")
Back to Journals » Journal of Inflammation Research » Volume 17
PM2.5 Promotes Macrophage-Mediated Inflammatory Response Through Airway Epithelial Cell-Derived Exosomal miR-155-5p
Authors Xu H, Li X, Liu K, Huang P, Liu XJ
Received 7 August 2024
Accepted for publication 1 November 2024
Published 9 November 2024 Volume 2024:17 Pages 8555—8567
DOI https://doi.org/10.2147/JIR.S482509
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Hui Xu,1,2 Xin Li,1 Kai Liu,1 Ping Huang,1 Xiao-Ju Liu1,2
1The First School of Clinical Medicine, Lanzhou University, Lanzhou, Gansu, People’s Republic of China; 2The First Hospital of Lanzhou University, Lanzhou, Gansu, People’s Republic of China
Correspondence: Xiao-Ju Liu, Email [email protected]
Background: Airway epithelial cells (AECs) and alveolar macrophages are involved in airway inflammation. The direct effects of atmospheric fine-particulate-matter (PM2.5) on airway cells, such as AECs and alveolar macrophages, have been widely investigated, but the effect of cell-cell interaction on inflammatory response remains unclear. Exosomes play a crucial role in intercellular communication. However, the cellular interaction of exosomes in PM2.5-induced airway inflammation is unclear.
Methods: The PM2.5-induced human bronchial epithelial (BEAS-2B) cells and phorbol 12-myristate 13-acetate-induced macrophages (M&phis;) were co-cultured and then the expression of IL-6, IL-1β, TNF-α and miRNA-155-5p were detected. Exosomes from PM2.5-exposed BEAS-2B cells were then co-cultured with M&phis; to detect the expression of miR-155-5p and inflammatory cytokines, as well as cytokine signaling inhibitor-1 (SOCS1)/NFκB, and to detect the effect of the exosome inhibitor GW4869.
Results: After the co-culture of PM2.5-induced BEAS-2B cells and M&phis;, the expression of M&phis;-derived IL-6, IL-1β, and TNF-α, as well as miRNA-155-5p were upregulated. The expression of miRNA-155-5p was upregulated in BEAS-2B and BEAS-2B cell-derived exosomes after exposure to PM2.5. Furthermore, co-culturing exosomes derived from PM2.5-exposed BEAS-2B cells with M&phis;, upregulated miR-155-5p and inflammatory cytokines, decreased cytokine signaling inhibitor-1 (SOCS1) expression, and activated NF-κB. In addition, adding exosome inhibitor GW4869 to PM2.5-interfered BEAS-2B cells co-culture with M&phis; downregulated miRNA-155-5p expression, inhibited NF-κB, and reduced the levels of inflammatory factors.
Conclusion: PM2.5 promotes M&phis; inflammation by upregulating miRNA-155-5P in exosomes obtained from BEAS-2B cells through miR-155-5P/SOCS1/NF-κB pathway. Exosomal miRNAs mediate cellular communication between BEAS-2B cells and M&phis;, which may be a new mechanism of PM2.5-stimulated pulmonary inflammatory response.
Keywords: PM2.5, exosome, microRNA, lung inflammation
Graphical Abstract:

Introduction
Airway inflammation is the major driver of chronic obstructive pulmonary disease (COPD) and asthma. Airway epithelial cells (AECs) and alveolar Mφ play a vital role in airway inflammation. AECs serve as the primary barrier against inhaled particles and pathogens and can directly respond to harmful substances, secrete pro-inflammatory factors and chemokines, initiate the immune response and attract inflammatory cells.1 Activated alveolar macrophages (AMs) can release various cytokines and inflammatory mediators.2 The direct impacts of PM2.5 on AECs and alveolar macrophages have been studied, but the regulatory mechanism of cell-cell interactions on inflammatory response is not clear.
