")
Back to Journals » Clinical Ophthalmology » Volume 19
Outcomes of Treatment in Ocular Myasthenia Gravis Based on Minimal Manifestation: A Real-World Retrospective Cohort Study
Authors Kemchoknatee P , Santitamrongvtit B, Srisombut T
Received 19 February 2025
Accepted for publication 27 April 2025
Published 7 May 2025 Volume 2025:19 Pages 1505—1513
DOI https://doi.org/10.2147/OPTH.S520136
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Parinee Kemchoknatee,1 Boonravee Santitamrongvtit,2 Thansit Srisombut3
1Department of Ophthalmology, Rajavithi Hospital, Rangsit University, Bangkok, Thailand; 2Department of Medicine, Phra Nang Klao Hospital, Nonthaburi, Thailand; 3Department of Ophthalmology, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Correspondence: Parinee Kemchoknatee, Department of Ophthalmology, Rajavithi Hospital, Rangsit University, 2, Phayathai Road, Bangkok, 10400, Thailand, Email [email protected]
Background: Myasthenia gravis (MG) is a chronic autoimmune disorder characterized by muscle weakness caused by autoantibodies targeting acetylcholine receptors. Prednisolones improve symptoms by reducing autoantibody activity, but its optimal use, particularly in ocular myasthenia gravis (OMG), remains unclear due to limited trials and variable responses.
Objective: To evaluate the clinical efficacy, optimal dosing, and predictors of first minimal manifestation (MM) with oral prednisolone in myasthenia gravis, based on the Myasthenia Gravis Foundation of America Post-Intervention Status (MGFA-PIS).
Methods: Patients with OMG treated with oral prednisolones between 1st January 2015 and 31st December 2022 at Rajavithi Hospital were retrospectively reviewed. Baseline data, dosing, efficacy, and outcomes were analyzed using Cox regression and survival curves to identify predictors and time to first MM.
Results: Of the 101 OMG, 81 patients (80.2%) achieved MM. The mean age was 46.6 ± 6.9 years in the MM group and 46.2 ± 6.0 years in the non-MM group. The mean daily dose of pyridostigmine was significant higher in the MM group (152.68 ± 32.38 mg/day) compared to the non-MM group (133 ± 34.50 mg/day) (p = 0.018). The prednisolone dosage was comparable between the two groups. Seropositivity of AChRAb, thymoma, thymectomy, concurrent autoimmune diseases were notably observed in non-MM group (p < 0.05, respectively). Cox proportional hazards analysis and survival curves revealed higher pyridostigmine dosage (HR 1.713, 95% CI: 1.029– 2.849, p = 0.038). Prednisolone initiation at 6– 12 months reduced MM (HR 0.527, 95% CI: 0.297– 0.936, p = 0.029), with further reduction observed beyond 12 months (HR 0.165, 95% CI: 0.073– 0.368, p < 0.001). The presence of AChRAb, thymoma, prednisolones dosage and ophthalmic manifestations demonstrated no significant association with the achievement of minimal manifestation.
Conclusion: Early prednisolone initiation (within 6 months) and higher pyridostigmine doses were linked to remission, highlighting treatment factors over demographics in OMG outcomes.
Keywords: myasthenia gravis, ocular myasthenia gravis, OMG, oral prednisolones, remission, minimal manifestation
Introduction
Myasthenia gravis (MG) is a chronic autoimmune disorder characterized by impaired neuromuscular transmission, leading to fluctuating muscle weakness and fatigue, primarily mediated by autoantibodies targeting acetylcholine receptors at the neuromuscular junction, disrupting synaptic signaling.1,2
Prednisolones effectively reduce the activity of pathogenic autoantibodies and modulates immune responses, resulting in symptom alleviation and despite the widespread use of oral prednisolones in managing MG, including ocular myasthenia gravis (OMG), there is no universally accepted consensus from major organizations such as the Myasthenia Gravis Foundation of America (MGFA) on dosage, duration, or tapering regimen.3 While low-dose prednisolones (15 mg/day) with gradual escalation have been demonstrated to be safe and effective with a median time 14 weeks for the achievement minimal manifestation status of OMG, no report of long-term treatment outcome has been addressed.4 Achieving optimal disease control remains challenging on the optimal dosage and duration of treatment for long-term symptom control.5 Clinical practice varies widely, with some clinicians favoring a low-dose, slow-taper strategy to mitigate side effects, while others advocate for higher initial doses to achieve rapid disease control. Moreover, the clinical response to prednisolones varies among individuals, influenced by factors, such as age or gender,6 ophthalmic symptoms,7 disease severity8,9 or antibody subtype.3 Patients with late-onset disease were more likely to have been treated with lower doses of prednisolones.10 This highlights the importance of personalized treatment strategies.
