11 may lead to DNA double-strand break repair (DSBR) defects.271 Thus, α-syn interfered with the DNA repair system in microglia leading to the accumulation of DNA damage, and this may also trigger pro-inflammatory and neurotoxic signals through stimulation of the cGAS–STING pathway. Given the potential deleterious effects of pro-inflammatory factors and pathogenic α-syn on astrocytic genomic DNA,265,272 DNA damage may be an important driver of astrocyte activation, contributing to neurotoxic inflammation in these glial cells.273,274 Inflammatory factors secreted by glial cells can also cause DNA damage in the neuronal genome.256,265 Recent evidence suggests that DNA damage in neurons is also an important source of brain inflammation, engaging the inflammatory signalling triggered by the cGAS–STING pathway.250 Summarizing, α-syn might be a key initiating factor of the cGAS-STING-mediated neuroinflammation and neurodegeneration (Table 1).
 |
Table 1 Direct and Indirect Evidence Suggesting the Possibility of α-Syn to Induce the cGAS–STING Pathway Activity, and Thus Neuroinflammation |
Limitations
Despite intensive research conducted in the last few years, several critical questions remain to be answered. The major outstanding questions:
- To what extent the α-syn–induced neuroinflammation is the result of stimulation of the cGAS–STING pathway and to what extent do other signalling cascades, which recognize endogenous danger signals (DAMPs) operating in the brain, including TLR (TLR9, TLR4) and NLR signalling (NLRP3)? What are the relative contributions of these pathways to the modulation of inflammatory responses as a consequence of α-syn–induced stress, as compared to the cGAS–STING pathway?
- To what extent α-syn is able to activate the cGAS–STING pathway in neuronal cells and glial cells, respectively? What are the relative proportions of neurons, astrocytes and microglia in different brain regions capable of activating the α-syn–evoked cGAS–STING pathway? It appears that genetic activation of cGAS–STING in different cell types will be required to determine the contributions of STING activation within glial cells and neurons.
- Does STING–induced neurodegeneration of dopaminergic neurons could initiate degeneration of further neuron populations? Are some brain regions and neuron populations more susceptible to STING–induced damage than others? An area which merits further investigation is the analysis of the individual immune response of different brain regions, which could provide new information about the differential susceptibility of the various brain regions to STING–evoked damages, as a result of stress induced by α-syn.
- While activation of immunostimulatory pathways is required for the normal functioning of the brain, persistent engagement of these responses can prove detrimental. What must be the magnitude and scale of α–syn–induced pathology, which is necessary for cGAS–STING pathway activation to such an extent that to tip the balance towards neuroinflammation?
- In response to various stresses in dynamic microenvironments, cGAS–STING can stimulate the process of apoptosis, autophagy/mitophagy, necroptosis, pyroptosis, and ferroptosis. Which type of cell death is triggered due to α–syn–induced cGAS–STING pathway stimulation?
- What is the exact sequence of molecular events linking α-syn oligomerization/aggregation to Parkin–mediated mitochondrial dysfunction and ultimately cGAS–STING activation? This field needs to be more rigorously characterized. Elucidation of the molecular mechanisms underlying immune activation in the CNS would yield valuable clues to develop novel therapeutics.
- Which processes in the cascade of α-syn–induced pathological events leading to inflammation are crucial in the pathogenesis of PD? At what stage of pathological molecular events would therapeutic intervention be effective? (Parkin dysfunction/mitochondria damage/DNA damage and repair abnormalities/cGAS–STING overstimulation?). Clarifying the molecular events in the α-syn–induced cascade of pathological events and the relationships between them might open new avenues for understanding the pathogenesis of PD and provide fertile ground for future drug development; identifying a potential point of therapeutic intervention would be possible.
- One of the mtDAMPS, cytosolic mtDNA, triggers inflammatory responses. Proposed are several routes by which mtDNA is released into the cytosol to mediate inflammatory responses. It is not well elucidated how mtDNA leaks exactly into the cytosol and whether mtDNA present in the cytosol could be a therapeutic target.
- How effective and afford neuroprotection will be the action on the deregulated cGAS–STING itself (inhibitors of STING, cGAS–targeted inhibitors), and to what extent would multi-point therapeutic intervention (in addition to cGAS–STING inhibition) including for example Parkin–targeted activators, mitochondrial therapies, and therapeutic strategies targeting DNA damage repair would be necessary in the case of α-syn–induced neurotoxicity?
Additionally, the studies of the interplay between cells of the blood-brain barrier and the component cells of the brain, which comprise the neurovascular unit, could prove helpful in the analysis of α-syn–induced pathology, and lead to the discovery of the potential communication channels between the brain and periphery, which may be targeted to identification of potential biomarkers of PD (blood- or CSF-based biomarkers).
Conclusions and Future Directions
Taken together, in light of the available data, it is reasonable to conclude that α-syn–induced mitochondrial pathology has been consistently associated with the release of mtDAMPs, genomic and mitochondrial DNA damage with simultaneous impairment of the DNA repair system, which may collectively cause cGAS–STING–driven neuroinflammation ultimately resulting in progressive neurodegeneration (Figures 1 and 2). In particular, emerging evidence suggests that mitochondria play an important role in the control of inflammatory responses, offering a unique platform for mtDAMPs redistribution, PRRs signalling and inflammation in the context of failing adaptation to cellular stress. Considering the important role of mitochondria in pro-inflammatory processes, we suggest that therapeutic manipulations aiming to prevent mitochondrial damage, and/or disruption of mitophagy may prevent mtDAMPs–induced cGAS–STING activation and detrimental “mito-inflammation”. Triggered by α-syn, the cGAS–STING–driven “mito-inflammation” may be a key mechanism of progressive neurodegeneration in PD, opening doors to new therapeutic approaches targeting the innate immune response in PD. We hypothesise that the cGAS–STING pathway can be activated during the progression of PD and through this activation a neurotoxic environment could be created. Therefore, cGAS or STING inhibition may serve as a viable therapeutic approach for slowing down the progression of PD. To investigate the mechanisms, genetically modified animals lacking either cGAS or STING expression crossed with α-syn overexpression lines in order to uncover the role of α-syn-dependent pathways could be a useful research tool. A better understanding of the innate immune signalling events taking place during PD onset and progression could reveal new possible therapeutic targets.
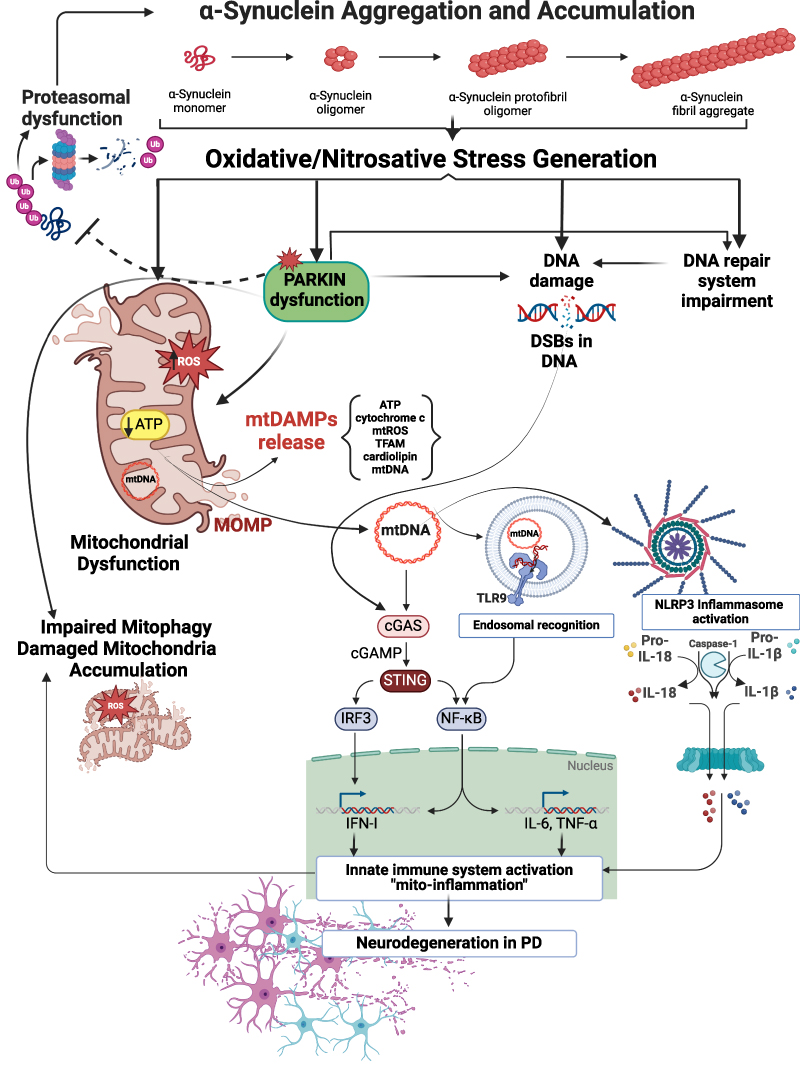 |
