")
Back to Journals » Journal of Inflammation Research » Volume 17
Novel Insights into the Kallikrein–Kinin System in Fulminant Myocarditis: Physiological Basis and Potential Therapeutic Advances
Authors Ji M , Ran X, Zuo H, Zhang Q
Received 23 July 2024
Accepted for publication 8 October 2024
Published 15 October 2024 Volume 2024:17 Pages 7347—7360
DOI https://doi.org/10.2147/JIR.S488237
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Ning Quan
Mengmeng Ji,1,* Xiao Ran,2,3,* Houjuan Zuo,1 Qin Zhang4
1Division of Cardiology, Department of Internal Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China; 2Department of Critical-Care Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China; 3Department of Emergency Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 4Department of Anesthesiology, Hubei Key Laboratory of Geriatric Anesthesia and Perioperative Brain Health, and Wuhan Clinical Research Center for Geriatric Anesthesia, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qin Zhang, Department of Anesthesiology, Hubei Key Laboratory of Geriatric Anesthesia and Perioperative Brain Health, and Wuhan Clinical Research Center for Geriatric Anesthesia, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095# Jiefang Ave, Wuhan, 430030, People’s Republic of China, Tel +86-15717154768, Email [email protected] Houjuan Zuo, Division of Cardiology, Department of Internal Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095# Jiefang Ave, Wuhan, 430030, People’s Republic of China, Tel +86-83663280, Email [email protected]
Abstract: Fulminant myocarditis (FM) is characterized by rapid cardiac deterioration often instigated by an inflammatory cytokine storm. The kallikrein–kinin system (KKS) is a metabolic cascade known for releasing vasoactive kinins, such as bradykinin-related peptides, possessing diverse pharmacological activities that include inflammation, regulation of vascular permeability, endothelial barrier dysfunction, and blood pressure modulation. The type 1 and type 2 bradykinin receptors (B1R and B2R), integral components of the KKS system, mediate the primary biological effects of kinin peptides. This review aims to offer a comprehensive overview of the primary mechanisms of the KKS in FM, including an examination of the structural components, regulatory activation, and downstream signaling pathways of the KKS. Furthermore, it explores the involvement of the tissue kallikrein/B1R/inducible nitric oxide synthase (TK/B1R/iNOS) pathway in myocyte dysfunction, modulation of the immune response, and preservation of endothelial barrier integrity. The potential therapeutic advances targeting the inhibition of the KKS in managing FM will be discussed, providing valuable insights for the development of clinical treatment strategies.
Keywords: fulminant myocarditis, Kallikrein–Kinin system, inducible nitric oxide synthase, nitric oxide, inflammatory
Graphical Abstract:

Background
Fulminant myocarditis (FM) is a rare yet severe cardiac inflammatory disease characterized by inflammatory infiltration and myocardial damage. It is often triggered by infectious agents or autoimmune disorders, particularly viral infectious.1–3 This review focuses primarily on viral myocarditis. The detrimental effects of infectious pathogens can hyperactivate the immune response, accelerating disease progression.4 This excessive immune activation can cause severe myocardial injury, leading to FM. Inflammatory infiltration and myocardial damage represent the primary pathogenic mechanisms of FM, with immunomodulatory therapy emerging as an effective and dependable treatment option.5–7 Nonetheless, the limited understanding of the pathophysiological mechanisms underlying FM has impeded the development of comprehensive therapeutic strategies that could intervene effectively at various disease mediators.
The Kallikrein–Kinin System (KKS) is an enzymatic cascade of molecules intricately linked to the activation of the coagulation and renin-angiotensin-aldosterone system (RAAS) pathways, influencing vascular permeability and inflammation.8 The KKS is an important endothelial autacoid system that exerts several beneficial protective actions on the endothelium, such as preventing the formation of thrombi and possibly the formation of atherosclerotic plaques.9 Kinins exert their effects through two types of kinin receptors: the predominant type 2 bradykinin receptor (B2R) and the injury-induced type 1 bradykinin receptor (B1R). Bradykinin receptor (BKR) signaling within the KKS is associated with vasodilation, hypotension, increased vascular permeability, edema formation, angiogenesis, and pain, commonly observed during viral infections. Cell death from viral infection and activation of innate immune cells can trigger the release of KKS activators, leading to the production of vasoactive kinin peptides. Our research indicates that the B1R of the KKS can activate inducible nitric oxide synthase (iNOS) through the p21ras/rapidly accelerated fibrosarcoma protein kinase/mitogen-activated protein kinase extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase/iNOS/nitric oxide (Ras/Raf/MEK/ERK/iNOS/NO) signaling pathway, eliciting robust and sustained responses that release significant amounts of NO, mediating inflammatory responses and enhancing vascular permeability.10 iNOS, a key enzyme in NO production under pathological conditions,11 is increasingly associated with progressive myocardial damage in myocarditis.12,13 Upregulation of iNOS production plays a crucial role during cardiac stress, such as in myocarditis, bridging the gap between the pathophysiological changes in myocarditis and the molecular alterations of the KKS. Therefore, the tissue kallikrein (TK) /B1R/iNOS pathway is emerging as a significant route involved in inducing proinflammatory responses triggered by viruses.
The purpose of this review is to reveal the potential correlation between the inflammatory cascade in FM and the activation of the KKS. It is proposed that the activation of the TK/B1R/iNOS signaling pathway could play a crucial role in driving the inflammatory response in FM. Furthermore, the review discusses the therapeutic implications of targeting the B1R/iNOS signaling axis in the clinical management of FM.