Therefore, the present study aims to investigate the role of oral prednisolones in the management of OMG, with a focus on its clinical efficacy, optimal dosing strategies, and the predictors of achievement minimal manifestation status of OMG at Rajavithi Hospital assessed through time-to-event analysis.
Materials and Methods
Patient Selection and Baseline Characteristic
A retrospective cohort study was conducted in patients diagnosed with MG who received immunosuppressive therapy between 1st January 2015 and 31st December 2022 at Rajavithi Hospital, Bangkok, Thailand. The inclusion criteria were age 18 to 80 years and diagnosis of MG.The diagnosis of OMG was based on the criteria established by Osserman and Genkins,11 requiring patients to present with persistent symptoms of extraocular muscle weakness, such as diplopia, ptosis, or both, and to meet at least one of the following laboratory criteria: (1) positive acetylcholine receptor antibody (AChRAb) titer, (2) positive single-fiber electromyography, (3) clinical response to edrophonium chloride (Tensilon test), or (4) positive repetitive nerve stimulation (RNS) test. Eligible patients were required to have attended follow-up appointments for a minimum of 24 months. Exclusion criteria consisted of OMG patients with follow-up appointments for less than 24 months, uncertain prednisolone initiation dates or incomplete records or received the therapy in another medical institution or abroad were excluded from analyses requiring precise timing of treatment initiation.
Data Collection
Data were collected from electronic medical records, including demographic information, clinical characteristics, baseline severity of OMG, disease onset, AChRAb status. Dosage of cholinesterase inhibitors, specifically pyridostigmine, was categorized into two groups for analysis based on the Myasthenia Gravis Foundation of America Post-Intervention Status (MGFA-PIS): low dosage (<120 mg/day) and high dosage (≥120 mg/day).12 Corticosteroid therapy was generally considered for patients with OMG who had an inadequate response to pyridostigmine, presence of diplopia, risk of progression to GMG, or preference for rapid symptom control due to significant impact on daily life. The dosage of prednisolones was categorized into two groups for analysis: low dosage (<15 mg/day) and moderate dosage (≥15 mg/day) to facilitate the investigation of potential predictors of achieving minimal manifestation in OMG. The timing of prednisolones initiation was stratified into three categories for analysis: within 6 months, between 6 and 12 months, and beyond 12 months. Imaging findings, where applicable, such as chest Computed Tomography (CT) or magnetic resonance imaging (MRI) for thymoma or thymic hyperplasia detection.
In our study, follow-up intervals were scheduled which ranged from 3 to 6 months, depending on the clinical status and individual patient needs. Primary outcomes include the time to achieving the first minimal manifestation (MM) as defined by MGFA-PIS3,12 which classified into MM-0 to MM-3 based on treatment status: no treatment, immunosuppressant only, low-dose pyridostigmine only, or both treatments, respectively. Conclusively, patients without orbicularis oculi weakness on forced eyelid closure during physical examination were classified as having MM and were identified predictors associated with achieving MM in OMG.
Ethical Approval
The study was approved by the Rajavithi Hospital Research Ethics Committee (certificate number 143/67), and all participants provided informed consent prior to the collection. The study was conducted in accordance with the principles outlined in the Declaration of Helsinki.