Figure 1 Proposed mechanisms of α-syn neurotoxicity in PD. Pathological α-syn aggregates evoke oxidative/nitrosative stress leading to mitochondrial damage, S-nitrosylation and inactivation of E3 ubiquitin ligase Parkin, generation and accumulation of DNA damage, and DNA repair system impairment. α-syn–evoked Parkin down-regulation promotes mitochondrial dysfunction and impairment mitophagy in neuronal cells, which results in the accumulation of damaged mitochondria. Moreover, α-syn–induced Parkin dysfunction disturbs proteasomal degradation of misfolded proteins leading to the accumulation of defective proteins, including α-syn. Parkin deregulation may also lead to exacerbation of oxidative genomic DNA damages induced by α-syn itself. Parkin deficiency, combined with mitochondrial damage and mtDAMPs release, including mtDNA, might modulate the inflammatory response by stimulating NLRP3, triggering endosomal TLR9 receptors, or by activation of the DNA-induced cGAS–STING pathway. NLRP3 stimulation elicits robust caspase-1 activation and consequent proteolytic maturation of IL-1β and IL-18. mtDNA can be bound by TLR9 in the endosome promoting the activation of a MyD88 pathway that ultimately leads to the expression of pro-inflammatory cytokines. In turn, the induction of cGAS–STING downstream effectors such as IRF3 and NF-κB turns on robust transcription of IFN-I and a number of pro-inflammatory factors (IL-6 and TNF-α) to enhance innate immune response. The cGAS–STING pathway might be also stimulated by α-syn–induced genomic DNA damages in the context of nuclear dysfunction, particularly when micronucleation and DNA breaks are present. Finally, evoked by α-syn the innate immune system activation and chronic inflammation (“mito-inflammation”) may promote neurodegeneration in PD and exacerbate mitochondrial damage, thus generating a vicious cycle of neurotoxic events. This Figure was partially created using BioRender dynamic assets and retrieved from https://app.biorender.com/biorender-templates. |
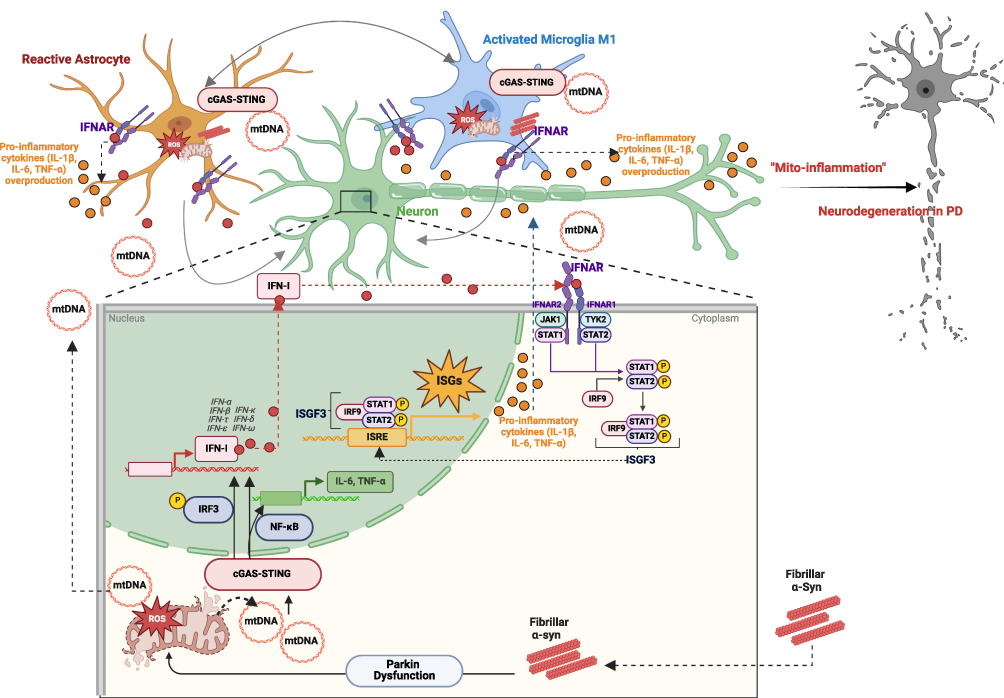 |
Figure 2 Schematic representation of the α-syn–induced events (Parkin-mediated mitochondrial dysfunction and excessive stimulation of cGAS–STING pathway together with IFN-I signalling) contributing to neurodegeneration in PD. α-syn–induced Parkin dysfunction promotes mitochondrial failure and impairment mitophagy in neuronal cells, leading to the accumulation of damaged mitochondria and the release of the various mtDAMPs. The release of mtDAMPs, including mtDNA, can induce inflammatory responses by activating the innate immune system in neurons and glial cells. A portion of the released mtDNA binds to cGAS, activating the intracellular cGAS–STING pathway. Activation of cGAS–STING leads to the induction of downstream effectors such as IRF3 and NF-κB, triggering robust transcription of genes encoding IFN-I and other pro-inflammatory factors, including IL-6 and TNF-α. IFN-I, once secreted, acts through IFNAR on target cells (neurons, microglia and astrocytes), comprising IFNAR1 and IFNAR2 subunits, associated with TYK2 and JAK1, respectively. In turn, stimulation of TYK2 and JAK1 promotes the activation of STAT1 and STAT2 proteins, which bind to the IRF9 forming an ISGF3. The ISGF3 complex migrates to the nucleus and then by binding genes harboring the ISRE stimulates the transcription of numerous ISGs, including pro-inflammatory IL-1β, IL-6, and TNF-α. Additionally, mtDNA released by neuronal cells activates the cGAS–STING pathway in neighboring glial cells, thereby enhancing IFN-I–mediated immune responses. Prolonged overactivation of glial cells and excessive engagement of the cGAS–STING signaling can lead to chronic neuroinflammation, contributing to neurodegeneration. Long-term overstimulation of glia results in the overproduction and release of harmful stress–inducing pro-inflammatory factors, aggravating the inflammatory environment and inducing neuronal cell death and tissue damage, thereby promoting neurodegeneration in PD. Thus, the aberrant cGAS–STING-triggered inflammatory response to pathological α-syn may be one of the key mechanisms driving the progression of neurodegeneration in PD. This Figure was partially created using BioRender dynamic assets and retrieved from https://app.biorender.com/biorender-templates. |
Abbreviations
AAV, adeno-associated virus; AIM2, absent in melanoma-2; ALS, amyotrophic lateral sclerosis; AMBRA1, autophagy and beclin 1 regulator 1; AMPK, AMP-activated protein kinase; AP-1, activator protein 1; APE1, DNA (apurinic/apyrimidinic site) endonuclease 1; ASC, apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain; ATM, ataxia telangiectasia mutated kinase; ATP, adenosine triphosphate; BAK1, Bcl-2 antagonist/killer 1; BAX, Bcl-2-associated X; Bcl-2, B-cell lymphoma-2; BER pathway, base excision repair pathway; CCL20, C-C Motif Chemokine Ligand 20; cGAMP, 2′3′- cyclic GMP-AMP; cGAS, cyclic GMP-AMP synthase; cGAS–STING, cyclic GMP-AMP synthase (cGAS)–stimulator of interferon genes (STING); CNS, central nervous system; COPII, coatomer protein complex II; CXCL10, C-X-C Motif Chemokine Ligand 10; CXCL2, C-X-C Motif Chemokine Ligand 2; DA, dopaminergic; DAMPs, damage-associated molecular patterns; DDR, DNA damage response; DLB, dementia with Lewy bodies; Drp1, dynamin-related protein 1; DSBR, DNA double-strand break repair; DSBs, double-strand breaks; dsDNA, double-stranded DNA; ER, endoplasmic reticulum; ERGIC, ER–Golgi intermediate compartment; ESCs, embryonic stem cells; ETC, mitochondrial electron transport chain; EVs, extracellular vesicles; Fis1, mitochondrial fission protein 1; GBA1, glucocerebrosidase beta 1; GTP, guanosine triphosphate; H2AX, H2A Histone Family Member X; H2O2, hydrogen peroxide; HIF-1, hypoxia-inducible factor 1; HMGB, high mobility group box; IFI16, interferon-inducible protein 16; IFNAR, type I interferon receptor/or interferon-α/β receptor; IFNAR1, interferon-α/β receptor alpha chain; IFNAR2, interferon-α/β receptor beta chain; IFN-I, type I interferons; IFN-α, interferon-alpha; IFN-β, interferon-beta; IKK, IκB kinase; IL-18, interleukin-18; IL-1β, interleukin-1β; IL-6, interleukin-6; iNOS, inducible nitric oxide synthase; iPSCs, induced pluripotent stem cells; IRF3, interferon regulatory factor 3; IRF9, interferon regulatory factor 9; ISGF3, interferon-stimulated gene factor-3; ISGs, interferon-stimulated genes; ISRE, interferon-sensitive response element; JAK1, Janus kinase 1; LB, Lewy bodies; LLPS, liquid-liquid phase separation; LPS, lipopolysaccharide; LRRK2, leucine-rich repeat kinase 2; MAPK, mitogen-activated protein kinase; MAPK/JNK, mitogen-activated protein kinase/c-Jun N-terminal kinase; MDVs, mitochondria-derived vesicles; Mff, mitochondrial fission factor; Mfn1, mitofusin 1; Mfn2, mitofusin 2; MOMP, mitochondrial outer membrane permeabilisation; mPTP, mitochondrial permeability transition pore; MQC, mitochondrial quality control; MRE11, double-strand break repair protein MRE11; MSA, multiple system atrophy; mtDAMPs, mitochondrial-derived damage-associated molecular patterns; mtDNA, mitochondrial DNA; mTOR, mammalian target of rapamycin; mTORC1, mTOR complex 1; mtROS, mitochondrial-derived reactive oxygen species; MyD88, myeloid differentiation primary response 88; NBS1, nibrin 1; NDP52, nuclear dot protein 52; NER, nucleotide excision repair; NFAT, nuclear factor of activated T-cells; NF-κB, nuclear factor κB; NHEJ, non-homologous end-joining reaction; NLR, (NOD)-like receptor; NLRP3, nucleotide-binding oligomerization domain (NOD)-like receptor family pyrin domain containing protein 3; NLRs, NOD-like receptors; NO, nitric oxide; NOS, nitric oxide synthase; NOX, mitochondrial NADPH oxidase; NPCs, neural lineage progenitor cells; NRF2, nuclear factor-erythroid 2 related factor 2; OB, human olfactory bulb; OB/AON, olfactory bulb/anterior olfactory nucleus; Opa-1, optical atrophy 1; OPTN, optineurin; PAF, pure autonomic failure; PAMPs, pathogen-associated molecular patterns; Panx1, Pannexin 1; PARIS, Parkin-interacting substrate; PARK2, Parkinson Disease Protein 2; PCNA, proliferating cell nuclear antigen; PD, Parkinson’s disease; pDCs, plasmacytoid dendritic cells; PGC-1, peroxisome proliferator-activated receptor gamma coactivator 1; PINK1, PTEN-induced putative