Pathophysiological Mechanism of FM
FM is primarily triggered by viral infections, which induce viral injury and evoke an immune response, contributing to its development.14 Viral replication in cardiac myocytes directly results in tissue injury15 (Figure 1). Initially, viral replication and transcription result in cardiomyocyte destruction, causing cellular damage and the release of intracellular components.16 This activates the innate immune response through pattern recognition receptors such as toll-like receptors (TLRs) (Figure 1). For instance, Coxsackievirus B3 (CVB3) can stimulate neutrophils via TLR8, initiating nuclear factor kappa-B (NF-κB) activation17 (Figure 1) and subsequent production of inflammatory cytokines, thus triggering an immune response.18
 |
Figure 1 Pathophysiological mechanism of fulminant myocarditis (FM) and the cytokine storm cascade. FM triggers an intense inflammatory response driven by various factors, notably infections. The process initiates with the activation of inflammatory pathways such as Toll-like receptors (TLRs), leading to the generation of pro-inflammatory cytokines like IL-1, IL-6, and TNF-α. This surge in cytokines amplifies inflammation, activating immune cells such as macrophages and neutrophils. Inflammation within cardiomyocytes results in mitochondrial dysfunction, accumulation of reactive oxygen species (ROS), and cell death, consequently affecting heart function and potentially leading to heart failure. Endothelial cells play a crucial role in this mechanism by producing nitric oxide (NO). The release of internal components due to cell death triggers a subsequent wave of cytokine release, exacerbating inflammation. Therapeutic approaches, such as the use of neutralizing antibodies targeting vital cytokines, are designed to mitigate the cytokine storm, reduce inflammation, and safeguard cardiac function. |
The innate immune system further activates the acquired immune response by initiating the activation and proliferation of T- and B-cells (Figure 1). Neutralizing antibodies play a vital role in inhibiting viral replication in the heart and other organs. Concurrently, autoantibodies, particularly those targeting mitochondrial and contractile proteins, are produced by activated B cells to assist in viral clearance. Nevertheless, autoantibodies can detrimentally affect myocyte function.19 Proinflammatory cytokines and infiltration of antigen-specific T lymphocytes additionally aggravate myocardial inflammation and necrosis, ultimately leading to ventricular dysfunction20 (Figure 1). In cases of persistent viral replication, a continuous cycle of cell-mediated immunity activation attracts a variety of immune cells to the site of injury. The excessive production of inflammatory cytokines, along with antibodies targeting viral and cardiac proteins, worsens cardiac damage and impairs systolic function.
Besides cytokines, NO plays a significant role in FM pathogenesis, particularly in immunocompromised individuals. Excessive NO production, facilitated by iNOS, can exacerbate the severity of myocarditis (Figure 1).21 Interventions targeting iNOS activity have displayed promise in preserving contractile function during ischemic conditions, underscoring the intricate interplay of factors in myocarditis pathophysiology. Extensive research has shed light on the mechanisms involved in FM; however, the in-depth comprehension of the pathophysiological processes associated with FM remains limited and warrants further investigation. The significant contribution of iNOS/NO to the pathogenesis of FM emphasizes the crucial need to comprehend and target this mechanism to improve clinical management strategies.
Immune Response and Inflammatory Cytokines in FM
In FM, inflammatory infiltration and myocardial damage are significant pathogenic mechanisms influenced by various inflammatory cytokines. Cytokines and chemokines act as essential mediators in the inflammatory response by quickly responding to infection. Elevated levels of proinflammatory cytokines like tumor necrosis factor-α (TNF-α), Interleukin-1β (IL-1β), and Interleukin-6 (IL-6) are found in FM patients, present in the myocardium during infection.22 Initially triggering a protective and healing process, prolonged expression of these cytokines, along with immune cell infiltration, can have harmful effects.
The term “cytokine storm” describes the disrupted immune balance in FM, impacting myocardial contraction and function4 (Figure 1). Cytokines such as IL-1, IL-2, TNF-α, and Interferon-γ (IFN-γ) negatively affect heart function by reducing muscle contractility and promoting cell death.23 Inflammatory activation is common in various types of myocardial damage,24 with evidence of immune cell infiltration in necrotic areas.4
The severity of myocardial damage and distribution of inflammatory cells in FM are closely linked, suggesting that extensive myocyte damage from inflammation contributes to the rapid progression of myocarditis. Inflammatory markers, erythrocyte sedimentation rate (ESR) and high-sensitive c-reactive protein (hs-CRP), played the most important role in diagnosing myocarditis in the Diagnosis of Acute Myocarditis in Emergency (DAME) score,25 which corroborates our viewpoint. Overstimulation of the immune response by infectious agents accelerates disease progression, resulting in sustained inflammation, endothelial dysfunction, tissue damage, and heart remodeling.26 Our recent research has shown that the TK/B1R/iNOS pathway plays a significant role in cardiac inflammation and endothelial dysfunction following reperfusion injury, potentially exacerbating inflammation and endothelial permeability.10 It is hypothesized that the upregulation of proinflammatory mediators in FM may trigger this pathway, resulting in increased NO production, further worsening inflammation and ventricular injury. Subsequently, the activation of downstream signaling cascades may amplify the release of proinflammatory factors, establishing a feedback loop that contributes to cytokine storms, worsening myocardial injury, and hastening the progression of FM (Figure 1).
Role of the KKS in Inflammation and Cardiovascular System
The KKS is a part of the humoral defense system that participate in the inflammatory response. In mild, acute insults, kallikreins and kinins play a salutary role recruiting to the extravascular milieu proteases, acute phase proteins, and neutrophils. In severe inflammation, however, the same system amplifies the inflammatory cascade, and contributes to tissue destruction and chronic inflammation.
The KKS involves a series of enzymatic reactions that generate bioactive kinin peptides.27 When activated, the KKS releases kinin peptides from low/high-molecular-weight kininogen, which are further processed by kallikreins to generate BK and Lys-BK in response to factors like infection, tissue trauma, or inflammation. Kininase I (carboxypeptidase N, CPN) and carboxypeptidase M(CPM) remove arginine from the carboxyl terminus of BK and kallidin, producing des-Arg9-bradykinin(des-Arg9-BK) and des-Arg10-kallidin, respectively27 (Figure 2). These derivatives exhibit affinity for receptors such as B1R, whereas BK predominantly binds to B2R. Both receptor types, B1R and B2R, are G-protein coupled receptors that play BK crucial roles in inflammation, vascular function, blood pressure regulation, and pain response.