Statistical Analysis
Descriptive statistics will summarize patient demographics, clinical characteristics, and treatment details. Continuous Variables, Independent t-tests or Mann–Whitney U-tests were used for normally and non-normally distributed data, respectively. Categorical Variables, Chi-square or Fisher’s exact tests were employed as appropriate. Univariable and multivariable Cox proportional hazards regression models were used to identify factors associated with the first MM and time to achievement between subgroups were reported as survival curves. Key clinical variables known from previous literature to be relevant to disease progression and treatment response both clinical relevance and statistical considerations in univariable analysis. Variables with a p-value<0.2 in univariable analysis were considered for inclusion in the multivariable model to identify potential predictors associated with the achievement of MM. Proportional hazards assumption was checked using Schoenfeld residual after each model. Hazard ratios (HRs) with 95% confidence intervals (CIs) were reported for each variable. A p-value of <0.05 was be considered statistically significant. All analyses were performed using a standard statistical software package.
Results
Clinical Characteristics of OMG Patients
In total, one hundred and seventy-two patients were diagnosed with OMG within this timeframe. Seventy-one were excluded. Thirty-two were excluded due to incomplete medical records, twenty-five were insufficient follow-up duration, fourteen were received prior corticosteroids or immunotherapy before the study period. In all, one hundred and one patients who fulfilled the inclusion criteria were enrolled and subsequently included in the final analysis of the study.
Table 1 demonstrated the key differences between OMG patients achieving and not achieving MM. Mean age was 46.59 ± 6.95 years in MM group and there was a comparable difference in non-MM group with a mean age of 46.15 ± 5.99 years (p = 0.794). Significant variables included seropositivity of AChRAb, thymoma, thymectomy, concurrent autoimmune diseases (all p < 0.05). The mean daily dose of pyridostigmine was higher in the MM group (152.68 ± 32.38 mg/day) compared to the non-MM group (133 ± 34.50 mg/day, p = 0.018). Time to prednisolones initiation (<6 months) was notably observed in the MM group (p < 0.001). The mean duration of prednisolone