kinase 1; PKC/MAPK, protein kinase C/mitogen-activated protein kinase; PRC, PGC-1-related coactivator; PRRs, pattern-recognition receptors; PYHIN, pyrin and hematopoietic interferon-inducible nuclear (HIN) domain; RAD50, RAD50 double strand break repair protein; RAGE, receptor for advanced glycation endproducts; RBD, REM sleep behaviour disorder; RNS, reactive nitrogen species; ROS, reactive oxygen species; SAR1A, secretion associated, Ras related GTPase 1A; SEC24C, SEC24-related protein C; SNpc, substantia nigra pars compacta; STAT1 and STAT2, signal transducer and activator of transcription 1 and 2; STIM1, stromal interaction molecule 1; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; TFAM, mitochondrial transcription factor A; ThioS, thioflavin S; TLR9, Toll-like receptor 9; TLRs, Toll-like receptors; TNFR, tumour necrosis factor-α (TNF-α) receptor; TNF-α, tumour necrosis factor-α; TYK2, Tyrosine kinase 2; ULK1, Unc-51-like kinase 1; UPRmt, mitochondrial unfolded protein response; VDAC1, voltage-dependent anion channel 1; α-syn, alpha-synuclein; α-syn-PFF, alpha-synuclein preformed fibrils; γH2A.X, serine139-phosphorylated gamma histone H2A.X; 53BP1, p53-binding protein 1.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was funded by National Science Centre, Poland (https://www.ncn.gov.pl) by grant number 2020/39/I/NZ4/01031 for A.A. and by German research foundation DFG CU43/17-1 and Flexi-Lung-Brain funds to CC.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Haque ME, Akther M, Azam S, et al. Targeting α-synuclein aggregation and its role in mitochondrial dysfunction in Parkinson’s disease. Br J Pharmacol. 2022;179(1):23–45. doi:10.1111/bph.15684
2. Calabresi P, Mechelli A, Natale G, Volpicelli-Daley L, Di Lazzaro G, Ghiglieri V. Alpha-synuclein in Parkinson’s disease and other synucleinopathies: from overt neurodegeneration back to early synaptic dysfunction. Cell Death Dis. 2023;14(3):176. doi:10.1038/s41419-023-05672-9
3. Gelders G, Baekelandt V, Van der Perren A. Linking neuroinflammation and neurodegeneration in Parkinson’s Disease. J Immunol Res. 2018;2018:4784268. doi:10.1155/2018/4784268
4. Chen H, O’Reilly EJ, Schwarzschild MA, Ascherio A. Peripheral inflammatory biomarkers and risk of Parkinson’s disease. Am J Epidemiol. 2008;167(1):90–95. doi:10.1093/aje/kwm260
5. Wilkaniec A, Lenkiewicz AM, Czapski GA, et al. Extracellular alpha-synuclein oligomers induce Parkin S-nitrosylation: relevance to sporadic Parkinson’s disease etiopathology. Mol Neurobiol. 2019;56(1):125–140. doi:10.1007/s12035-018-1082-0
6. Jęśko H, Lenkiewicz AM, Wilkaniec A, Adamczyk A. The interplay between parkin and alpha-synuclein; possible implications for the pathogenesis of Parkinson’s disease. Acta Neurobiol Exp. 2019;79(3):276–289.
7. Wilkaniec A, Lenkiewicz AM, Babiec L, et al. Exogenous alpha-synuclein evoked parkin downregulation promotes mitochondrial dysfunction in neuronal cells. implications for Parkinson’s disease pathology. Front Aging Neurosci. 2021;13:591475. doi:10.3389/fnagi.2021.591475
8. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795–803. doi:10.1083/jcb.200809125
9. Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111(9):1208–1221. doi:10.1161/CIRCRESAHA.112.265819
10. Feany MB, Pallanck LJ. Parkin: a multipurpose neuroprotective agent? Neuron. 2003;38(1):13–16. doi:10.1016/s0896-6273(03)00201-0
11. Frank-Cannon TC, Tran T, Ruhn KA, et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci off J Soc Neurosci. 2008;28(43):10825–10834. doi:10.1523/JNEUROSCI.3001-08.2008
12. Paterna JC, Leng A, Weber E, Feldon J, Büeler H. DJ-1 and Parkin modulate dopamine-dependent behavior and inhibit MPTP-induced nigral dopamine neuron loss in mice. Mol Ther J Am Soc Gene Ther. 2007;15(4):698–704. doi:10.1038/sj.mt.6300067
13. Vercammen L, Van der Perren A, Vaudano E, et al. Parkin protects against neurotoxicity in the 6-hydroxydopamine rat model for Parkinson’s disease. Mol Ther J Am Soc Gene Ther. 2006;14(5):716–723. doi:10.1016/j.ymthe.2006.06.009
14. Yamada M, Mizuno Y, Mochizuki H. Parkin gene therapy for alpha-synucleinopathy: a rat model of Parkinson’s disease. Hum Gene Ther. 2005;16(2):262–270. doi:10.1089/hum.2005.16.262
15. Lo Bianco C, Schneider BL, Bauer M, et al. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101(50):17510–17515. doi:10.1073/pnas.0405313101
16. Palacino JJ, Sagi D, Goldberg MS, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279(18):18614–18622. doi:10.1074/jbc.M401135200
17. Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A. Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron. 2003;37(5):735–749. doi:10.1016/s0896-6273(03)00084-9
18. Petrucelli L, O’Farrell C, Lockhart PJ, et al. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36(6):1007–1019. doi:10.1016/s0896-6273(02)01125-x
19. Chung JY, Park HR, Lee SJ, et al. Elevated TRAF2/6 expression in Parkinson’s disease is caused by the loss of Parkin E3 ligase activity. Lab Investig J Tech Methods Pathol. 2013;93(6):663–676. doi:10.1038/labinvest.2013.60
20. Dionísio PEA, Oliveira SR, Amaral JSJD, Rodrigues CMP. Loss of Microglial Parkin Inhibits Necroptosis and Contributes to Neuroinflammation. Mol Neurobiol. 2019;56(4):2990–3004. doi:10.1007/s12035-018-1264-9
21. Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164(5):896–910. doi:10.1016/j.cell.2015.12.057
22. Riley JS, Tait SW. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020;21(4):e49799. doi:10.15252/embr.201949799
23. He Y, Hara H, Núñez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci. 2016;41(12):1012–1021. doi:10.1016/j.tibs.2016.09.002
24. Matheoud D, Sugiura A, Bellemare-Pelletier A, et al. Parkinson’s disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell. 2016;166(2):314–327. doi:10.1016/j.cell.2016.05.039
25. Rodríguez-Navarro JA, Casarejos MJ, Menéndez J, et al. Mortality, oxidative stress and tau accumulation during ageing in parkin null mice. J Neurochem. 2007;103(1):98–114. doi:10.1111/j.1471-4159.2007.04762.x
26. Casarejos MJ, Menéndez J, Solano RM, Rodríguez-Navarro JA, García de Yébenes J, Mena MA. Susceptibility to rotenone is increased in neurons from parkin null mice and is reduced by minocycline. J Neurochem. 2006;97(4):934–946. doi:10.1111/j.1471-4159.2006.03777.x
27. Tran TA, Nguyen AD, Chang J, Goldberg MS, Lee JK, Tansey MG. Lipopolysaccharide and tumor necrosis factor regulate Parkin expression via nuclear factor-kappa B. PLoS One. 2011;6(8):e23660. doi:10.1371/journal.pone.0023660
28. Moehlman AT, Kanfer G, Youle RJ. Loss of STING in parkin mutant flies suppresses muscle defects and mitochondria damage. PLoS Genet. 2023;19(7):e1010828. doi:10.1371/journal.pgen.1010828
29. Jiménez-Loygorri JI, Villarejo-Zori B, Viedma-Poyatos Á, et al. Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging. Nat Commun. 2024;15(1):830. doi:10.1038/s41467-024-45044-1
30. Huang Y, Liu B, Sinha SC, Amin S, Gan L. Mechanism and therapeutic potential of targeting cGAS-STING signaling in neurological disorders. Mol Neurodegener. 2023;18(1):79. doi:10.1186/s13024-023-00672-x
31. Luo Y, Zhao Z, Hu X. cGAS-STING-mediated inflammation and neurodegeneration as a strategy for the treatment of neurodegenerative diseases. Acta Biochim Biophys Sin. 2023;56(1):148–150. doi:10.3724/abbs.2023258
32. Yu L, Liu P. Cytosolic DNA sensing by cGAS: regulation, function, and human diseases. Signal Transduct Target Ther. 2021;6:170. doi:10.1038/s41392-021-00554-y
33. Chowdhury A, Witte S, Aich A. Role of Mitochondrial Nucleic Acid Sensing Pathways in Health and Patho-Physiology. Front Cell Dev Biol. 2022;10. doi:10.3389/fcell.2022.796066
34. Bader V, Winklhofer KF. Mitochondria at the interface between neurodegeneration and neuroinflammation. Semin Cell Dev Biol. 2020;99:163–171. doi:10.1016/j.semcdb.2019.05.028
35. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. 2021;21(9):548–569. doi:10.1038/s41577-021-00524-z
36. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi:10.1146/annurev-immunol-032713-120231
37. Subhramanyam CS, Wang C, Hu Q, Dheen ST. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol. 2019;94:112–120. doi:10.1016/j.semcdb.2019.05.004
38. Lyman M, Lloyd DG, Ji X, Vizcaychipi MP, Ma D. Neuroinflammation: the role and consequences. Neurosci Res. 2014;79:1–12. doi:10.1016/j.neures.2013.10.004
39. Lin MM, Liu N, Qin ZH, Wang Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol Sin. 2022;43(10):2439–2447. doi:10.1038/s41401-022-00879-6
40. Zhou X, Wang J, Yu L, et al. Mitophagy and cGAS–STING crosstalk in neuroinflammation. Acta Pharm Sin B. 2024. doi:10.1016/j.apsb.2024.05.012
41. Gulen MF, Samson N, Keller A, et al. cGAS–STING drives ageing-related inflammation and neurodegeneration. Nature. 2023;620(7973):374–380. doi:10.1038/s41586-023-06373-1
42. Paul BD, Snyder SH, Bohr VA. Signaling by cGAS-STING in neurodegeneration, neuroinflammation, and aging. Trends Neurosci. 2021;44(2):83–96. doi:10.1016/j.tins.2020.10.008
43. Weindel CG, Bell SL, Vail KJ, West KO, Patrick KL, Watson RO. LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. eLife. 2020;9:e51071. doi:10.7554/eLife.51071
44. Hinkle JT, Patel J, Panicker N, et al. STING mediates neurodegeneration and neuroinflammation in nigrostriatal α-synucleinopathy. Proc Natl Acad Sci U S A. 2022;119(15):e2118819119. doi:10.1073/pnas.2118819119
45. Szego EM, Malz L, Bernhardt N, Rösen-Wolff A, Falkenburger BH, Luksch H. Constitutively active STING causes neuroinflammation and degeneration of dopaminergic neurons in mice. eLife. 2022;11:e81943. doi:10.7554/eLife.81943
46. Rodríguez-Nuevo A, Zorzano A. The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress. 2019;3(6):195–207. doi:10.15698/cst2019.06.190
47. Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med. 2010;31(3):227–285. doi:10.1016/j.mam.2010.03.002
48. Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112(4):481–490. doi:10.1016/s0092-8674(03)00116-8
49. Risiglione P, Zinghirino F, Di Rosa MC, Magrì A, Messina A. Alpha-synuclein and mitochondrial dysfunction in Parkinson’s disease: the emerging role of VDAC. Biomolecules. 2021;11(5):718. doi:10.3390/biom11050718
50. Zanon A, Kalvakuri S, Rakovic A, et al. SLP-2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in Parkin-deficient human iPSC-derived neurons and Drosophila. Hum Mol Genet. 2017;26(13):2412–2425. doi:10.1093/hmg/ddx132
51. Picca A, Guerra F, Calvani R, et al. Mitochondrial dysfunction and aging: insights from the analysis of extracellular vesicles. Int J Mol Sci. 2019;20(4):805. doi:10.3390/ijms20040805
52. Hou Y, Dan X, Babbar M, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15(10):565–581. doi:10.1038/s41582-019-0244-7
53. Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9(11):1750–1757. doi:10.4161/auto.26122
54. Chan NC, Salazar AM, Pham AH, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20(9):1726–1737. doi:10.1093/hmg/ddr048
55. Sarraf SA, Raman M, Guarani-Pereira V, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496(7445):372–376. doi:10.1038/nature12043
56. Vincow ES, Merrihew G, Thomas RE, et al. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A. 2013;110(16):6400–6405. doi:10.1073/pnas.1221132110
57. Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–131. doi:10.1038/ncb2012
58. Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107(1):378–383. doi:10.1073/pnas.0911187107
59. Matsuda N, Sato S, Shiba K, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189(2):211–221. doi:10.1083/jcb.200910140
60. Clark IE, Dodson MW, Jiang C, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441(7097):1162–1166. doi:10.1038/nature04779
61. Kim Y, Park J, Kim S, et al. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377(3):975–980. doi:10.1016/j.bbrc.2008.10.104
62. Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–608. doi:10.1038/33416
63. Valente EM, Salvi S, Ialongo T, et al. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol. 2004;56(3):336–341. doi:10.1002/ana.20256
64. Doherty KM, Silveira-Moriyama L, Parkkinen L, et al. Parkin disease: a clinicopathologic entity? JAMA Neurol. 2013;70(5):571–579. doi:10.1001/jamaneurol.2013.172
65. Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22(2):320–333. doi:10.1016/j.devcel.2011.12.014
66. Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy. 2013;9(11):1758–1769. doi:10.4161/auto.24633
67. Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLOS Biol. 2010;8(1):e1000298. doi:10.1371/journal.pbio.1000298
68. Chen Y, Dorn GW. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340(6131):471–475. doi:10.1126/science.1231031
69. Caulfield TR, Fiesel FC, Moussaud-Lamodière EL, Dourado DFAR, Flores SC, Springer W. Phosphorylation by PINK1 releases the UBL domain and initializes the conformational opening of the E3 ubiquitin ligase Parkin. PLOS Comput Biol. 2014;10(11):e1003935. doi:10.1371/journal.pcbi.1003935
70. Gladkova C, Maslen SL, Skehel JM, Komander D. Mechanism of parkin activation by PINK1. Nature. 2018;559(7714):410–414. doi:10.1038/s41586-018-0224-x
71. Iguchi M, Kujuro Y, Okatsu K, et al. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J Biol Chem. 2013;288(30):22019–22032. doi:10.1074/jbc.M113.467530
72. Kondapalli C, Kazlauskaite A, Zhang N, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2(5):120080. doi:10.1098/rsob.120080
73. McWilliams TG, Barini E, Pohjolan-Pirhonen R, et al. Phosphorylation of Parkin at serine 65 is essential for its activation in vivo. Open Biol. 2018;8(11):180108. doi:10.1098/rsob.180108
74. Trempe JF, Sauvé V, Grenier K, et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science. 2013;340(6139):1451–1455. doi:10.1126/science.1237908
75. Kane LA, Lazarou M, Fogel AI, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205(2):143–153. doi:10.1083/jcb.201402104
76. Kazlauskaite A, Kondapalli C, Gourlay R, et al. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014;460(1):127–139. doi:10.1042/BJ20140334
77. Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510(7503):162–166. doi:10.1038/nature13392
78. Ordureau A, Sarraf SA, Duda DM, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56(3):360–375. doi:10.1016/j.molcel.2014.09.007
79. Wauer T, Swatek KN, Wagstaff JL, et al. Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 2015;34(3):307–325. doi:10.15252/embj.201489847
80. Okatsu K, Koyano F, Kimura M, et al. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol. 2015;209(1):111–128. doi:10.1083/jcb.201410050
81. Shiba-Fukushima K, Arano T, Matsumoto G, et al. Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLOS Genet. 2014;10(12):e1004861. doi:10.1371/journal.pgen.1004861
82. Ham SJ, Lee D, Yoo H, Jun K, Shin H, Chung J. Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc Natl Acad Sci U S A. 2020;117(8):4281–4291. doi:10.1073/pnas.1909814117
83. Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. doi:10.1038/nature14893
84. Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 2014;37(6):315–324. doi:10.1016/j.tins.2014.03.004
85. Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32(1):2–11. doi:10.1128/MCB.06159-11
86. Hung CM, Lombardo PS, Malik N, et al. AMPK/ULK1-mediated phosphorylation of Parkin ACT domain mediates an early step in mitophagy. Sci Adv. 2021;7(15):eabg4544. doi:10.1126/sciadv.abg4544
87. Park D, Lee MN, Jeong H, et al. Parkin ubiquitinates mTOR to regulate mTORC1 activity under mitochondrial stress. Cell Signal. 2014;26(10):2122–2130. doi:10.1016/j.cellsig.2014.06.010
88. Van Humbeeck C, Cornelissen T, Hofkens H, et al. Parkin interacts with Ambra1 to induce mitophagy. J Neurosci. 2011;31(28):10249–10261. doi:10.1523/JNEUROSCI.1917-11.2011