 |
Figure 2 The Kallikrein-Kinin System (KKS) and the Inflammatory Cascade. The figure primarily elucidates the KKS and its associated signaling pathways that regulate inflammation, with a specific focus on the B1R/iNOS pathway in fulminant myocarditis. The pathways controlled by B1R and iNOS are emphasized, demonstrating their roles in modulating crucial inflammatory processes. B1R activation triggers the production of reactive oxygen species (ROS) and subsequent mitochondrial phosphorylation, thus contributing to the inflammatory response. The signaling cascade involves Gai, MAPK, and ERK1/2, culminating in iNOS expression and the formation of peroxynitrite, which exerts negative inotropic effects and enhances cytotoxicity. Furthermore, interactions with pathways regulated by EGF and TGF-β are indicated, elucidating the intricate network of interactions within the KKS system responsible for modulating myocardial inflammation. Additionally, the figure underscores the significance of related signaling pathways and molecules like B2R, PI3K, eNOS, PLC, PLA2, cAMP, COX-2, PGE2, and MMP-9 in processes such as vascular relaxation, permeability, cytokine release, and cardiac fibrosis. |
B2R is widely distributed and exerts a crucial function in various physiological processes by activating diverse signaling pathways, including the synthesis of NO and prostaglandins(PGs).27 Upon binding with BK, B2R interacts with multiple G proteins, such as Gαqs, Gαq/Gα11, Gαi1, and Gβ1γ2, leading to the activation of Gαq and Gαi proteins (Figure 2). This interaction triggers a series of events involving the activation of molecules like phospholipases, protein kinase C (PKC), and phosphoinositide 3-kinase/ protein kinase B (PI3K/AKT), as well as the release of secondary messengers such as inositol-1,4,5-trisphosphate and diacylglycerol.28,29 These pathways can activate endothelial NO synthase (eNOS) through Akt phosphorylation at Ser117730 (Figure 2) and induce the expression of NF-κB and Cyclooxygenase-2 (COX-2) through the cyclic adenosine monophosphate (cAMP) response element and the Ras/Raf-1/ERK pathway. The BK-B2R signaling pathway plays a vital role in protecting the heart from ischemic damage, as evidenced by the cardioprotective effects of TK or kinin infusion against cardiac remodeling, apoptosis, and fibrosis mediated by B2R-NO. However, it is crucial to note that BK can also stimulate the production of inflammatory mediators through NF-κB and cAMP response element-induced COX-2, leading to potentially adverse outcomes (Figure 2). These signaling pathways are implicated in the inflammatory side effects associated with B2R signaling. Moreover, B2R can undergo temporary desensitization through phosphorylation, which induces endocytosis followed by receptor recycling. In contrast, B1R lacks this desensitization mechanism, as it is not susceptible to phosphorylation, potentially resulting in prolonged signaling duration.31
B1Rs are typically sparse or absent in normal tissues, but exhibit heightened responsiveness and rapid upregulation following exposure to inflammatory stimuli, infections, trauma, or agents like lipopolysaccharide endotoxin.32 These receptors mainly modulate vasodilation and endothelial permeability. Studies employing specific agonists and antagonists have shown that B1R activation induces vascular leakage in diverse peripheral organs.33,34 An important role of B1R is its contribution to the recruitment of inflammatory cells. Recent investigations, conducted both in vivo and in vitro, indicate that stimulating B1R in neutrophils during inflammation promotes their attachment and movement to nearby tissues. The interaction of des-Arg9-BK with B1R leads to heightened vascular permeability,35 enhanced neutrophil attraction, and triggers broncho- and vasoconstriction, sparking inflammation.36
BK is acknowledged for its critical involvement in specific cardiovascular disorders.37 It has been implicated in the pathogenesis of septic shock syndrome due to its elevated production following infection and its established effects in inducing hypotension and plasma extravasation.
The excessive presence of BK can induce vasodilation, heightened vascular permeability, and hypotension, ultimately culminating in severe manifestations such as lung edema and cardiovascular dysfunction. Consequently, BK is believed to primarily impact the cardiovascular system through vasodilation and plasma leakage, actively contributing to the inflammatory cascade. Collectively, the KKS serves as a pivotal regulator of inflammatory pathways and is influenced by innate immune response elements.35 Previous research has highlighted the pivotal role of an inflammatory cytokine storm in the pathogenesis of FM.4 Elevated levels of BK have been consistently associated with heightened inflammation, oxidative stress, endothelial dysfunction, and fibrosis, all of which are prominent clinical features of FM.
Inflammatory Cascade and KKS Activation in FM
The initiation of the inflammatory cascade in FM involves the activation of the inflammasome within the innate immune response, induced by diverse pathogens or stimuli. This activation facilitates the transmission of signals from pathogen-associated molecular patterns and external stimuli into the cell, where they are amplified through the MAPK pathway.38 ERK, a crucial member of MAPK family, has been implicated in the development of inflammatory diseases, including cardiac injury. Studies have shown that the proteolytic cleavage of Ras GTPase-activating protein RasGAP by CVB3 leads to the activation of the MAPK ERK-1/2 pathway via a Raf-1/MEK-1-dependent mechanism39 (Figures 1 and 2). Additionally, ERK has been observed to phosphorylate NF-κB, resulting in the transcription of various inflammatory molecules. Inhibition of ERK1/2 has been shown to block virus replication, highlighting its significance in the inflammatory response.40
Furthermore, the production of reactive oxygen species (ROS), predominantly derived from mitochondrial oxidative phosphorylation, additionally stimulates NF-κB, sustaining the inflammatory cascade41 (Figure 2). Another important protein kinase involved in cell survival and cellular balance is Akt, which is activated in cardiomyocytes during CVB3 infection through a specific pathway involving PI3K and integrin-linked kinase. This activation of Akt has been studied for its role in the regulation of downstream effectors in the PI3K/Akt pathway.42
Interestingly, these signaling pathways intersect significantly with the KKS, where BK-induced activation of MAPKs contributes to the upregulation of various cytokines.43,44 This suggests a pivotal role for BK in the activation of inflammatory responses. The KKS can be activated on endothelial cell surfaces, along with the coagulation cascade, with the B1R showing pro-inflammatory characteristics that promote inflammation by increasing cytokine production and immune cell migration, as well as inducing vascular permeability (Figure 2). Transcription factors like NF-κB and activator protein 1 (AP-1), along with the TGF-β1/MAPKs signaling pathway, regulate the expression of the B1R during inflammatory processes.45 Research has shown that inhibitors targeting different MAPKs and NF-κB can reduce the overexpression of B1Rs46,47 (Table 1).10–12,22,35,47–51 Various stimuli, including inflammatory cytokines, innate immune system activators, growth factors, and phorbol esters activator of PKC, can trigger an increase in B1R expression in vascular cells.52 Activation of the B1R has been linked to oxidative stress through nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase activation in the vasculature.50 Notably, B1R knockout has been shown to provide protection against hypotension, pain sensitivity, and injury in different experimental models.53 It can also lead to the post-translational activation of iNOS through specific intracellular pathways, suggesting a significant role in myocardial inflammation and other biological effects mediated by kinins (Figure 2).
 |
Table 1 The B1R/iNOS Pathway of KKS in the Pathogenesis of Inflammation and Cardiovascular Diseases |
NO Production and iNOS Activation in Myocarditis
Myocarditis, a condition characterized by myocardial inflammation, is associated with the upregulation of proinflammatory cytokines that contribute to tissue damage and modulation of immune responses. NO functions as a pivotal mediator in this pathophysiological cascade, serving as a signaling molecule in the host’s antiviral defense mechanisms.