use was 8.2 months, with a slightly longer average duration observed in the MM group (8.9 months). None of the patients in the OMG cohort had underlying hepatic or renal disease, and none received corticosteroids via the intravenous route. Among the 14 patients diagnosed with thymoma, nine (64.3%) underwent thymectomy. Overall, ten patients progressed to secondary generalized myasthenia gravis (GMG).
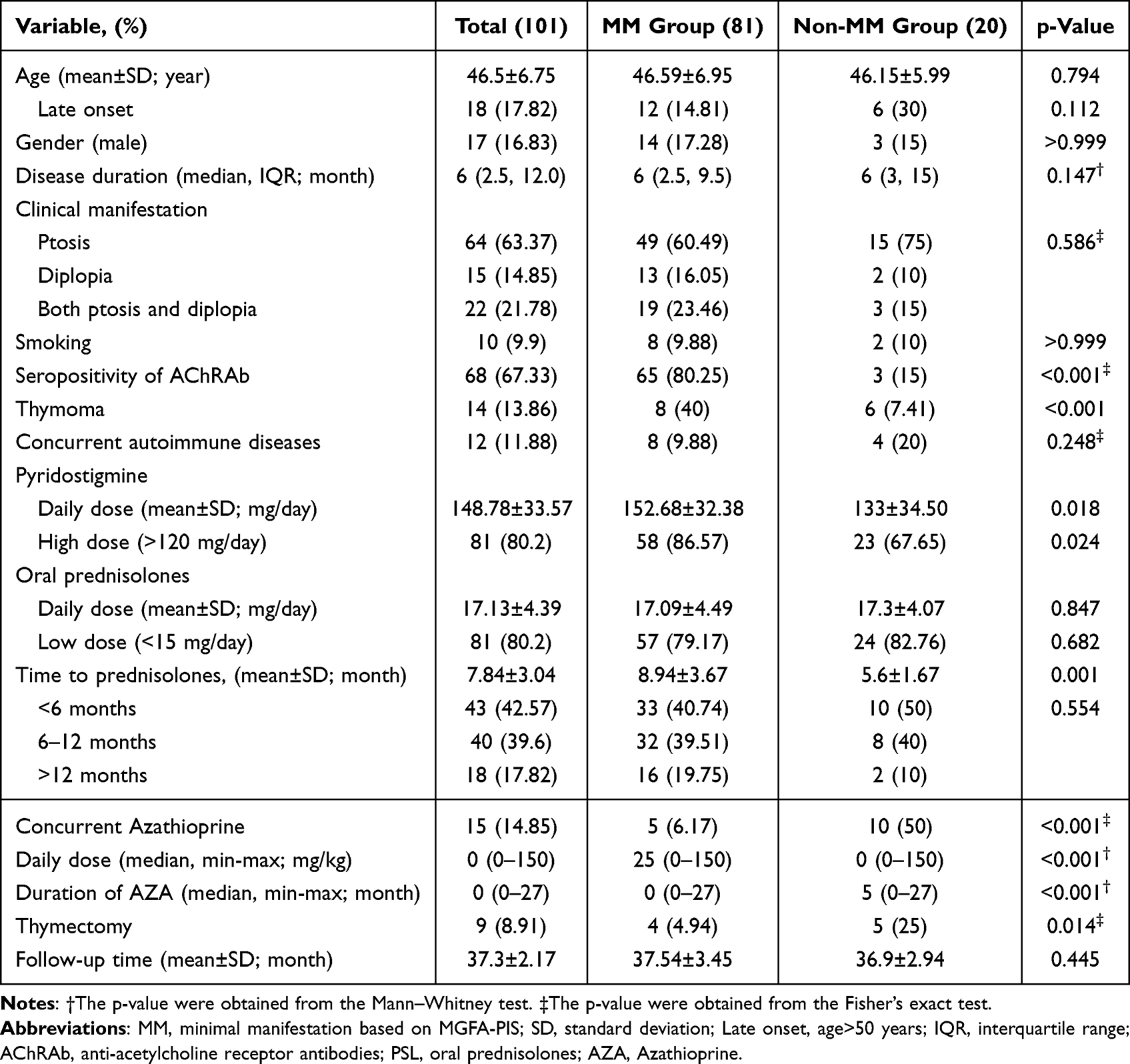 |
Table 1 Baseline Characteristic of All OMG Patients Classified by Achievement of MM Outcome |
Figure 1A illustrated the cumulative rate of achieving MM over a 48-month follow-up period in the whole cohort. The curve demonstrates a gradual increase in the rate of MM achievement, with a marked rise within the first 24 months (average time to remission was 12.2±3.1 months), followed by a steady progression thereafter.
 |
Figure 1 Predictors of first MM achievement based on demographic and clinical characteristics in ocular myasthenia gravis. (A) Overall rate of minimal manifestation achievement; (B) Ocular symptoms, ptosis or diplopia or both (C) AChRAb, anti-acetylcholine receptor antibodies; (D) Presence of thymoma. |
Outcome of Minimal Manifestation Achievement Based on Baseline Characteristics, Seropositivity and Thymoma
Table 2, various factors associated with achieving MM in OMG patients were analyzed. Age at onset, gender, and the presence of autoimmune diseases between the two groups of MM achievement were comparable. While diplopia and ptosis, seropositivity for AChRAb and the presence of thymoma were significant in the univariable analysis, they loss statistical significance in the multivariable model and survival curve analysis (Figure 1B–D).
 |
Table 2 Cox- Proportional Hazards Model for Predictors of Minimal Manifestation Achievement† |
Outcome of Minimal Manifestation Achievement Based on Treatments (Pyridostigmine, Immunosuppressants Dosage, and Duration)
Higher pyridostigmine dosage (>120 mg/day) remained a predictor of MM achievement (HR 1.713, 95% CI 1.029–2.849, p = 0.038, Figure 2A). Prednisolone initiation at 6–12 months reduced MM achievement (HR 0.527, 95% CI: 0.297–0.936, p = 0.029), with further reduction observed beyond 12 months (HR 0.165, 95% CI: 0.073–0.368, p < 0.001). Rate of MM achievement stratified by prednisolones dosage group (<15mg/day or ≥15 mg/day) or azathioprine therapy were comparable (Figure 2B–D).