PM2.5 is the main harmful pollutant in smog.3,4 PM2.5 can significantly increase the incidence of asthma and COPD, impair lung function, and increase the mortality rate of lung cancer.5–7 PM2.5 exposure strongly induces airway inflammation in chronic airway diseases.8–10 PM2.5 acts on airway epithelial cells, leading to mitochondrial dysfunction and inflammatory cytokine release.11 PM2.5 regulates PI3K/Akt/mTOR signaling pathway by upregulating interleukin17A. It also inhibits bronchial epithelial cell autophagy and promotes pulmonary inflammation.12 PM2.5 induces airway inflammation by activating NLRP3 inflammasome in bronchial epithelial cells.13 Exposure to PM2.5 can increase the production of reactive oxygen species in alveolar macrophages, activate the stimulating NF-κB signal transduction, and induce airway inflammation.14 The direct effects of PM2.5 on airway cells, such as AECs and alveolar macrophages, have been widely reported, but how PM2.5 affects macrophages remains unclear.
Exosomes are nanoscale extracellular vesicles, typically measuring between 40 and 160 nanometers in diameter, with an average size of 100 nanometers. They carry bioactive molecules, such as proteins, RNAs, microRNAs (miRNAs), DNAs, etc. Exosomal miRNAs can be transported to neighboring cells or enter recipient cells via body fluids to maintain their biological activity and regulate protein expression in recipient cells through transcription, translation or direct effects at the protein level.15,16 Pulmonary exosomes can be derived from different types of cells, including bronchial epithelial cells, lung macrophages, and dendritic cells.17 Studies have reported that exosomal miRNAs are responsible for the onset of chronic airway inflammatory disorders, including asthma and COPD.18,19 Extracellular vesicles inhibit NLRP3 inflammasome by delivering miR-223/142, thereby inhibiting macrophage activation and pulmonary inflammation.20 Alveolar epithelial exosomal miR-92a-3p promotes sepsis-induced acute lung injury by activating alveolar macrophages.21 Macrophages deliver exosomal miRNA-142-3p to lung fibroblasts and AECs, inhibiting the progression of pulmonary fibrosis.22 The exosome inhibitor GW4869 is a symmetric dihydroimidazolamide compound with cell permeability, which can significantly reduce the release of exosomes, and has been widely used in experimental studies.23
miRNA-155 is a versatile microRNA known for its pivotal role in modulating inflammatory pathways across different cellular types and organs.24,25 Jiang and co-workers26 found that serum exosomes of mice with acute lung injury can activate NF-κB by delivering miR-155 to macrophages, thereby targeting SHIP1 and SOCS1. miR-155 can upregulate TNF-α and IL-6, inducing macrophage proliferation and inflammatory response. Inhibition of miR-155 alleviated sepsis-induced acute lung injury via the IRF2BP2/NFAT1 axis.27 miRNA-155-5p expression was increased in the AMs of smokers and COPD patients and in the AMs of cigarette smoke-exposed mice. Besides, miR-155-5p inhibition significantly attenuated inflammatory response in cigarette smoke-exposed mice and alveolar macrophages.28 miR-155 has various target genes, such as SOCS1 and SHIP1 genes, which mainly affect inflammation.29 SOCS1 modulates the inflammatory response by suppressing NF-κB signal transduction.30,31
This study investigated how PM2.5 regulates macrophage-mediated inflammatory response through the bronchial epithelial exosome miR-155-5p.
Methods
Human Bronchial Epithelial (BEAS-2B) Cell Culture
BEAS-2B cell line was supplied by Fu Heng Biology (Shanghai, China) and cultured in DMEM containing 10% FBS, 1% penicillin/streptomycin. The cells were maintained at 37°C and 5% CO2.
PM2.5-Induced BEAS-2B Cells
First, 5 μg/μL PM2.5 (SRM 1648a, NIST, USA) stock solution was prepared with DMEM containing 10% FBS. BEAS-2B cells were placed in 175T culture bottles, and the final concentration of PM2.5 was 0, 100, 200, and 400 μg/mL for approximately 48 hours per generation. Culture cycles were repeated for 5 generations to simulate chronic exposure.32 After reaching the 5th generation, the medium was renewed with DMEM without FBS, and after 24h, the cell culture supernatant was collected for follow-up experiments.
Acquisition and Identification of Mφ
The human monocytic cell line (THP-1) was obtained from Fu Heng Biology. Cells were cultured in RPMI-1640 medium containing 10% FBS, 1% penicillin/streptomycin, and maintained at 37°C and 5% CO2.