The vasodilatory and antioxidant properties of NO have been demonstrated to mitigate mitochondrial oxidant damage in adult rat cardiomyocytes.54 Furthermore, NO can inhibit the production of neutrophil superoxide anions by directly acting on the membrane components of NADPH oxidase and the assembly of dihydronicotinamide adenine dinucleotide (NADH)/NADPH oxidase subunits.55 It also mediates the effects of TK in improving cardiac function and limiting post-infarction remodeling by inhibiting inflammation.56 Activation of the B2R/eNOS pathway leads to a transient release of NO, contributing to improved cardiac function.9
Conversely, activation of the B1R/iNOS signaling pathway in response to acute inflammation leads to persistent generation of high levels of NO, resulting in edema, inflammation, pain, and excessive ROS production. The presence of activated macrophages and neutrophils in inflammatory and autoimmune lesions can exacerbate NO production, further damaging tissues57 (Figure 1). Recent animal studies have underscored the detrimental effects of increased NO production, driven by proinflammatory cytokines, on heart muscle function, leading to reduced contractility and impaired coronary autoregulation and oxygen utilization.58–60
iNOS is primarily regulated at the expression level, induced in response to inflammatory mediators such as lipopolysaccharide (LPS) or cytokines like IL-1β, IL-6, and IFN-γ.61 Activation of iNOS through B1R mediated MAPK ERK signaling significantly increases NO production, indicating an alternative regulatory mechanism beyond conventional modes of regulation49 (Figure 2).
Various mechanisms regulate iNOS activity, including post-translational modifications.61 S-nitrosylation can inhibit iNOS activity,62 while interaction with heat shock protein 90 (hsp90) can enhance it.63 Once iNOS is expressed, it continuously produces NO until undergoing degradation.61 This unregulated production of NO by iNOS can have cytotoxic effects, particularly in the context of the host defense response.
Role of B1R/iNOS Pathway in FM
Recent research has shed light on the connection between iNOS and the KKS in FM pathophysiology. Studies indicate that inhibiting iNOS can reduce autophagy, apoptosis, and oxidative stress while promoting cell proliferation in cardiomyocytes infected with CVB3. Additionally, it has been found that reducing iNOS activity leads to a decrease in oxidative stress and improvement in cardiac function affected by cytokines.48 Moreover, research indicates that overproduction of NO due to iNOS expression can hinder mitochondrial function and contribute to reduced mechanical performance during prolonged cytokine exposure58 (Figures 1 and 2). In agreement with this, mutant mice lacking iNOS were resistant to LPS-induced mortality.64 Furthermore, studies have revealed that the integrity of the blood-brain barrier (BBB) can be influenced by the TK via B1R/iNOS signaling pathway, which can impact various cellular processes.65
Activation of B1R has been linked to the induction of Matrix metallopeptidase 9 (MMP-9) through the ERK MAP kinase pathway66 (Figures 1 and 2), which plays a crucial role in neurovascular damage and BBB disruption. Excessive MMP-9 expression can lead to BBB injury and inflammation by promoting inflammatory cell migration It has been suggested that ERK/NF-κB/MMP-9 pathway may be downstream of the B1R signal in FM pathogenesis.67,68
Nevertheless, some studies have presented conflicting results, low to moderate doses of iNOS inhibitors restore myocardial contractility in hearts exposed to proinflammatory cytokines, whereas at higher doses, the effect reverses itself. This finding may indicate that small amounts of NO produced by iNOS may be necessary to maintain contractility.58,69 NO donors may reduce inflammation by limiting cytokine-induced endothelial activation, leukocyte adherence, and microvascular permeability alterations70 (Figure 2). The effect caused by iNOS may be depend on the amount of NO produced. Collectively, recent studies have deepened our understanding of how the B1R/iNOS pathway contributes to inflammation in FM. These findings suggest that B1R-induced leucocyte recruitment and increased NO production are key factors in inflammatory cardiovascular diseases. The continuous self-amplification of this pathway after the initial inflammatory trigger may sustain and aggravate the inflammatory response.
Therapeutic Implications of Targeting KKS in FM
Kinins have long been recognized for inducing various features of acute inflammation, including microvascular permeability, leukocyte migration, and apoptosis71 (Table 2).67,72–77 This leads to a wide range of potential therapeutic targets, such as hypertension9,78 ischemic diseases, diabetic complications, hereditary or acquired inflammatory diseases,79 and brain diseases.75 Hara et al demonstrated that selective, orally active, nonpeptide BR1 antagonist SSR240612 significantly reduced intestinal tissue damage and neutrophil influx in a mouse model of colitis.77 Moreover, a selective B2R agonist showed cardioprotective effects in post-acute myocardial infarction.80 Although the clinical development of these drugs is in the early stages and limited human clinical studies have been reported so far.