 |
Figure 2 Predictors of first MM achievement based on treatments in ocular myasthenia gravis. (A) Pyridostigmine, categorized on dosage <120 or ≥120 mg/day (p=0.038); (B) prednisolones dosage categorized on daily dosage low (<15 mg/day) or moderate (≥15 mg/day) (p>0.05); (C) Time initiation oral prednisolones categorized on early initiation within 6, 6–12, or >12 months (p=0.029,p<0.001); (D) Azathioprine use was categorized as azathioprine-treated versus non-azathioprine groups (p>0.05). Statistical differences analyzed by cox proportional hazards regression model. |
Discussion
Our study identified several clinical characteristics that influence treatment response in patients with ocular myasthenia gravis. Significant predictors of achieving minimal manifestation included higher doses of pyridostigmine, and early initiation of prednisolones.
Timely initiation of corticosteroids in OMG has been associated with better outcomes, including a reduced risk of GMG progression.13 Early treatment can mitigate immune-mediated damage and stabilize ocular symptoms, diplopia or ptosis.9,14 Whereas delayed therapy may face challenges in reversing established pathology.9 Patients with recent-onset OMG may exhibit a better response to corticosteroids compared to those with a longer disease duration. Regarding tapering strategies, while Miano et al proposed that a slower rate of corticosteroids tapering, combined with a higher maintenance dose of prednisones, increases the likelihood of achieving successful symptom control.15 A recent clinical trial reported that rapidly tapering prednisolones has been found to be safe, well tolerated, and favorable treatment outcome in patients with moderate to severe GMG.16 However, no significant difference in dosage was observed in relation to treatment outcomes. We have proposed several explanations to account for these findings. Firstly, Disease Heterogeneity in OMG presents with diverse clinical manifestations and severities, leading to individualized responses to treatment.17 Ptosis, or combination of ptosis and diplopia represents more severe manifestations and it is often managed with higher doses of prednisolones.18 This severity may act as a confounding factor, potentially influencing the interpretation of dosage efficacy. Secondly, A concept of “ceiling effect” with prednisolones dosage in OMG suggests that beyond a certain dose, increasing the amount does not lead to additional therapeutic benefits.19 In addition, the optimal dosage of prednisolone for the treatment of OMG remains inconclusive. The EPITOME study demonstrated that low-dose prednisolone (15 mg/day) with gradual escalation was effective in achieving minimal manifestation status in OMG.4 Verma et al suggested that long-term administration of low-dose prednisolones (≤7.5 mg/day) may offer benefits in preserving binocular vision in OMG patients.20 The variability in the frequency of specific MHC alleles and polymorphisms in glucocorticoid receptor genes across populations can influence treatment efficacy. For example, the rs17209237 polymorphism in the glucocorticoid receptor gene has been identified as an independent factor affecting glucocorticoid efficacy in MG patients within Chinese populations.21 This variability contributes to differences in treatment response and dosage requirements,22 warranting further investigation to better understand the underlying mechanisms and progression of the disease. The impact of early initiation and sustained administration of prednisolones on achieving MM, compared to the specific dosage, warrants further evaluation through prospective studies. Balancing efficacy with the risk of side effects through individualized dosing strategies and combination therapies is essential for optimizing patient outcomes.23
While our findings suggest a positive association between higher pyridostigmine doses and MM achievement. It is mostly used in conjunction with corticosteroids or immunosuppressants. The synergistic effects of these therapies might reduce the reliance on higher pyridostigmine doses, as other treatments address the underlying autoimmune pathology and decreased risk of generalization.9 Ocular symptoms have influenced treatment response.18 Combined symptoms of ptosis and diplopia or severe symptoms may require more aggressive treatment approaches. While either symptom of ptosis or diplopia generally respond well to timely and appropriate therapy.7 This complex interplay poses challenges in isolating the specific contribution of pyridostigmine dosage to the achievement of MM. Due to variability in patient responses, the need for individualized treatment is warrant. In the present study, the lack of impact of azathioprine may due to several explanations. First, While OMG typically presents with milder disease severity, which may not necessitate potent immunosuppression corticosteroids alone or in combination with acetylcholinesterase inhibitors may suffice for symptom control, patients who treated with azathioprine presented with more disease severity resulting in a decrease of the added benefit of azathioprine.24 Second, small sample sizes or variability in dosing and adherence among patients treated with azathioprine may hinder the detection of a significant association. These factors could obscure the true therapeutic effect of azathioprine.