THP-1 cells (5×105 /well) were grown 6-well plates containing 10 ng/mL phorbol 12-myristate 13-acetate (PMA, Apexbio, USA) for 48h to differentiate into adherent Mφ. Subsequently, the culture medium was renewed, and cells were incubated for 24h for subsequent experiments. Cell surface CD11b (Elabscience, Wuhan, China) was detected by flow cytometry to identify Mφ.
PM2.5-BEAS-2B Co-Cultured with Mφ
According to the final concentration of PM2.5 (0, 100, 200, and 400 μg/mL), the experiment was divided into four groups: Group A (control group): PM2.50-BEAS-2B+Mφ, Group B: PM2.5100-BEAS-2B+Mφ, Group C: PM2.5200-BEAS-2B+Mφ, and Group D: PM2.5400-BEAS-2B+Mφ. BEAS-2B cells exposed to diverse PM2.5 concentrations were co-cultured with Mφ in the Transwell chamber (Getter Biotechnology Co., Guangzhou, China) at 37°C and 5% CO2. The upper layer consisted of PM2.5-stimulated BEAS-2B cells, and the lower layer consisted of Mφ. The supernatant of the lower cell culture was collected for measuring inflammatory factors. The total RNA and protein of macrophages were extracted for subsequent experiments.
Exosome Extraction, Characterization, and Mφ Uptake of Exosomes
Exosome Extraction
According to the experimental methods defined by Xu et al33 and He et al,34 exosomes (BEAS-2B-Exo) were extracted from BEAS-2B cell culture supernatant using ExoQuick-TCTM (System Biosciences, USA). Briefly, the supernatant was centrifuged at 3000×g for 30 min to eliminate dead cells and cellular debris, followed by an additional 10,000×g for 20 min to eliminate other large vesicles. The centrifuged supernatants were subjected to ultrafiltration using a 100-kDa ultrafiltration device (Amicon Ultra-15, Millipore, USA) to remove microparticles. Then, the supernatant was transferred to a sterile vessel. ExoQuick-TCTM was mixed with the bio-fluid and kept at +4°C overnight. Next, the supernatant and ExoQuick-TCTM mixture were centrifuged at 1500×g for 30 min. The supernatant was aspirated, and the residual solution was centrifuged at 3000×g for 5 min. Following centrifugation, exosomes formed a beige pellet at the vessel’s base. The supernatant was carefully aspirated, and exosomes were finally resuspended in 200 μL PBS for subsequent analyses.
Exosome Characterization
Exosome morphologies were examined via transmission electron microscopy (TEM) (HT7800, Hitachi, Japan). Exosome particle size was detected by Zeta potential and particle size analyzer (90Plus Pals, Brookhaven Instruments, USA). Heat shock protein 70 (HSP70) (Boster, Wuhan, China) and CD81 (Boster, Wuhan, China) were detected by Western blotting.
BEAS-2B-Exo Uptake by Mφ
To measure the BEAS-2B-Exo internalization by Mφ, staining of BEAS-2B-Exo with PKH67 (Mei5bio, Beijing, China) was conducted. These exosomes were re-isolated using the same exosome isolation approach employing ExoQuick-TC™ (System Biosciences). BEAS-2B-Exo was finally resuspended in 100 μL PBS. Stained BEAS-2B-Exo was incubated with Mφ at 37°C and 5% CO2 for 6h. Mφ was rinsed twice with PBS, and each well was fixed with 250 μL of 4% paraformaldehyde for 20 min. Mφ were rinsed thrice with PBS. Subsequently, 200 μL of DAPI (Boster, Wuhan, China) solution was added, dark incubated for 10 minutes, and rinsed thrice with PBS. Intracellular green fluorescence was examined under a fluorescence microscope (IX73, Olympus, Japan) and photographed.