 |
Table 2 Summary of Therapeutic Effects of Pharmacological B1 or B2 Receptor Agonists and Antagonists in Inflammation and Cardiovascular Diseases |
Kinins, as inflammatory mediators implicated in vascular responses, play a crucial role in inflammation as observed in FM. The dual effects of kinin receptors, particularly B1R, raise an unsettled issue on the therapeutic value of B1R agonists versus antagonists in cardiovascular diseases. Targeting kinin receptors, especially the B1R, presents a promising therapeutic approach for FM (Figure 3). By blocking the B1R/iNOS pathway and regulating NO production, it may be possible to alleviate cardiac inflammation and endothelial dysfunction associated with FM. In addition to B1R antagonists, inhibiting CPM to reduce B1R agonists generation could serve as a novel therapeutic strategy (Figure 3). This approach may enhance the cardioprotective and anti-diabetic effects of the B2R by preventing the cleavage of certain kinins. Developing kininase 1 inhibitors could also be beneficial for pharmacotherapy in FM9 (Figure 3). Research indicates that inhibiting MAPK-associated pathways and reducing iNOS expression or activity could help in reducing fibrosis and improving cardiac inflammation. Strengthening the effects of the B2R, which may protect against heart disorders from oxidative stress and reduce ROS, while simultaneously inhibiting the B1R, could be advantageous for managing cardiovascular diseases.81,82
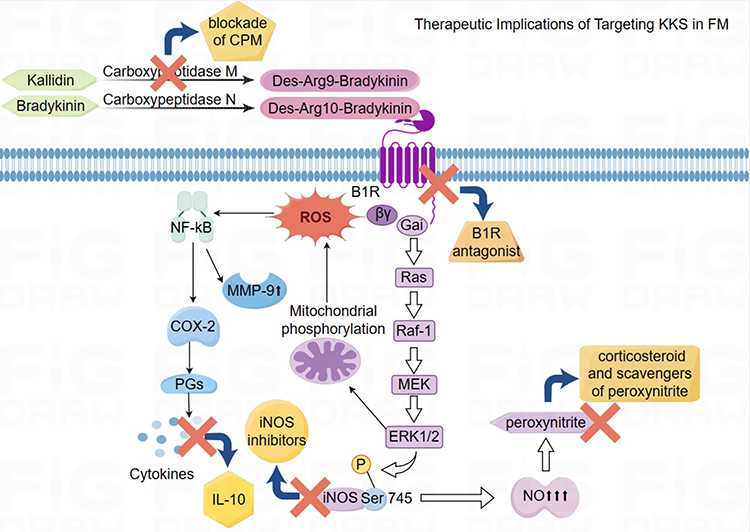 |
Figure 3 Therapeutic Strategies Targeting Kallikrein–Kinin System (KKS) in Fulminant Myocarditis (FM). The figure illustrates key treatments for FM targeting the KKS, focusing on B1 receptor (B1R) and inducible nitric oxide synthase (iNOS) signaling. The use of B1R antagonists and selective iNOS blockers is crucial for regulating the KKS and reducing nitric oxide production, which are essential for addressing inflammation and oxidative stress in FM. Moreover, carboxypeptidase enzymes aid in decreasing inflammation levels by breaking down kinins. The combination of corticosteroids and peroxynitrite scavengers, which counteract harmful peroxynitrite effects, helps reduce inflammation and maintain cardiac function in FM cases. Additionally, the inclusion of interleukin-10 (IL-10) assists in resolving inflammation linked to FM. A comprehensive therapeutic approach centered on B1R inhibition is critical for minimizing reactive oxygen species (ROS) generation and modulating downstream signaling pathways, such as the Ras-Raf-MEK-ERK1/2 axis, in FM. |
To safeguard the positive impacts of the KKS while minimizing adverse effects, selective iNOS inhibitors like aminoguanidine could provide protection against the harmful effects of NO and PGs production.83 Additionally, the use of corticosteroids and NO scavengers may be beneficial in reducing cardiac damage resulting from cytokine exposure in FM patients. It is worth to note that elevated cytokine levels, including IL-10, in FM patients present opportunities for cytokine-targeted treatments. IL-10 has shown to regulate iNOS mRNA production, preventing ongoing myocardial injury. Studies suggest that IL-10 plays a protective role in myocarditis and can modulate macrophage activities to limit cardiomyocyte destruction.84 Further exploration into the therapeutic implications of IL-10 in inflammatory cardiomyopathy, including administration methods and potential side effects, is warranted.85
Conclusion and Prospect
The upregulation of iNOS is a critical factor in the pathogenesis of FM, affecting myocardial function and contributing to inflammation and compromised endothelial barrier integrity. The KKS was considered to participate in the inflammatory cascade of FM by activating the B1R/iNOS signaling pathway. Activation of the B1R-dependent iNOS results in excessive NO production. This pathway is influenced by inflammatory signals triggered by the host’s response to viral infection, modulating the release of inflammatory molecules and balancing anti-inflammatory responses against myocardial damage. Future investigations could explore the complex molecular mechanisms and pathways related to B1R/iNOS in FM, potentially yielding novel insights into diagnostic and therapeutic strategies for improving FM management.
Abbreviations
FM, Fulminant myocarditis; KKS, kallikrein–kinin system; TK, tissue kallikrein; BK, bradykinin; BKR, bradykinin receptor; B1R, type 1 bradykinin receptor; B2R, type 2 bradykinin receptor; iNOS, inducible nitric oxide synthase; RAAS, renin-angiotensin-aldosterone system; Ras, p21ras; Raf, rapidly accelerated fibrosarcoma protein kinase; MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; MEK, MAPK ERK kinase; NO, nitric oxide; TLRs, toll-like receptors; CVB3, Coxsackievirus B3; CPN, carboxypeptidase N; CPM, carboxypeptidase M; PGs, prostaglandins; eNOS, endothelial NO synthase; PKC, protein kinase C; PI3K/Akt, Phosphatidylinositol 3-kinase/protein kinase B; MAPK, mitogen-activated protein kinase; ERKs, extracellular signal-regulated kinases; ROS, reactive oxygen species; AP-1, activator protein 1; NADPH, nicotinamide adenine dinucleotide phosphate hydrogen; NADH, dihydronicotinamide adenine dinucleotide; hsp90, heat shock protein 90; TNF, tumor necrosis factor; IFN-γ, Interferon-γ; ESR, erythrocyte sedimentation rate; hs-CRP, high-sensitive c-reactive protein; DAME score, the Diagnosis of Acute Myocarditis in Emergency score; PAF, platelet-activating factor; BBB, Blood–Brain Barrier; NF-κb, nuclear factor kappa-B; MMP-9, Matrix metallopeptidase 9; LPS, Lipopolysaccharide; IL, Interleukin; GTP, Guanosine triphosphate; mRNA, messenger Ribonucleic Acid; cAMP, Cyclic adenosine monophosphate; COX-2, Cyclooxygenase-2; TGF-β1, transforming growth factor-β1.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by funding from National Natural Science Foundation of China (82271358) and Natural Science Foundation of Hubei Province (2024AFB642).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Kociol RD, Cooper LT, Fang JC, et al. Recognition and Initial Management of Fulminant Myocarditis: a Scientific Statement From the American Heart Association. Circulation. 2020;141(6):e69–e92. doi:10.1161/CIR.0000000000000745
2. Ammirati E, Varrenti M, Veronese G, et al. Prevalence and outcome of patients with acute myocarditis and positive viral search on nasopharyngeal swab. Eur J Heart Fail. 2021;23(7):1242–1245. doi:10.1002/ejhf.2247
3. Ammirati E, Moslehi JJ. Diagnosis and Treatment of Acute Myocarditis: a Review. JAMA. 2023;329(13):1098–1113. doi:10.1001/jama.2023.3371
4. Hang W, Chen C, Seubert JM, Wang DW. Fulminant myocarditis: a comprehensive review from etiology to treatments and outcomes. Signal Transduct Target Ther. 2020;5(1):287. doi:10.1038/s41392-020-00360-y