The levels of AChRAb may correlate with the severity of myasthenia gravis, with higher titers often indicating a more aggressive disease course.6 While seropositivity is a marginal predictor of disease severity and treatment response in our univariable model, the time to achieve minimal manifestation was comparable. This finding may be influenced by factors such as advanced age, gender, and a diagnosis of GMG, which are known to significantly impact the likelihood of AChRAb seropositivity.25 Consequently, it is crucial to consider demographic and clinical characteristics when interpreting antibody test results in MG patients.
Thymoma is frequently associated with more severe phenotypes including a higher prevalence of GMG and an increased likelihood of relapsing treatment outcomes. Consistent with this, the present study observed a lower proportion of patients achieving MM in the thymoma group.26 Despite this, the response to treatment in thymoma-associated MG may be comparable to non-thymoma MG once appropriate therapy is initiated, resulting in comparable times to MM achievement in hazard model. Regarding thymectomy on remission outcome, while these interventions aim to control disease progression, they do not directly correlate with faster symptom resolution or achievement of MM, as the effect of thymectomy on symptom improvement cannot be immediately.27 A small sample size may also reduce the statistical power to detect significant associations with thymectomy benefit.
Although late-onset ocular myasthenia gravis has been associated with worse treatment outcomes in previous studies.10 This observation may vary depending on factors such as comorbidities, treatment adherence, and the use of immunosuppressive therapies. In the present study, a comparable age distribution was observed between the two groups based on MM status. Acetylcholinesterase inhibitors, and immunosuppressive therapies target the underlying autoimmune mechanism of the disease leading to comparable outcomes regardless of age at onset.28 In addition, localized manifestations may respond uniformly or minimize the impact of age-related differences in immune function or treatment tolerance.10 Early diagnosis and timely initiation of therapy can mitigate the effects of age-related factors. Regarding the influence of gender on treatment outcome, while estrogen is known to modulate immune responses and contribute to the higher prevalence of autoimmune diseases in females, its impact may be less pronounced in OMG due to its localized nature compared to systemic autoimmune disease.29
This study has several limitations that may impact the interpretation and generalizability of its findings. First, being conducted in a referral and teaching medical center, the study is susceptible to selection bias. As a retrospective analysis, the study is subject to inherent biases, including selection bias and incomplete data collection. Variability in patient characteristics, such as differences in disease severity, antibody status, and treatment regimens, could introduce confounding factors, complicating the interpretation of results. Specific timing of dose reduction following achievement of minimal manifestation were not consistently available. The decision to taper prednisolone was based on individual physician discretion, which may introduce variability in treatment exposure and limit the generalizability of our findings regarding long-term steroid management. In our OMG cohort, though baseline liver and kidney function tests were not routinely performed before initiating pyridostigmine, except in patients on azathioprine, for whom CBC and LFT were monitored periodically. Instead, clinical history and available laboratory results were used to assess organ function. Notably, no patients had liver or kidney disease. Further study with larger sample sizes of liver and kidney function cases should be considered as potential confounders in multivariable analysis. Although the findings may not be fully applicable to diverse populations due to differences in demographic, genetic, or environmental factors across regions and juvenile myasthenia gravis or pediatric populations. To our knowledge, this is the first study in Thai population evaluating the impact of treatment timing, duration of prednisolones providing insights into optimizing therapeutic strategies for achieving MM. The study’s follow-up period with 36 months may be sufficient to capture long-term outcomes, particularly for conditions with delayed therapeutic responses. In summary, optimizing pyridostigmine dosing and initiating corticosteroid therapy at an early stage are essential strategies for effective disease management.