Exosomes Derived from PM2.5-Exposed BEAS-2B Cells (PM2.5-BEAS-2B-Exo) Co-Cultured with Mφ
According to the final intervention concentration of PM2.5 (0, 100, 200, and 400 μg/mL), the cells were assigned to 4 groups: Group E (control group): PM2.50-BEAS-2B-Exo+Mφ, Group F: PM2.5100-BEAS-2B-Exo+Mφ, Group G: PM2.5200-BEAS-2B-Exo+Mφ, and Group H: PM2.5400-BEAS-2B-Exo+Mφ. 200 μL of PM2.5-BEAS-2B-Exo was added to Mφ. Mφ were cultured at 37°C and 5% CO2 for 48h. The supernatant was harvested for measuring inflammatory factors. Total RNA and protein of Mφ were isolated for subsequent experiments.
The Role of Exosome Inhibitor GW4869
To verify whether miR-155-5p is transmitted between BEAS-2B and Mφ through the exosome pathway, cells were divided into three groups: Group A: PM2.50-BEAS-2B+Mφ, Group C: PM2.5200-BEAS-2B+Mφ, and Group I: PM2.5200-BEAS-2B+GW4869+Mφ. A co-culture system was established, with PM2.5-BEAS-2B cells and Mφ in the upper and bottom layers, respectively. In Group I, 10 μM of GW4869 (Topscience, Shanghai, China) was added to the upper layer and incubated at 37°C and 5% CO2 for 48h. The subculture supernatant was collected for measuring inflammatory factors. The total RNA and protein of Mφ were extracted for subsequent experiments.
Expression of miRNA-155-5p, SOCS1, and NF-κB mRNA
The expressions of miRNA-155-5p, SOCS1, and NF-κB mRNA in BEAS-2B-Exo, BEAS-2B cells, and Mφ were determined through qRT-PCR. RNA extraction and cDNA synthesis were conducted using miRNA-1st-Strand-cDNA-Synthesis-Kit and Evo-M-MLV-RT-Mix-Kit-with-gDNA-Clean for qPCR Ver.2 (Accurate Biotechnology, Hunan, China). Primer pairs for mRNA and miRNA (Table 1) were purchased from Accurate Biotechnology (Hunan, China). qRT-PCR was performed using the SYBR®-Green-Premix-Pro-Taq-HS-qPCR-Kit and the SYBR®-Green-Premix-Pro-Taq-HS-qPCR-Kit-II (Accurate Biotechnology, Hunan, China). The relative expression of mRNA or miRNA was computed using the 2-ΔΔCT approach and normalized to GAPDH or U6, respectively.
 |
Table 1 Primers Sequence |
The Expression of HSP70, CD81, and SOCS1, NF-κB p65 (P65), Phospho-NF-κB p65 (PP65) Were Detected in Mφ
Western blotting was conducted to determine the protein levels of HSP70, CD81, SOCS1, P65, and PP65 in Mφ. Total proteins were extracted from exosomes and Mφ using RIPA lysis buffer (Solarbio, Beijing, China). The protein specimen was separated through SDS-PAGE and transferred onto PVDF membranes. After blocking with protein-free rapid-blocking solution (Boster, Wuhan, China) for 1h, the membranes were exposed to rabbit anti-SOCS1 polyclonal antibody (Boster), rabbit anti-P65 polyclonal antibody (Boster), rabbit anti-PP65 monoclonal antibody (CellSignalingTechnology), rabbit anti-β-tublin polyclonal antibody (Boster), and rabbit anti-GAPDH polyclonal antibody (Boster) at 4°C overnight. After washing with TBST buffer, the membranes were exposed to goat anti-rabbit IgG (Signalway Antibody) for 1 hour. Lastly, bands were visualized using an enhanced chemiluminescence kit, and GAPDH or β-tubulin was utilized as the internal control.
Detection of IL-1β, IL-6, and TNF-α
The levels of IL-1β, IL-6, and TNF-α in supernatant were determined using an ELISA kit (Feiya Biotechnology, Jiangsu, China) following the manufacturers’ instructions. Absorbance was read at 450 nm using an enzyme-labeled instrument.
Statistical Analysis
All experiments were conducted utilizing a minimum of 3 independent assays. Data are expressed as mean±standard deviation. Statistical tests were performed with SPSS 19.0 software. One-way ANOVA was employed to compare means among multiple groups, followed by post hoc LSD tests for inter-group comparisons. Statistical significance was defined as P<0.05.