5. Ammirati E, Veronese G, Bottiroli M, et al. Update on acute myocarditis. Trends Cardiovasc Med. 2021;31(6):370–379. doi:10.1016/j.tcm.2020.05.008
6. Wang D, Li S, Jiang J, et al. Chinese society of cardiology expert consensus statement on the diagnosis and treatment of adult fulminant myocarditis. Sci China Life Sci. 2019;62(2):187–202. doi:10.1007/s11427-018-9385-3
7. He W, Zhou L, Xu K, et al. Immunopathogenesis and immunomodulatory therapy for myocarditis. Sci China Life Sci. 2023;66(9):2112–2137. doi:10.1007/s11427-022-2273-3
8. Nokkari A, Abou-El-Hassan H, Mechref Y, et al. Implication of the Kallikrein-Kinin system in neurological disorders: quest for potential biomarkers and mechanisms. Prog Neurobiol. 2018;165–167:26–50. doi:10.1016/j.pneurobio.2018.01.003
9. Regoli D, Gobeil F. Critical insights into the beneficial and protective actions of the kallikrein-kinin system. Vascul Pharmacol. 2015;64:1–10. doi:10.1016/j.vph.2014.12.003
10. Zhang Q, Ruan H, Wang X, Luo A, Ran X. Ulinastatin attenuated cardiac ischaemia/reperfusion injury by suppressing activation of the tissue kallikrein-kinin system. Br J Pharmacol. 2024. doi:10.1111/bph.16477
11. Hua W, Chen Q, Gong F, Xie C, Zhou S, Gao L. Cardioprotection of H2S by downregulating iNOS and upregulating HO-1 expression in mice with CVB3-induced myocarditis. Life Sci. 2013;93(24):949–954. doi:10.1016/j.lfs.2013.10.007
12. Ishiyama S, Hiroe M, Nishikawa T, et al. Nitric oxide contributes to the progression of myocardial damage in experimental autoimmune myocarditis in rats. Circulation. 1997;95(2):489–496. doi:10.1161/01.CIR.95.2.489
13. Wang WZ, Matsumori A, Yamada T, et al. Beneficial Effects of Amlodipine in a Murine Model of Congestive Heart Failure Induced by Viral Myocarditis. Circulation. 1997;95(1):245–251. doi:10.1161/01.CIR.95.1.245
14. Tschöpe C, Ammirati E, Bozkurt B, et al. Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol. 2021;18(3):169–193. doi:10.1038/s41569-020-00435-x
15. Esfandiarei M, McManus BM. Molecular biology and pathogenesis of viral myocarditis. Annu Rev Pathol. 2008;3(1):127–155. doi:10.1146/annurev.pathmechdis.3.121806.151534
16. Martin U, Nestler M, Munder T, Zell R, Sigusch HH, Henke A. Characterization of coxsackievirus B3-caused apoptosis under in vitro conditions. Med Microbiol Immunol. 2004;193(2–3):133–139. doi:10.1007/s00430-003-0197-7
17. Rivadeneyra L, Charó N, Kviatcovsky D, de la Barrera S, Gómez RM, Schattner M. Role of neutrophils in CVB3 infection and viral myocarditis. J Mol Cell Cardiol. 2018;125:149–161. doi:10.1016/j.yjmcc.2018.08.029
18. Shi Y, Fukuoka M, Li G, et al. Regulatory T cells protect mice against coxsackievirus-induced myocarditis through the transforming growth factor beta-coxsackie-adenovirus receptor pathway. Circulation. 2010;121(24):2624–2634. doi:10.1161/CIRCULATIONAHA.109.893248
19. Kaya Z, Afanasyeva M, Wang Y, et al. Contribution of the innate immune system to autoimmune myocarditis: a role for complement. Nat Immunol. 2001;2(8):739–745. doi:10.1038/90686
20. Fuse K, Chan G, Liu Y, et al. Myeloid differentiation factor-88 plays a crucial role in the pathogenesis of Coxsackievirus B3-induced myocarditis and influences type I interferon production. Circulation. 2005;112(15):2276–2285. doi:10.1161/CIRCULATIONAHA.105.536433
21. Kamijo R, Harada H, Matsuyama T, et al. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science. 1994;263(5153):1612–1615. doi:10.1126/science.7510419
22. Freeman GL, Colston JT, Zabalgoitia M, Chandrasekar B. Contractile depression and expression of proinflammatory cytokines and iNOS in viral myocarditis. Am J Physiol. 1998;274(1):H249–258.
23. Hollenberg SM, Cunnion RE, Parrillo JE. The effect of tumor necrosis factor on vascular smooth muscle. In vitro studies using rat aortic rings. Chest. 1991;100(4):1133–1137. doi:10.1378/chest.100.4.1133
24. Trachtenberg BH, Hare JM. Inflammatory Cardiomyopathic Syndromes. Circ Res. 2017;121(7):803–818. doi:10.1161/CIRCRESAHA.117.310221
25. Scicchitano P, Grazioli Gauthier L, D’Agostino C, et al. The Diagnosis of Acute Myocarditis in Emergency (DAME) score: improving diagnostics within the emergency department. Eur J Intern Med. 2021;85:56–62. doi:10.1016/j.ejim.2021.01.011
26. Dennert R, Crijns HJ, Heymans S. Acute viral myocarditis. Eur Heart J. 2008;29(17):2073–2082. doi:10.1093/eurheartj/ehn296
27. Leeb-Lundberg LM, Marceau F, Müller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57(1):27–77. doi:10.1124/pr.57.1.2
28. Liao JK, Homcy CJ. The G proteins of the G alpha i and G alpha q family couple the bradykinin receptor to the release of endothelium-derived relaxing factor. J Clin Invest. 1993;92(5):2168–2172. doi:10.1172/JCI116818
29. Quitterer U, Lohse MJ. Crosstalk between Galpha(i)- and Galpha(q)-coupled receptors is mediated by Gbetagamma exchange. Proc Natl Acad Sci U S A. 1999;96(19):10626–10631. doi:10.1073/pnas.96.19.10626
30. Fontana J, Fulton D, Chen Y, et al. Domain mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circ Res. 2002;90(8):866–873. doi:10.1161/01.RES.0000016837.26733.BE
31. Marceau F, Bachelard H, Bouthillier J, et al. Bradykinin receptors: agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. Int Immunopharmacol. 2020;82:106305. doi:10.1016/j.intimp.2020.106305
32. Campos MM, Leal PC, Yunes RA, Calixto JB. Non-peptide antagonists for kinin B1 receptors: new insights into their therapeutic potential for the management of inflammation and pain. Trends Pharmacol Sci. 2006;27(12):646–651. doi:10.1016/j.tips.2006.10.007
33. Cruwys SC, Garrett NE, Perkins MN, Blake DR, Kidd BL. The role of bradykinin B1 receptors in the maintenance of intra-articular plasma extravasation in chronic antigen-induced arthritis. Br J Pharmacol. 1994;113(3):940–944. doi:10.1111/j.1476-5381.1994.tb17083.x
34. Abdouh M, Talbot S, Couture R, Hasséssian HM. Retinal plasma extravasation in streptozotocin-diabetic rats mediated by kinin B(1) and B(2) receptors. Br J Pharmacol. 2008;154(1):136–143. doi:10.1038/bjp.2008.48
35. Li H, Zhang M, Zhao Q, et al. Self-recruited neutrophils trigger over-activated innate immune response and phenotypic change of cardiomyocytes in fulminant viral myocarditis. Cell Discov. 2023;9(1):103. doi:10.1038/s41421-023-00593-5
36. van de Veerdonk FL, Netea MG, van Deuren M, et al. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. Elife. 2020;9. doi:10.7554/eLife.57555
37. Blaes N, Girolami JP. Targeting the ‘Janus face’ of the B2-bradykinin receptor. Expert Opin Ther Targets. 2013;17(10):1145–1166. doi:10.1517/14728222.2013.827664
38. Ai X, Yan J, Carrillo E, Ding W. The Stress-Response MAP Kinase Signaling in Cardiac Arrhythmias. Rev Physiol Biochem Pharmacol. 2016;172:77–100.