Conclusion
Early prednisolone initiation (within 6 months) and higher pyridostigmine doses were linked to remission, highlighting treatment factors over demographics in OMG outcomes.
Consent Statement
All participants gave written informed consent before the data collection process began.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding was received for this research.
Disclosure
None of the authors have any conflict of interest to disclose for this work.
References
1. Gilhus NE. Myasthenia Gravis. N Engl J Med. 2016;375(26):2570–2581. doi:10.1056/NEJMra1602678
2. Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol. 2015;14(10):1023–1036. doi:10.1016/s1474-4422(15)00145-3
3. Jaretzki III A, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology. 2000;55(1):16–23. doi:10.1212/wnl.55.1.16
4. Benatar M, McDermott MP, Sanders DB, et al. Efficacy of prednisone for the treatment of ocular myasthenia (EPITOME): a randomized, controlled trial. Muscle Nerve. 2016;53(3):363–369. doi:10.1002/mus.24769
5. Skeie GO, Apostolski S, Evoli A, et al. Guidelines for treatment of autoimmune neuromuscular transmission disorders. Eur J Neurol. 2010;17(7):893–902. doi:10.1111/j.1468-1331.2010.03019.x
6. Mazzoli M, Ariatti A, Valzania F, et al. Factors affecting outcome in ocular myasthenia gravis. Int J Neurosci. 2018;128(1):15–24. doi:10.1080/00207454.2017.1344237
7. Kupersmith MJ, Ying G. Ocular motor dysfunction and ptosis in ocular myasthenia gravis: effects of treatment. Br J Ophthalmol. 2005;89(10):1330–1334. doi:10.1136/bjo.2004.063404
8. Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized Trial of Thymectomy in Myasthenia Gravis. N Engl J Med. 2016;375(6):511–522. doi:10.1056/NEJMoa1602489
9. Sommer N, Sigg B, Melms A, et al. Ocular myasthenia gravis: response to long-term immunosuppressive treatment. J Neurol Neurosurg Psychiatry. 1997;62(2):156–162. doi:10.1136/jnnp.62.2.156
10. Kemchoknatee P, Armornpetchsathaporn A, Tangon D, Srisombut T. Age of onset and factors affecting treatment responses in ocular myasthenia gravis. Int Ophthalmol. 2023;43(8):2777–2785. doi:10.1007/s10792-023-02676-4
11. Osserman KE, Genkins G. Studies in myasthenia gravis: review of a twenty-year experience in over 1200 patients. Mt Sinai J Med. 1971;38(6):497–537.