Results
PM2.5-BEAS-2B Was Co-Cultured with Mφ to Promote the Inflammation of Mφ Through the miR-155-5p /SOCS1/NF-κB Pathway
PM2.5-BEAS-2B was co-cultured with Mφ (Figure 1A), the results showed that miR-155-5p levels in Mφ increased with the increase of PM2.5 concentration, SOCS1 mRNA and protein expression decreased, and PP65 protein and PP65/P65 ratio increased. The levels and mRNA expression of inflammatory cytokines IL-1β, IL-6 and TNF-α were increased (Figure 1B–G). The results showed that PM2.5-BEAS-2B co-culture with Mφ can promote the inflammatory response of Mφ, which may be related to the miR-155-5p /SOCS1/NF-κB pathway.
 |
Figure 1 PM2.5-BEAS-2B co-cultured with Mφ. (A) PM2.5-BEAS-2B cell co-cultured with Mφ mode diagram; (B) Cellular supernatant IL-6, IL-1β, TNF-α contents of Mφ co-cultured with PM2.5-BEAS-2B cells; (C) mRNA expression levels of IL-6, IL-1β, TNF-α in Mφ co-cultured with PM2.5-BEAS-2B cells; (D) miR-155-5p expression level in Mφ co-cultured with PM2.5-BEAS-2B cells; (E) mRNA expression levels of SOCS1, NF-κB in Mφ co-cultured with PM2.5-BEAS-2B cells; (F and G) Protein expression level of SOCS1, P65, PP65 in Mφ co-cultured with PM2.5-BEAS-2B cells as detected by Western blot analysis as well as statistical analysis of gray values. Experimental group: group A PM2.50-BEAS-2B+Mφ, group B PM2.5100-BEAS-2B+Mφ, group C PM2.5200-BEAS-2B+Mφ, group D PM2.5400-BEAS-2B+Mφ. **P<0.01, *P<0.05 vs group A. |
Characterization of Exosomes and Uptake of BEAS-2B-Exo by Mφ
TEM showed that exosomes were spherical (Figure 2A). The size distribution of exosomes shows a diameter in the 50–150nm range (Figure 2B). The expression of exosome surface markers HSP70 and CD81 protein was positive (Figure 2C).