89. Hammerling BC, Najor RH, Cortez MQ, et al. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat Commun. 2017;8:14050. doi:10.1038/ncomms14050
90. McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014;33(4):282–295. doi:10.1002/embj.201385902
91. Bingol B, Tea JS, Phu L, et al. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510(7505):370–375. doi:10.1038/nature13418
92. Cunningham CN, Baughman JM, Phu L, et al. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol. 2015;17(2):160–169. doi:10.1038/ncb3097
93. Glauser L, Sonnay S, Stafa K, Moore DJ. Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J Neurochem. 2011;118(4):636–645. doi:10.1111/j.1471-4159.2011.07318.x
94. Rakovic A, Grünewald A, Kottwitz J, et al. Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS One. 2011;6(3):e16746. doi:10.1371/journal.pone.0016746
95. Tanaka A, Cleland MM, Xu S, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191(7):1367–1380. doi:10.1083/jcb.201007013
96. Westermann B. Bioenergetic role of mitochondrial fusion and fission. Biochim Biophys Acta. 2012;1817(10):1833–1838. doi:10.1016/j.bbabio.2012.02.033
97. Tondera D, Grandemange S, Jourdain A, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28(11):1589–1600. doi:10.1038/emboj.2009.89
98. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–1065. doi:10.1126/science.1219855
99. Yu W, Sun Y, Guo S, Lu B. The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum Mol Genet. 2011;20(16):3227–3240. doi:10.1093/hmg/ddr235
100. Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AHV, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19(24):4861–4870. doi:10.1093/hmg/ddq419
101. Ziviani E, Tao RN, Whitworth AJ. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A. 2010;107(11):5018–5023. doi:10.1073/pnas.0913485107
102. Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One. 2010;5(4):e10054. doi:10.1371/journal.pone.0010054
103. Wang X, Winter D, Ashrafi G, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147(4):893–906. doi:10.1016/j.cell.2011.10.018
104. Jannig PR, Dumesic PA, Spiegelman BM, Ruas JL. SnapShot: regulation and biology of PGC-1α. Cell. 2022;185(8):1444–1444.e1. doi:10.1016/j.cell.2022.03.027
105. Shin JH, Ko HS, Kang H, et al. Paris (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144(5):689–702. doi:10.1016/j.cell.2011.02.010
106. Lee Y, Stevens DA, Kang SU, et al. PINK1 primes parkin-mediated ubiquitination of Paris in dopaminergic neuronal survival. Cell Rep. 2017;18(4):918–932. doi:10.1016/j.celrep.2016.12.090
107. Stevens DA, Lee Y, Kang HC, et al. Parkin loss leads to Paris-dependent declines in mitochondrial mass and respiration. Proc Natl Acad Sci U S A. 2015;112(37):11696–11701. doi:10.1073/pnas.1500624112
108. Siddiqui A, Rane A, Rajagopalan S, Chinta SJ, Andersen JK. Detrimental effects of oxidative losses in parkin activity in a model of sporadic Parkinson’s disease are attenuated by restoration of PGC1alpha. Neurobiol Dis. 2016;93:115–120. doi:10.1016/j.nbd.2016.05.009
109. LaPak KM, Saeidi S, Bok I, et al. Proximity proteomic analysis of the NRF family reveals the Parkinson’s disease protein ZNF746/Paris as a co-complexed repressor of NRF2. Sci Signal. 2023;16(815):eadi9018. doi:10.1126/scisignal.adi9018
110. Magalhães JD, Cardoso SM. Mitochondrial signaling on innate immunity activation in Parkinson disease. Curr Opin Neurobiol. 2023;78:102664. doi:10.1016/j.conb.2022.102664
111. Patergnani S, Morciano G, Carinci M, Leo S, Pinton P, Rimessi A. The “mitochondrial stress responses”: the “Dr. Jekyll and Mr. Hyde” of neuronal disorders. Neural Regen Res. 2022;17(12):2563–2575. doi:10.4103/1673-5374.339473
112. Picca A, Calvani R, Coelho-Junior HJ, Landi F, Bernabei R, Marzetti E. Mitochondrial dysfunction, oxidative stress, and neuroinflammation: intertwined roads to neurodegeneration. Antioxid Basel Switz. 2020;9(8):647. doi:10.3390/antiox9080647
113. Fleshner M, Frank M, Maier SF. Danger signals and inflammasomes: stress-evoked sterile inflammation in mood disorders. Neuropsychopharmacol off Publ Am Coll Neuropsychopharmacol. 2017;42(1):36–45. doi:10.1038/npp.2016.125
114. Nakahira K, Haspel JA, Rathinam VAK, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230. doi:10.1038/ni.1980
115. Sadatomi D, Nakashioya K, Mamiya S, et al. Mitochondrial function is required for extracellular ATP-induced NLRP3 inflammasome activation. J Biochem. 2017;161(6):503–512. doi:10.1093/jb/mvw098
116. Patergnani S, Bouhamida E, Leo S, Pinton P, Rimessi A. Mitochondrial oxidative stress and “Mito-Inflammation”: actors in the diseases. Biomedicines. 2021;9(2):216. doi:10.3390/biomedicines9020216
117. Grazioli S, Pugin J. Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front Immunol. 2018;9:832. doi:10.3389/fimmu.2018.00832
118. Garg M, Johri S, Chakraborty K. Immunomodulatory role of mitochondrial DAMPs: a missing link in pathology? FEBS J. 2023;290(18):4395–4418. doi:10.1111/febs.16563
119. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20(2):95–112. doi:10.1038/s41577-019-0215-7
120. Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–305. doi:10.1126/science.1071059
121. Martin WF, Garg S, Zimorski V. Endosymbiotic theories for eukaryote origin. Philos Trans R Soc Lond B Biol Sci. 2015;370(1678):20140330. doi:10.1098/rstb.2014.0330
122. Wilkins HM, Swerdlow RH. Relationships between mitochondria and neuroinflammation: implications for Alzheimer’s disease. Curr Top Med Chem. 2016;16(8):849–857. doi:10.2174/1568026615666150827095102
123. Boguszewska K, Szewczuk M, Kaźmierczak-Barańska J, Karwowski BT. The similarities between human mitochondria and bacteria in the context of structure, genome, and base excision repair system. Mol. 2020;25(12):2857. doi:10.3390/molecules25122857
124. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21(10):1359–1369. doi:10.1038/s41593-018-0242-x
125. Han QQ, Le W. NLRP3 inflammasome-mediated neuroinflammation and related mitochondrial impairment in Parkinson’s disease. Neurosci Bull. 2023;39(5):832–844. doi:10.1007/s12264-023-01023-y
126. Donnelly CR, Chen O, Ji RR. How do sensory neurons sense danger signals? Trends Neurosci. 2020;43(10):822–838. doi:10.1016/j.tins.2020.07.008
127. Zhang X, Wu X, Hu Q, et al. Mitochondrial DNA in liver inflammation and oxidative stress. Life Sci. 2019;236:116464. doi:10.1016/j.lfs.2019.05.020
128. Yu Y, Feng S, Wei S, et al. Extracellular ATP activates P2X7R-NF-κB (p65) pathway to promote the maturation of bone marrow-derived dendritic cells of mice. Cytokine. 2019;119:175–181. doi:10.1016/j.cyto.2019.03.019
129. Schindler SM, Frank MG, Annis JL, Maier SF, Klegeris A. Pattern recognition receptors mediate pro-inflammatory effects of extracellular mitochondrial transcription factor A (TFAM). Mol Cell Neurosci. 2018;89:71–79. doi:10.1016/j.mcn.2018.04.005
130. Iyer SS, He Q, Janczy JR, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39(2):311–323. doi:10.1016/j.immuni.2013.08.001
131. Pointer CB, Wenzel TJ, Klegeris A. Extracellular cardiolipin regulates select immune functions of microglia and microglia-like cells. Brain Res Bull. 2019;146:153–163. doi:10.1016/j.brainresbull.2019.01.002
132. Gouveia A, Bajwa E, Klegeris A. Extracellular cytochrome c as an intercellular signaling molecule regulating microglial functions. Biochim Biophys Acta Gen Subj. 2017;1861(9):2274–2281. doi:10.1016/j.bbagen.2017.06.017
133. Wenzel TJ, Bajwa E, Klegeris A. Cytochrome c can be released into extracellular space and modulate functions of human astrocytes in a toll-like receptor 4-dependent manner. Biochim Biophys Acta Gen Subj. 2019;1863(11):129400. doi:10.1016/j.bbagen.2019.07.009
134. Pérez-Treviño P, Velásquez M, García N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim Biophys Acta Mol Basis Dis. 2020;1866(6):165761. doi:10.1016/j.bbadis.2020.165761