12. Narayanaswami P, Sanders DB, Wolfe G, et al. International Consensus Guidance for Management of Myasthenia Gravis: 2020 Update. Neurology. 2021;96(3):114–122. doi:10.1212/wnl.0000000000011124
13. Hong YH, Kwon SB, Kim BJ, et al. Prognosis of ocular myasthenia in Korea: a retrospective multicenter analysis of 202 patients. J Neurol Sci. 2008;273(1–2):10–14. doi:10.1016/j.jns.2008.05.023
14. Wakata N, Iguchi H, Sugimoto H, Nomoto N, Kurihara T. Relapse of ocular symptoms after remission of myasthenia gravis--a comparison of relapsed and complete remission cases. Clin Neurol Neurosurg. 2003;105(2):75–77. doi:10.1016/s0303-8467(02)00104-x
15. Miano MA, Bosley TM, Heiman-Patterson TD, et al. Factors influencing outcome of prednisone dose reduction in myasthenia gravis. Neurology. 1991;41(6):919–921. doi:10.1212/wnl.41.6.919
16. Sharshar T, Porcher R, Demeret S, et al. Comparison of Corticosteroid Tapering Regimens in Myasthenia Gravis: a Randomized Clinical Trial. JAMA Neurol. 2021;78(4):426–433. doi:10.1001/jamaneurol.2020.5407
17. Threetong T, Poonyathalang A, Preechawat P, Jindahra P, Padungkiatsagul T, Vanikieti K. Initial Treatment Response in Ocular Myasthenia Gravis: a Comparison Between Low and Moderate Doses of Prednisolone. Clin Ophthalmol. 2020;14:2051–2056. doi:10.2147/opth.S261259
18. Feng X, Huan X, Yan C, et al. Adult Ocular Myasthenia Gravis Conversion: a Single-Center Retrospective Analysis in China. Eur Neurol. 2020;83(2):182–188. doi:10.1159/000507853
19. Sussman J, Farrugia ME, Maddison P, Hill M, Leite MI, Hilton-Jones D. Myasthenia gravis: association of British Neurologists’ management guidelines. Pract Neurol. 2015;15(3):199–206. doi:10.1136/practneurol-2015-001126
20. Verma R, Wolfe GI, Kupersmith MJ. Ocular myasthenia gravis - How effective is low dose prednisone long term? J Neurol Sci. 2021;420:117274. doi:10.1016/j.jns.2020.117274
21. Xie Y, Meng Y, Li HF, et al. GR gene polymorphism is associated with inter-subject variability in response to glucocorticoids in patients with myasthenia gravis. Eur J Neurol. 2016;23(8):1372–1379. doi:10.1111/ene.13040
22. Varade J, Wang N, Lim CK, et al. Novel genetic loci associated HLA-B*08:01 positive myasthenia gravis. J Autoimmun. 2018;88:43–49. doi:10.1016/j.jaut.2017.10.002
23. Benatar M, Sanders DB, Wolfe GI, McDermott MP, Tawil R. Design of the efficacy of prednisone in the treatment of ocular myasthenia (EPITOME) trial. Ann N Y Acad Sci. 2012;1275(1):17–22. doi:10.1111/j.1749-6632.2012.06780.x
24. Axelsen KH, Kjær Andersen R, Andersen LK, Vissing J, Witting N. Ocular versus generalized myasthenia gravis: a continuum associated with acetylcholine receptor antibody titers. Neuromuscul Disord. 2024;43:39–43. doi:10.1016/j.nmd.2024.07.002
25. Peeler CE, De Lott LB, Nagia L, Lemos J, Eggenberger ER, Cornblath WT. Clinical Utility of Acetylcholine Receptor Antibody Testing in Ocular Myasthenia Gravis. JAMA Neurol. 2015;72(10):1170–1174. doi:10.1001/jamaneurol.2015.1444
26. Evoli A, Batocchi AP, Palmisani MT, Lo Monaco M, Tonali P. Long-term results of corticosteroid therapy in patients with myasthenia gravis. Eur Neurol. 1992;32(1):37–43. doi:10.1159/000116785
27. Cataneo AJM, Felisberto G Jr, Cataneo DC. Thymectomy in nonthymomatous myasthenia gravis - systematic review and meta-analysis. Orphanet J Rare Dis. 2018;13(1):99. doi:10.1186/s13023-018-0837-z
28. Evoli A, Batocchi AP, Minisci C, Di Schino C, Tonali P. Clinical characteristics and prognosis of myasthenia gravis in older people. J Am Geriatr Soc. 2000;48(11):1442–1448. doi:10.1111/j.1532-5415.2000.tb02635.x
29. Lasrado N, Jia T, Massilamany C, Franco R, Illes Z, Reddy J. Mechanisms of sex hormones in autoimmunity: focus on EAE. Biol Sex Differ. 2020;11(1):50. doi:10.1186/s13293-020-00325-4
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.