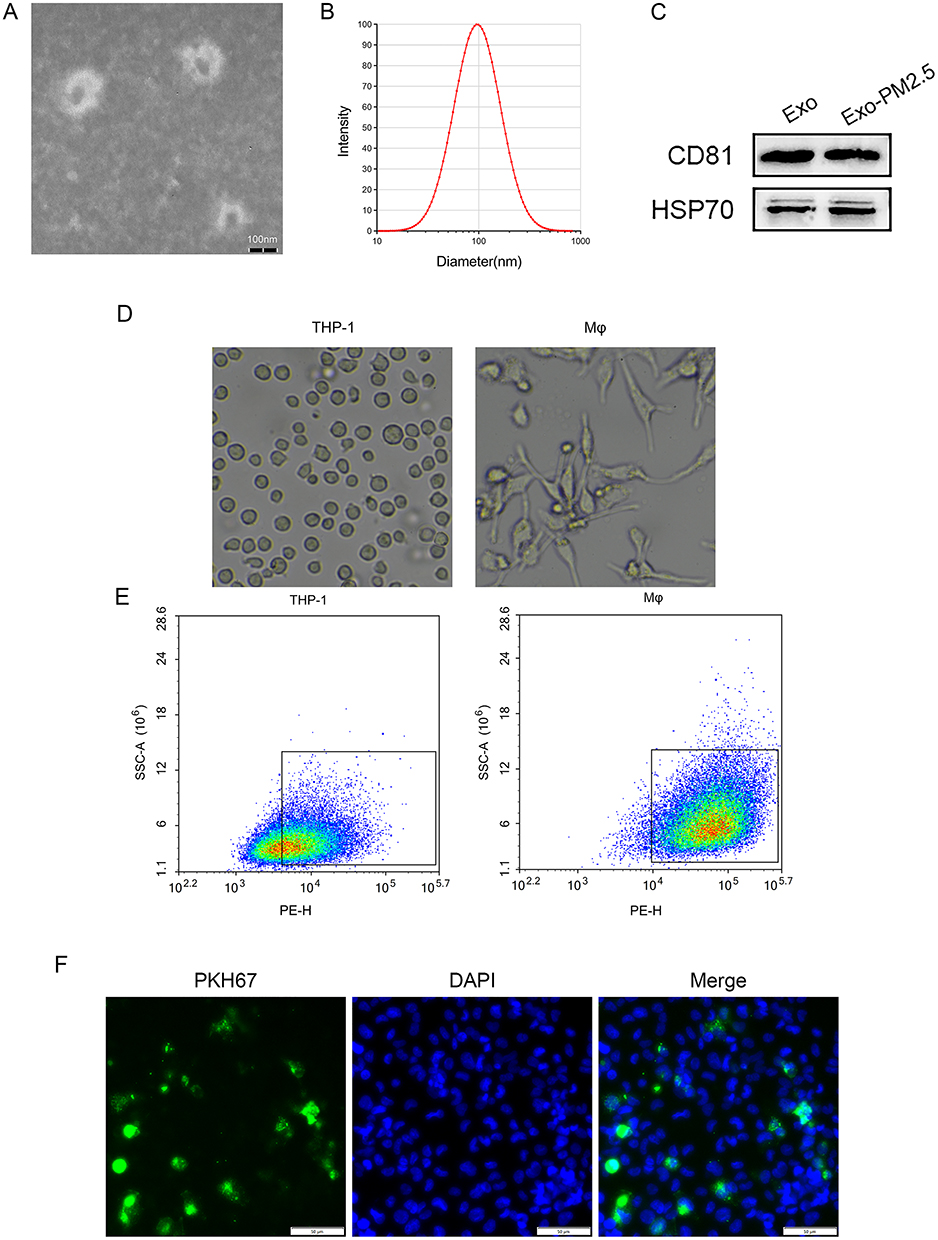 |
Figure 2 Characteristics and of exosomes and macrophages take up BEAS-2B-Exo. (A) A representative image of exosomes isolated from the BEAS-2B cells. Scale bar: 100 nm; (B) Histogram showing the size distribution of exosomes analysed by nanoparticle tracking analysis; (C) The expression levels of the exosome-related markers CD81 and HSP70 were detected by Western blot analysis. Exosomes were isolated from the control BEAS-2B cells (Exo) and the BEAS-2B cells exposed to PM2.5 (Exo-PM2.5); (D) THP-1cellular morphology and Mφcellular morphology; (E) Flow cytometry of THP-1 cells and Mφ surface marker CD11b; (F) Fluorescence microscopy analysis of PKH67-labeled BEAS-Exos internalization by Mφ. The green-labeled exosomes were visible in the perinuclear region of recipient cells. Scale bar: 50 μm. |
Mφ: The round and suspended THP-1 cells were differentiated by PMA, and the cells changed from suspension to adhesion state. Some of the cells had pseudopods protruding and showed fusiform (Figure 2D). Flow cytometry showed that CD11b expression in Mφ was higher than that in THP-1 [(96.7±4.7) % vs (66.1±4.6) %], the difference was statistically significant (p < 0.01), indicating that THP-1 cells were induced to become Mφ (Figure 2E).
PKH67 fluorescently labeled BEAS-2B-Exo was incubated with Mφ for 6 hours, and the co-localization of BEAS-2B-Exo and Mφ could be seen under fluorescence microscopy, indicating that BEAS-2B-Exo could be ingested by Mφ (Figure 2F).
Expression of miR-155-5p in PM2.5-BEAS-2B and PM2.5-BEAS-2B-Exo
The expression of miR-155-5p in PM2.5-BEAS-2B and PM2.5-BEAS-2B-Exo increased with the increase of PM2.5 concentration (Figure 3A and B).