135. Andreeva L, Hiller B, Kostrewa D, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature. 2017;549(7672):394–398. doi:10.1038/nature23890
136. West AP, Khoury-Hanold W, Staron M, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–557. doi:10.1038/nature14156
137. Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215(5):1287–1299. doi:10.1084/jem.20180139
138. Riley JS, Quarato G, Cloix C, et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018;37(17):e99238. doi:10.15252/embj.201899238
139. McArthur K, Whitehead LW, Heddleston JM, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. 2018;359(6378):eaao6047. doi:10.1126/science.aao6047
140. Kim J, Gupta R, Blanco LP, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. 2019;366(6472):1531–1536. doi:10.1126/science.aav4011
141. Patrushev M, Kasymov V, Patrusheva V, Ushakova T, Gogvadze V, Gaziev A. Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell Mol Life Sci CMLS. 2004;61(24):3100–3103. doi:10.1007/s00018-004-4424-1
142. García N, Chávez E. Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci. 2007;81(14):1160–1166. doi:10.1016/j.lfs.2007.08.019
143. Moya GE, Rivera PD, Dittenhafer-Reed KE. Evidence for the Role of Mitochondrial DNA Release in the Inflammatory Response in Neurological Disorders. Int J Mol Sci. 2021;22(13):7030. doi:10.3390/ijms22137030
144. Picca A, Lezza AMS, Leeuwenburgh C, et al. Circulating mitochondrial DNA at the crossroads of mitochondrial dysfunction and inflammation during aging and muscle wasting disorders. Rejuvenation Res. 2018;21(4):350–359. doi:10.1089/rej.2017.1989
145. Cardon LR, Burge C, Clayton DA, Karlin S. Pervasive CpG suppression in animal mitochondrial genomes. Proc Natl Acad Sci U S A. 1994;91(9):3799–3803. doi:10.1073/pnas.91.9.3799
146. Pollack Y, Kasir J, Shemer R, Metzger S, Szyf M. Methylation pattern of mouse mitochondrial DNA. Nucleic Acids Res. 1984;12(12):4811–4824. doi:10.1093/nar/12.12.4811
147. Shimada K, Crother TR, Karlin J, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–414. doi:10.1016/j.immuni.2012.01.009
148. Zhong Z, Liang S, Sanchez-Lopez E, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560(7717):198–203. doi:10.1038/s41586-018-0372-z
149. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi:10.1016/j.cell.2010.01.022
150. Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. doi:10.1038/nature08780
151. Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock Augusta Ga. 2010;34(1):55–59. doi:10.1097/SHK.0b013e3181cd8c08
152. Zhong F, Liang S, Zhong Z. Emerging Role of Mitochondrial DNA as a Major Driver of Inflammation and Disease Progression. Trends Immunol. 2019;40(12):1120–1133. doi:10.1016/j.it.2019.10.008
153. Yu J, Nagasu H, Murakami T, et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci U S A. 2014;111(43):15514–15519. doi:10.1073/pnas.1414859111
154. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi:10.1038/nature09663
155. Luna-Sánchez M, Bianchi P, Quintana A. Mitochondria-Induced Immune Response as a Trigger for Neurodegeneration: a Pathogen from Within. Int J Mol Sci. 2021;22(16):8523. doi:10.3390/ijms22168523
156. Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol. 2023;23(3):159–173. doi:10.1038/s41577-022-00760-x
157. Todosenko N, Khaziakhmatova O, Malashchenko V, et al. Mitochondrial Dysfunction Associated with mtDNA in Metabolic Syndrome and Obesity. Int J Mol Sci. 2023;24(15):12012. doi:10.3390/ijms241512012
158. Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163(3):560–569. doi:10.1016/j.cell.2015.10.001
159. Park J, Min JS, Kim B, et al. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-κB pathways. Neurosci Lett. 2015;584:191–196. doi:10.1016/j.neulet.2014.10.016
160. Herb M, Gluschko A, Wiegmann K, et al. Mitochondrial reactive oxygen species enable proinflammatory signaling through disulfide linkage of NEMO. Sci Signal. 2019;12(568):eaar5926. doi:10.1126/scisignal.aar5926
161. Wang D, Malo D, Hekimi S. Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1alpha in long-lived Mclk1+/− mouse mutants. J Immunol Baltim. 2010;184(2):582–590. doi:10.4049/jimmunol.0902352
162. Checa J, Aran JM. Reactive oxygen Species: Drivers Of Physiological And Pathological Processes. J Inflamm Res. 2020;13:1057–1073. doi:10.2147/JIR.S275595
163. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278–286. doi:10.1038/nature10759
164. Garg C, Seo JH, Ramachandran J, Loh JM, Calderon F, Contreras JE. Trovafloxacin attenuates neuroinflammation and improves outcome after traumatic brain injury in mice. J Neuroinflammation. 2018;15(1):42. doi:10.1186/s12974-018-1069-9
165. Nakahira K, Hisata S, Choi AMK. The roles of mitochondrial damage-associated molecular patterns in diseases. Antioxid Redox Signal. 2015;23(17):1329–1350. doi:10.1089/ars.2015.6407
166. Illes P. P2X7 receptors amplify CNS damage in neurodegenerative diseases. Int J Mol Sci. 2020;21(17):5996. doi:10.3390/ijms21175996
167. Shiratori M, Tozaki-Saitoh H, Yoshitake M, Tsuda M, Inoue K. P2X7 receptor activation induces CXCL2 production in microglia through NFAT and PKC/MAPK pathways. J Neurochem. 2010;114(3):810–819. doi:10.1111/j.1471-4159.2010.06809.x
168. Ren M, Phoon CKL, Schlame M. Metabolism and function of mitochondrial cardiolipin. Prog Lipid Res. 2014;55:1–16. doi:10.1016/j.plipres.2014.04.001
169. Pizzuto M, Pelegrin P. Cardiolipin in immune signaling and cell death. Trends Cell Biol. 2020;30(11):892–903. doi:10.1016/j.tcb.2020.09.004
170. Balasubramanian K, Maeda A, Lee JS, et al. Dichotomous roles for externalized cardiolipin in extracellular signaling: promotion of phagocytosis and attenuation of innate immunity. Sci Signal. 2015;8(395):ra95. doi:10.1126/scisignal.aaa6179
171. Ott M, Zhivotovsky B, Orrenius S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ. 2007;14(7):1243–1247. doi:10.1038/sj.cdd.4402135
172. Au AK, Aneja RK, Bell MJ, et al. Cerebrospinal fluid levels of high-mobility group box 1 and cytochrome C predict outcome after pediatric traumatic brain injury. J Neurotrauma. 2012;29(11):2013–2021. doi:10.1089/neu.2011.2171
173. Pullerits R, Bokarewa M, Jonsson IM, Verdrengh M, Tarkowski A. Extracellular cytochrome c, a mitochondrial apoptosis-related protein, induces arthritis. Rheumatol Oxf Engl. 2005;44(1):32–39. doi:10.1093/rheumatology/keh406
174. Julian MW, Shao G, Bao S, et al. Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J Immunol Baltim. 2012;189(1):433–443. doi:10.4049/jimmunol.1101375
175. Julian MW, Shao G, Vangundy ZC, Papenfuss TL, Crouser ED. Mitochondrial transcription factor A, an endogenous danger signal, promotes TNFα release via RAGE- and TLR9-responsive plasmacytoid dendritic cells. PLoS One. 2013;8(8):e72354. doi:10.1371/journal.pone.0072354
176. Ablasser A, Chen ZJ. cGAS in action: expanding roles in immunity and inflammation. Science. 2019;363(6431):eaat8657. doi:10.1126/science.aat8657
177. Ablasser A. Structures of STING protein illuminate this key regulator of inflammation. Nature. 2019;567(7748):321–322. doi:10.1038/d41586-019-00707-8
178. Margolis SR, Wilson SC, Vance RE. Evolutionary origins of cGAS-STING signaling. Trends Immunol. 2017;38(10):733–743. doi:10.1016/j.it.2017.03.004
179. de Oliveira Mann CC, Hopfner KP. Nuclear cGAS: guard or prisoner? EMBO J. 2021;40(16):e108293. doi:10.15252/embj.2021108293
180. Xiong Y, Tang YD, Zheng C. The crosstalk between the caspase family and the cGAS‒STING signaling pathway. J Mol Cell Biol. 2021;13(10):739–747. doi:10.1093/jmcb/mjab071
181. Zhang M, Zou Y, Zhou X, Zhou J. Inhibitory targeting cGAS-STING-TBK1 axis: emerging strategies for autoimmune diseases therapy. Front Immunol. 2022;13:954129. doi:10.3389/fimmu.2022.954129
182. Zheng W, Chen N, Meurens F, Zheng W, Zhu J. How does cGAS avoid sensing self-DNA under normal physiological conditions? Int J Mol Sci. 2023;24(19):14738. doi:10.3390/ijms241914738