39. Huber M, Watson KA, Selinka HC, et al. Cleavage of RasGAP and phosphorylation of mitogen-activated protein kinase in the course of coxsackievirus B3 replication. J Virol. 1999;73(5):3587–3594. doi:10.1128/JVI.73.5.3587-3594.1999
40. Lim BK, Nam JH, Gil CO, et al. Coxsackievirus B3 replication is related to activation of the late extracellular signal-regulated kinase (ERK) signal. Virus Res. 2005;113(2):153–157. doi:10.1016/j.virusres.2005.04.018
41. Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and Age-Related Diseases: role of Inflammation Triggers and Cytokines. Front Immunol. 2018;9:586.
42. Esfandiarei M, Suarez A, Amaral A, et al. Novel role for integrin-linked kinase in modulation of coxsackievirus B3 replication and virus-induced cardiomyocyte injury. Circ Res. 2006;99(4):354–361. doi:10.1161/01.RES.0000237022.72726.01
43. Huang CD, Tliba O, Panettieri RA, Amrani Y. Bradykinin induces interleukin-6 production in human airway smooth muscle cells: modulation by Th2 cytokines and dexamethasone. Am J Respir Cell Mol Biol. 2003;28(3):330–338. doi:10.1165/rcmb.2002-0040OC
44. Meini S, Cucchi P, Catalani C, Bellucci F, Giuliani S, Maggi CA. Bradykinin and B₂ receptor antagonism in rat and human articular chondrocytes. Br J Pharmacol. 2011;162(3):611–622. doi:10.1111/j.1476-5381.2010.01062.x
45. Bachvarov DR, Hess JF, Menke JG, Larrivée JF, Marceau F. Structure and genomic organization of the human B1 receptor gene for kinins (BDKRB1). Genomics. 1996;33(3):374–381. doi:10.1006/geno.1996.0213
46. Sabourin T, Morissette G, Bouthillier J, Levesque L, Marceau F. Expression of kinin B(1) receptor in fresh or cultured rabbit aortic smooth muscle: role of NF-kappa B. Am J Physiol Heart Circ Physiol. 2002;283(1):H227–237. doi:10.1152/ajpheart.00978.2001
47. Passos GF, Fernandes ES, Campos MM, et al. Kinin B1 receptor up-regulation after lipopolysaccharide administration: role of proinflammatory cytokines and neutrophil influx. J Immunol. 2004;172(3):1839–1847. doi:10.4049/jimmunol.172.3.1839
48. Balligand JL, Ungureanu D, Kelly RA, et al. Abnormal contractile function due to induction of nitric oxide synthesis in rat cardiac myocytes follows exposure to activated macrophage-conditioned medium. J Clin Invest. 1993;91(5):2314–2319. doi:10.1172/JCI116461
49. Zhang Y, Brovkovych V, Brovkovych S, et al. Dynamic receptor-dependent activation of inducible nitric-oxide synthase by ERK-mediated phosphorylation of Ser745. J Biol Chem. 2007;282(44):32453–32461. doi:10.1074/jbc.M706242200
50. Dias JP, Talbot S, Sénécal J, Carayon P, Couture R. Kinin B1 receptor enhances the oxidative stress in a rat model of insulin resistance: outcome in hypertension, allodynia and metabolic complications. PLoS One. 2010;5(9):e12622. doi:10.1371/journal.pone.0012622
51. Lopatko Fagerström I, Ståhl AL, Mossberg M, et al. Blockade of the kallikrein-kinin system reduces endothelial complement activation in vascular inflammation. EBioMed. 2019;47:319–328. doi:10.1016/j.ebiom.2019.08.020
52. Koumbadinga GA, Petitclerc E, Bouthillier J, Adam A, Marceau F. Receptor tyrosine kinases as mediators of injury-induced bradykinin B1 receptor expression in rabbit aortic smooth muscle. Eur J Pharmacol. 2009;606(1–3):233–239. doi:10.1016/j.ejphar.2008.12.058
53. Pesquero JB, Bader M. Genetically altered animal models in the kallikrein-kinin system. Biol Chem. 2006;387(2):119–126. doi:10.1515/BC.2006.017
54. Xu Z, Park SS, Mueller RA, Bagnell RC, Patterson C, Boysen PG. Adenosine produces nitric oxide and prevents mitochondrial oxidant damage in rat cardiomyocytes. Cardiovasc Res. 2005;65(4):803–812. doi:10.1016/j.cardiores.2004.12.004
55. Clancy RM, Leszczynska-Piziak J, Abramson SB. Nitric oxide, an endothelial cell relaxation factor, inhibits neutrophil superoxide anion production via a direct action on the NADPH oxidase. J Clin Invest. 1992;90(3):1116–1121. doi:10.1172/JCI115929
56. Yin H, Chao L, Chao J. Nitric oxide mediates cardiac protection of tissue kallikrein by reducing inflammation and ventricular remodeling after myocardial ischemia/reperfusion. Life Sci. 2008;82(3–4):156–165. doi:10.1016/j.lfs.2007.10.021
57. Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19(1):683–765. doi:10.1146/annurev.immunol.19.1.683
58. Panas D, Khadour FH, Szabó C, Schulz R. Proinflammatory cytokines depress cardiac efficiency by a nitric oxide-dependent mechanism. Am J Physiol. 1998;275(3):H1016–1023. doi:10.1152/ajpheart.1998.275.3.H1016
59. Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257(5068):387–389. doi:10.1126/science.1631560
60. Avontuur JA, Bruining HA, Ince C. Inhibition of nitric oxide synthesis causes myocardial ischemia in endotoxemic rats. Circ Res. 1995;76(3):418–425. doi:10.1161/01.RES.76.3.418
61. Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol. 2004;500(1–3):255–266. doi:10.1016/j.ejphar.2004.07.030
62. Mitchell DA, Erwin PA, Michel T, Marletta MA. S-Nitrosation and regulation of inducible nitric oxide synthase. Biochemistry. 2005;44(12):4636–4647. doi:10.1021/bi0474463