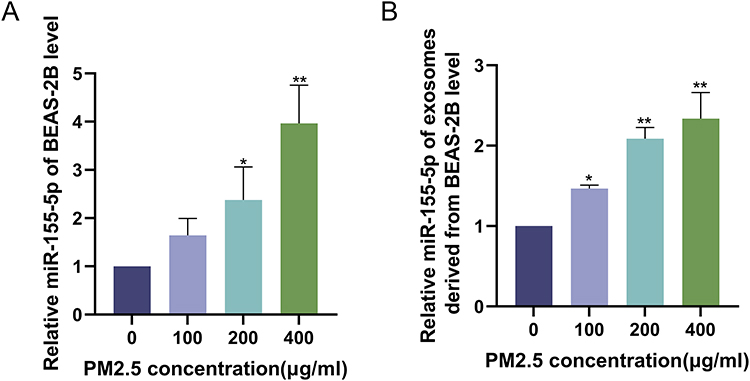 |
Figure 3 Expression of miR-155-5p in PM2.5-BEAS-2B and PM2.5-BEAS-2B-Exo. (A) miR-155-5p expression level in BEAS-2B cells exposed to different on concentration PM2.5 (0,100,200,400 μg/mL); (B) miR-155-5p expression level in exosomes derived from BEAS-2B cells exposed to different on concentration PM2.5 (0,100,200,400 μg/mL). **P<0.01, *P<0.05 vs the BEAS-2B cells exposed to PM2.5 (0 μg/mL). |
PM2.5-BEAS-2B-Exo Promotes Mφ Inflammation Through the miR-155-5p /SOCS1/NF-κB Pathway
PM2.5-BEAS-2B-Exo was co-cultured with Mφ (Figure 4A). The results showed that with the increase of PM2.5 intervention concentration, the expression of miR-155-5p in Mφ was increased, the expression of SOCS1 mRNA and protein was down-regulated, PP65 protein and the ratio of PP65/P65 was increased, and the contents and mRNA expression levels of inflammatory factors IL-1β, IL-6 and TNF-α were increased. All showed PM2.5 concentration dependence (Figure 4B–G). The results suggest that miR-155-5p /SOCS1/NF-κB pathway may be related to PM2.5-BEAS-2B-Exo promoting Mφ inflammatory response.
 |
Figure 4 PM2.5-BEAS-2B-Exo co-cultured with Mφ. (A) PM2.5-BEAS-2B-Exo co-cultured with Mφ mode diagram; (B) Cellular supernatant IL-6, IL-1β, TNF-α contents of Mφ incubated with PM2.5-BEAS-2B-Exo; (C) mRNA expression levels of IL-6, IL-1β, TNF-α in Mφ incubated with PM2.5-BEAS-2B-Exo; (D) miR-155-5p expression level in Mφ incubated with PM2.5-BEAS-2B-Exo; (E) mRNA expression levels of SOCS1, NF-κB in Mφ incubated with PM2.5-BEAS-2B-Exo; (F and G) Protein expression level of SOCS1, P65, PP65 in Mφ incubated with PM2.5-BEAS-2B-Exo as detected by Western blot analysis as well as statistical analysis of gray values. Experimental group: group E PM2.50-BEAS-2B-Exo+Mφ, group F PM2.5100-BEAS-2B-Exo+Mφ, group G PM2.5200-BEAS-2B-Exo+Mφ, group H PM2.5400-BEAS-2B-Exo+Mφ. **P<0.01, *P<0.05 vs group E. |
GW4869 Can Improve Mφ Inflammation
PM2.5200-BEAS-2B cells were treated with GW4869 and co-cultured with Mφ. The results showed that Mφ inflammatory cytokines IL-1β, IL-6 and TNF-α were significantly increased in group C (PM2.5–200-BEAS-2B+Mφ), and GW4869 could decrease the expression of inflammatory cytokines (Figure 5A and B). In group C, the level of miR-155-5p was significantly increased, SOCS1 expression was decreased, PP65 expression and PP65/P65 ratio were increased, and GW4869 could improve the effect of PM2.5 (Figure 5C–F).