183. Zhang X, Bai XC, Chen ZJ. Structures and mechanisms in the cGAS-STING innate immunity pathway. Immunity. 2020;53(1):43–53. doi:10.1016/j.immuni.2020.05.013
184. Zhou W, Whiteley AT, de Oliveira Mann CC, et al. Structure of the human cGAS-DNA complex reveals enhanced control of immune surveillance. Cell. 2018;174(2):300–311.e11. doi:10.1016/j.cell.2018.06.026
185. Civril F, Deimling T, de Oliveira Mann CC, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498(7454):332–337. doi:10.1038/nature12305
186. Luecke S, Holleufer A, Christensen MH, et al. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017;18(10):1707–1715. doi:10.15252/embr.201744017
187. Webb LG, Fernandez-Sesma A. RNA viruses and the cGAS-STING pathway: reframing our understanding of innate immune sensing. Curr Opin Virol. 2022;53:101206. doi:10.1016/j.coviro.2022.101206
188. Du M, Chen ZJ. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science. 2018;361(6403):704–709. doi:10.1126/science.aat1022
189. Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe. 2015;18(2):157–168. doi:10.1016/j.chom.2015.07.001
190. Fryer AL, Abdullah A, Taylor JM, Crack PJ. The complexity of the cGAS-STING pathway in CNS pathologies. Front Neurosci. 2021;15:621501. doi:10.3389/fnins.2021.621501
191. Ou L, Zhang A, Cheng Y, Chen Y. The cGAS-STING pathway: a promising immunotherapy target. Front Immunol. 2021;12:795048. doi:10.3389/fimmu.2021.795048
192. Gui X, Yang H, Li T, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;567(7747):262–266. doi:10.1038/s41586-019-1006-9
193. Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature. 2019;567(7748):394–398. doi:10.1038/s41586-019-1000-2
194. Zhao B, Du F, Xu P, et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature. 2019;569(7758):718–722. doi:10.1038/s41586-019-1228-x
195. Liu S, Cai X, Wu J, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347(6227):aaa2630. doi:10.1126/science.aaa2630
196. Wu J, Dobbs N, Yang K, Yan N. Interferon-independent activities of mammalian STING mediate antiviral response and tumor immune evasion. Immunity. 2020;53(1):115–126.e5. doi:10.1016/j.immuni.2020.06.009
197. Balka KR, Louis C, Saunders TL, et al. TBK1 and IKKε act redundantly to mediate STING-induced NF-κB responses in myeloid cells. Cell Rep. 2020;31(1):107492. doi:10.1016/j.celrep.2020.03.056
198. Yum S, Li M, Fang Y, Chen ZJ. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci U S A. 2021;118(14):e2100225118. doi:10.1073/pnas.2100225118
199. Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol. 2014;88(10):5328–5341. doi:10.1128/JVI.00037-14
200. Smale ST. Selective transcription in response to an inflammatory stimulus. Cell. 2010;140(6):833–844. doi:10.1016/j.cell.2010.01.037
201. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5(5):375–386. doi:10.1038/nri1604
202. Lousberg EL, Fraser CK, Tovey MG, Diener KR, Hayball JD. Type I interferons mediate the innate cytokine response to recombinant fowlpox virus but not the induction of plasmacytoid dendritic cell-dependent adaptive immunity. J Virol. 2010;84(13):6549–6563. doi:10.1128/JVI.02618-09
203. de Weerd NA, Nguyen T. The interferons and their receptors--distribution and regulation. Immunol Cell Biol. 2012;90(5):483–491. doi:10.1038/icb.2012.9
204. Yamada T, Horisberger MA, Kawaguchi N, Moroo I, Toyoda T. Immunohistochemistry using antibodies to alpha-interferon and its induced protein, MxA, in Alzheimer’s and Parkinson’s disease brain tissues. Neurosci Lett. 1994;181(1–2):61–64. doi:10.1016/0304-3940(94)90560-6
205. Owens T, Khorooshi R, Wlodarczyk A, Asgari N. Interferons in the central nervous system: a few instruments play many tunes. Glia. 2014;62(3):339–355. doi:10.1002/glia.22608
206. Brown GC, Vilalta A. How microglia kill neurons. Brain Res. 2015;1628(Pt B):288–297. doi:10.1016/j.brainres.2015.08.031
207. Baruch K, Deczkowska A, David E, et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. 2014;346(6205):89–93. doi:10.1126/science.1252945
208. Zheng W, Liu A, Xia N, Chen N, Meurens F, Zhu J. How the Innate Immune DNA Sensing cGAS-STING Pathway Is Involved in Apoptosis. Int J Mol Sci. 2023;24(3):3029. doi:10.3390/ijms24033029
209. Murthy AMV, Robinson N, Kumar S. Crosstalk between cGAS-STING signaling and cell death. Cell Death Differ. 2020;27(11):2989–3003. doi:10.1038/s41418-020-00624-8
210. Xu Y, Chen C, Liao Z, Xu P. cGAS-STING signaling in cell death: mechanisms of action and implications in pathologies. Eur J Immunol. 2023;53(9):e2350386. doi:10.1002/eji.202350386
211. Hu X, Zhang H, Zhang Q, Yao X, Ni W, Zhou K. Emerging role of STING signalling in CNS injury: inflammation, autophagy, necroptosis, ferroptosis and pyroptosis. J Neuroinflammation. 2022;19(1):242. doi:10.1186/s12974-022-02602-y
212. Wan D, Jiang W, Hao J. Research advances in how the cGAS-STING pathway controls the cellular inflammatory response. Front Immunol. 2020;11:615. doi:10.3389/fimmu.2020.00615
213. White AJ, Wijeyekoon RS, Scott KM, et al. The Peripheral Inflammatory Response to Alpha-Synuclein and Endotoxin in Parkinson’s Disease. Front Neurol. 2018;9:946. doi:10.3389/fneur.2018.00946
214. Cho B, Kim T, Huh YJ, Lee J, Lee YI. Amelioration of mitochondrial quality control and proteostasis by natural compounds in Parkinson’s disease models. Int J Mol Sci. 2019;20(20):5208. doi:10.3390/ijms20205208
215. Moehlman AT, Youle RJ. Mitochondrial quality control and restraining innate immunity. Annu Rev Cell Dev Biol. 2020;36:265–289. doi:10.1146/annurev-cellbio-021820-101354
216. Rocha EM, De Miranda B, Sanders LH. Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis. 2018;109(Pt B):249–257. doi:10.1016/j.nbd.2017.04.004
217. Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci. 2015;40(4):200–210. doi:10.1016/j.tibs.2015.02.003
218. Newman LE, Shadel GS. Pink1/Parkin link inflammation, mitochondrial stress, and neurodegeneration. J Cell Biol. 2018;217(10):3327–3329. doi:10.1083/jcb.201808118
219. Lin Q, Li S, Jiang N, et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019;26:101254. doi:10.1016/j.redox.2019.101254
220. Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85(2):257–273. doi:10.1016/j.neuron.2014.12.007
221. Oka T, Hikoso S, Yamaguchi O, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485(7397):251–255. doi:10.1038/nature10992
222. Ding Z, Liu S, Wang X, Khaidakov M, Dai Y, Mehta JL. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci Rep. 2013;3:1077. doi:10.1038/srep01077
223. Ding Z, Liu S, Wang X, et al. LOX-1, oxidant stress, mtDNA damage, autophagy, and immune response in atherosclerosis. Can J Physiol Pharmacol. 2014;92(7):524–530. doi:10.1139/cjpp-2013-0420
224. Picca A, Guerra F, Calvani R, et al. Mitochondrial signatures in circulating extracellular vesicles of older adults with Parkinson’s disease: results from the EXosomes in Parkinson’s Disease (EXPAND) Study. J Clin Med. 2020;9(2):504. doi:10.3390/jcm9020504
225. Trinh J, Hicks AA, König IR, et al. Mitochondrial DNA heteroplasmy distinguishes disease manifestation in PINK1/PRKN-linked Parkinson’s disease. Brain J Neurol. 2023;146(7):2753–2765. doi:10.1093/brain/awac464
226. Lood C, Blanco LP, Purmalek MM, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–153. doi:10.1038/nm.4027
227. Pereira SL, Grossmann D, Delcambre S, Hermann A, Grünewald A. Novel insights into Parkin-mediated mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Curr Opin Neurobiol. 2023;80:102720. doi:10.1016/j.conb.2023.102720
228. Wasner K, Smajic S, Ghelfi J, et al. Parkin deficiency impairs mitochondrial DNA dynamics and propagates inflammation. Mov Disord off J Mov Disord Soc. 2022;37(7):1405–1415. doi:10.1002/mds.29025
229. Matsui H, Ito J, Matsui N, Uechi T, Onodera O, Kakita A. Cytosolic dsDNA of mitochondrial origin induces cytotoxicity and neurodegeneration in cellular and zebrafish models of Parkinson’s disease. Nat Commun. 2021;12(1):3101. doi:10.1038/s41467-021-23452-x