63. Yoshida M, Xia Y. Heat shock protein 90 as an endogenous protein enhancer of inducible nitric-oxide synthase. J Biol Chem. 2003;278(38):36953–36958. doi:10.1074/jbc.M305214200
64. Wei XQ, Charles IG, Smith A, et al. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375(6530):408–411. doi:10.1038/375408a0
65. Zhang Q, Tan J, Wan L, et al. Increase in Blood-Brain Barrier Permeability is Modulated by Tissue Kallikrein via Activation of Bradykinin B1 and B2 Receptor-Mediated Signaling. J Inflamm Res. 2021;14:4283–4297. doi:10.2147/JIR.S322225
66. Arai K, Lee SR, Lo EH. Essential role for ERK mitogen-activated protein kinase in matrix metalloproteinase-9 regulation in rat cortical astrocytes. Glia. 2003;43(3):254–264. doi:10.1002/glia.10255
67. Sang H, Qiu Z, Cai J, et al. Early Increased Bradykinin 1 Receptor Contributes to Hemorrhagic Transformation After Ischemic Stroke in Type 1 Diabetic Rats. Transl Stroke Res. 2017;8(6):597–611. doi:10.1007/s12975-017-0552-4
68. Halade GV, Jin YF, Lindsey ML. Matrix metalloproteinase (MMP)-9: a proximal biomarker for cardiac remodeling and a distal biomarker for inflammation. Pharmacol Ther. 2013;139(1):32–40. doi:10.1016/j.pharmthera.2013.03.009
69. Potier L, Waeckel L, Vincent MP, et al. Selective kinin receptor agonists as cardioprotective agents in myocardial ischemia and diabetes. J Pharmacol Exp Ther. 2013;346(1):23–30. doi:10.1124/jpet.113.203927
70. De Caterina R, Libby P, Peng HB, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96(1):60–68. doi:10.1172/JCI118074
71. Kellermeyer RW, Graham RC. Kinins--possible physiologic and pathologic roles in man. N Engl J Med. 1968;279(14):754–759. doi:10.1056/NEJM196810032791406
72. Bartus RT, Elliott P, Hayward N, Dean R, McEwen EL, Fisher SK. Permeability of the blood brain barrier by the bradykinin agonist, RMP-7: evidence for a sensitive, auto-regulated, receptor-mediated system. Immunopharmacology. 1996;33(1–3):270–278. doi:10.1016/0162-3109(96)00070-7
73. Majima M, Hayashi I, Inamura N, Fujita T, Ogino M. A nonpeptide mimic of bradykinin blunts the development of hypertension in young spontaneously hypertensive rats. Hypertension. 2000;35(1):437–442. doi:10.1161/01.HYP.35.1.437
74. Emanueli C, Bonaria Salis M, Stacca T, et al. Targeting kinin B(1) receptor for therapeutic neovascularization. Circulation. 2002;105(3):360–366. doi:10.1161/hc0302.102142
75. Charignon D, Späth P, Martin L, Drouet C. Icatibant, the bradykinin B2 receptor antagonist with target to the interconnected kinin systems. Expert Opin Pharmacother. 2012;13(15):2233–2247. doi:10.1517/14656566.2012.723692
76. Marmarou A, Nichols J, Burgess J, et al. Effects of the bradykinin antagonist Bradycor (deltibant, CP-1027) in severe traumatic brain injury: results of a multi-center, randomized, placebo-controlled trial American Brain Injury Consortium Study Group. J Neurotrauma. 1999;16(6):431–444.
77. Hara DB, Leite DF, Fernandes ES, et al. The relevance of kinin B1 receptor upregulation in a mouse model of colitis. Br J Pharmacol. 2008;154(6):1276–1286. doi:10.1038/bjp.2008.212
78. Margolius HS. Tissue kallikreins and kinins: regulation and roles in hypertensive and diabetic diseases. Annu Rev Pharmacol Toxicol. 1989;29(1):343–364. doi:10.1146/annurev.pa.29.040189.002015
79. Stadnicki A. Intestinal tissue kallikrein-kinin system in inflammatory bowel disease. Inflamm Bowel Dis. 2011;17(2):645–654. doi:10.1002/ibd.21337
80. Marketou M, Kintsurashvili E, Papanicolaou KN, Lucero HA, Gavras I, Gavras H. Cardioprotective effects of a selective B(2) receptor agonist of bradykinin post-acute myocardial infarct. Am J Hypertens. 2010;23(5):562–568. doi:10.1038/ajh.2010.20
81. Manolis AJ, Marketou ME, Gavras I, Gavras H. Cardioprotective properties of bradykinin: role of the B(2) receptor. Hypertens Res. 2010;33(8):772–777. doi:10.1038/hr.2010.82
82. Mikrut K, Paluszak J, Kozlik J, Sosnowski P, Krauss H, Grześkowiak E. The effect of bradykinin on the oxidative state of rats with acute hyperglycaemia. Diabet Res Clin Pract. 2001;51(2):79–85. doi:10.1016/S0168-8227(00)00222-9
83. Szabó C, Ferrer-Sueta G, Zingarelli B, Southan GJ, Salzman AL, Radi R. Mercaptoethylguanidine and guanidine inhibitors of nitric-oxide synthase react with peroxynitrite and protect against peroxynitrite-induced oxidative damage. J Biol Chem. 1997;272(14):9030–9036. doi:10.1074/jbc.272.14.9030
84. Szalay G, Sauter M, Hald J, Weinzierl A, Kandolf R, Klingel K. Sustained nitric oxide synthesis contributes to immunopathology in ongoing myocarditis attributable to interleukin-10 disorders. Am J Pathol. 2006;169(6):2085–2093. doi:10.2353/ajpath.2006.060350
85. Nishio R, Matsumori A, Shioi T, Ishida H, Sasayama S. Treatment of experimental viral myocarditis with interleukin-10. Circulation. 1999;100(10):1102–1108. doi:10.1161/01.CIR.100.10.1102
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.