230. Cao T, Shao S, Li B, et al. Up-regulation of Interferon-inducible protein 16 contributes to psoriasis by modulating chemokine production in keratinocytes. Sci Rep. 2016;6:25381. doi:10.1038/srep25381
231. Pickrell AM, Huang CH, Kennedy SR, et al. Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron. 2015;87(2):371–381. doi:10.1016/j.neuron.2015.06.034
232. Borsche M, König IR, Delcambre S, et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain J Neurol. 2020;143(10):3041–3051. doi:10.1093/brain/awaa246
233. Liu H, Ho PW, Leung CT, et al. Aberrant mitochondrial morphology and function associated with impaired mitophagy and DNM1L-MAPK/ERK signaling are found in aged mutant Parkinsonian LRRK2R1441G mice. Autophagy. 2021;17(10):3196–3220. doi:10.1080/15548627.2020.1850008
234. Rodríguez-Nuevo A, Díaz-Ramos A, Noguera E, et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018;37(10):e96553. doi:10.15252/embj.201796553
235. Bae JH, Jo SI, Kim SJ, et al. Circulating cell-free mtDNA contributes to AIM2 inflammasome-mediated chronic inflammation in patients with type 2 diabetes. Cells. 2019;8(4):328. doi:10.3390/cells8040328
236. Silzer T, Barber R, Sun J, et al. Circulating mitochondrial DNA: new indices of type 2 diabetes-related cognitive impairment in Mexican Americans. PLoS One. 2019;14(3):e0213527. doi:10.1371/journal.pone.0213527
237. Caielli S, Athale S, Domic B, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. 2016;213(5):697–713. doi:10.1084/jem.20151876
238. Han X, Chen H, Gong H, et al. Autolysosomal degradation of cytosolic chromatin fragments antagonizes oxidative stress-induced senescence. J Biol Chem. 2020;295(14):4451–4463. doi:10.1074/jbc.RA119.010734
239. Singh F, Ganley IG. Parkinson’s disease and mitophagy: an emerging role for LRRK2. Biochem Soc Trans. 2021;49(2):551–562. doi:10.1042/BST20190236
240. Standaert DG, Childers GM. Alpha-synuclein-mediated DNA damage, STING activation, and neuroinflammation in Parkinson’s disease. Proc Natl Acad Sci U S A. 2022;119(17):e2204058119. doi:10.1073/pnas.2204058119
241. Kim C, Ho DH, Suk JE, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562. doi:10.1038/ncomms2534
242. Daniele SG, Béraud D, Davenport C, Cheng K, Yin H, Maguire-Zeiss KA. Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci Signal. 2015;8(376):ra45. doi:10.1126/scisignal.2005965
243. Panicker N, Sarkar S, Harischandra DS, et al. Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J Exp Med. 2019;216(6):1411–1430. doi:10.1084/jem.20182191
244. Sommer A, Marxreiter F, Krach F, et al. Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson’s disease. Cell Stem Cell. 2019;24(6):1006. doi:10.1016/j.stem.2019.04.019
245. Lindestam Arlehamn CS, Dhanwani R, Pham J, et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun. 2020;11(1):1875. doi:10.1038/s41467-020-15626-w
246. Gordon R, Albornoz EA, Christie DC, et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med. 2018;10(465):eaah4066. doi:10.1126/scitranslmed.aah4066
247. Fellner L, Irschick R, Schanda K, et al. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia. 2013;61(3):349–360. doi:10.1002/glia.22437
248. Mackenzie KJ, Carroll P, Martin CA, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548(7668):461–465. doi:10.1038/nature23449
249. Glück S, Guey B, Gulen MF, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19(9):1061–1070. doi:10.1038/ncb3586
250. Welch G, Tsai LH. Mechanisms of DNA damage-mediated neurotoxicity in neurodegenerative disease. EMBO Rep. 2022;23(6):e54217. doi:10.15252/embr.202154217
251. Gonzalez-Hunt CP, Sanders LH. DNA damage and repair in Parkinson’s disease: recent advances and new opportunities. J Neurosci Res. 2021;99(1):180–189. doi:10.1002/jnr.24592
252. Li YL, Wang ZX, Ying CZ, Zhang BR, Pu JL. Decoding the role of familial Parkinson’s disease-related genes in DNA damage and repair. Aging Dis. 2022;13(5):1405–1412. doi:10.14336/AD.2022.0216
253. Milanese C, Cerri S, Ulusoy A, et al. Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death Dis. 2018;9(8):1–12. doi:10.1038/s41419-018-0848-7
254. El-Saadi MW, Tian X, Grames M, et al. Tracing brain genotoxic stress in Parkinson’s disease with a novel single-cell genetic sensor. Sci Adv. 2022;8(15):eabd1700. doi:10.1126/sciadv.abd1700
255. Ludtmann MHR, Angelova PR, Horrocks MH, et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat Commun. 2018;9(1):2293. doi:10.1038/s41467-018-04422-2
256. Wang ZX, Li YL, Pu JL, Zhang BR. DNA Damage-Mediated Neurotoxicity in Parkinson’s Disease. Int J Mol Sci. 2023;24(7):6313. doi:10.3390/ijms24076313
257. Vasquez V, Mitra J, Hegde PM, et al. Chromatin-bound oxidized α-synuclein causes strand breaks in neuronal genomes in in vitro models of Parkinson’s disease. J Alzheimers Dis JAD. 2017;60(s1):S133–S150. doi:10.3233/JAD-170342
258. Kam TI, Mao X, Park H, et al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science. 2018;362(6414):eaat8407. doi:10.1126/science.aat8407
259. Jeltema D, Abbott K, Yan N. STING trafficking as a new dimension of immune signaling. J Exp Med. 2023;220(3):e20220990. doi:10.1084/jem.20220990
260. Ferecskó AS, Smallwood MJ, Moore A, et al. STING-triggered CNS inflammation in human neurodegenerative diseases. Biomedicines. 2023;11(5):1375. doi:10.3390/biomedicines11051375
261. Rothfuss O, Fischer H, Hasegawa T, et al. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum Mol Genet. 2009;18(20):3832–3850. doi:10.1093/hmg/ddp327
262. Kao SY. Regulation of DNA repair by parkin. Biochem Biophys Res Commun. 2009;382(2):321–325. doi:10.1016/j.bbrc.2009.03.048
263. Kao SY. DNA damage induces nuclear translocation of parkin. J Biomed Sci. 2009;16(1):67. doi:10.1186/1423-0127-16-67
264. Zhao Y, Simon M, Seluanov A, Gorbunova V. DNA damage and repair in age-related inflammation. Nat Rev Immunol. 2023;23(2):75–89. doi:10.1038/s41577-022-00751-y
265. Pezone A, Olivieri F, Napoli MV, Procopio A, Avvedimento EV, Gabrielli A. Inflammation and DNA damage: cause, effect or both. Nat Rev Rheumatol. 2023;19(4):200–211. doi:10.1038/s41584-022-00905-1
266. Schaser AJ, Osterberg VR, Dent SE, et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci Rep. 2019;9(1):10919. doi:10.1038/s41598-019-47227-z
267. Yoon YS, You JS, Kim TK, et al. Senescence and impaired DNA damage responses in alpha-synucleinopathy models. Exp Mol Med. 2022;54(2):115–128. doi:10.1038/s12276-022-00727-x
268. Abbotts R, Madhusudan S. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat Rev. 2010;36(5):425–435. doi:10.1016/j.ctrv.2009.12.006
269. Miner KM, Jamenis AS, Bhatia TN, et al. α-synucleinopathy exerts sex-dimorphic effects on the multipurpose DNA repair/redox protein APE1 in mice and humans. Prog Neurobiol. 2022;216:102307. doi:10.1016/j.pneurobio.2022.102307
270. Thakur S, Dhiman M, Tell G, Mantha AK. A review on protein-protein interaction network of APE1/Ref-1 and its associated biological functions. Cell Biochem Funct. 2015;33(3):101–112. doi:10.1002/cbf.3100
271. Reginato G, Cejka P. The MRE11 complex: a versatile toolkit for the repair of broken DNA. DNA Repair. 2020;91–92:102869. doi:10.1016/j.dnarep.2020.102869
272. Brandebura AN, Paumier A, Onur TS, Allen NJ. Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders. Nat Rev Neurosci. 2023;24(1):23–39. doi:10.1038/s41583-022-00641-1
273. Giordano AMS, Luciani M, Gatto F, et al. DNA damage contributes to neurotoxic inflammation in Aicardi-Goutières syndrome astrocytes. J Exp Med. 2022;219(4):e20211121. doi:10.1084/jem.20211121
274. Song X, Ma F, Herrup K. DNA damage-induced neuroinflammation in neurodegenerative disease. Alzheimers Dement. 2021;17(S3):e055175. doi:10.1002/alz.055175
 © 2024 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.
© 2024 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.