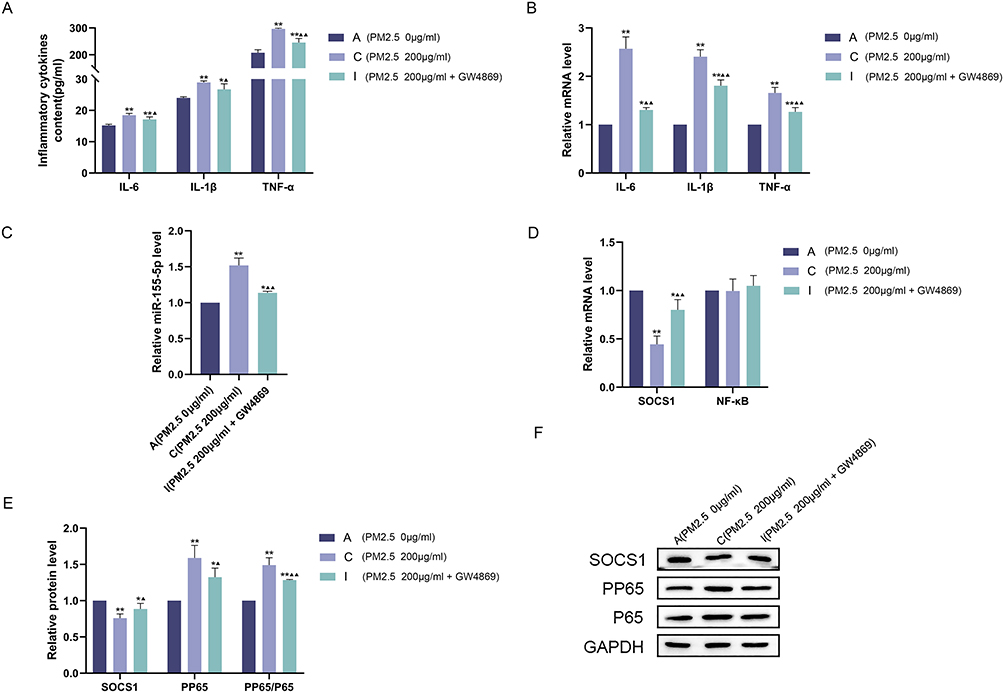 |
Figure 5 GW4869 improves inflammation in Mφ. PM2.5-BEAS-2B cells were pretreated with GW4869 and co-cultured with Mφ. (A) Cellular supernatant IL-6, IL-1β, TNF-α contents of Mφ; (B) mRNA expression levels of IL-6, IL-1β, TNF-α in Mφ; (C) miR-155-5p expression level in Mφ; (D) mRNA expression levels of SOCS1, NF-κB in Mφ; (E and F) Protein expression level of SOCS1, P65, PP65 in Mφ as detected by Western blot analysis as well as statistical analysis of gray values. Experimental group: group A PM2.50-BEAS-2B+Mφ, group C PM2.5200-BEAS-2B+Mφ, group I PM2.5200-BEAS-2B+GW4869+Mφ. **P<0.01, *P<0.05 vs group A;▲▲P<0.01,▲P<0.05 vs group C. |
Discussion
With the development of society, PM2.5 pollution has increasingly become a threat to human health. PM2.5 exposure most likely leads to direct toxicity, oxidative stress, and inflammation.
Jia et al6 showed that PM2.5 can cause lung inflammation in mice. BEAS-2B cell exposure to PM2.5 could elevate inflammatory cytokines. PM2.5 exposure can induce oxidative stress by activating NF-κB and MAPK signal transduction, enhance the expression of pro-inflammatory factors, reduce airway epithelial cell viability, trigger cell apoptosis, and results in airway barrier dysfunction.7 PM2.5 induces the release of IL-6, IL8, and IL-1β by human bronchial epithelial cells through the Wnt5a/Ror2 axis.35 Recently, the primary focus of research has been on the mechanism of airway epithelial cell inflammation after PM2.5 exposure. In our work, it was found that co-culture of PM2.5-stimulated BEAS-2B cells with Mφ can promote Mφ-mediated inflammatory response, suggesting that PM2.5 can indirectly affect Mφ-induced inflammation through the communication between BEAS-2B cells and Mφ. Further studies showed that miR-155-5p is overexpressed in PM2.5-stimulated BEAS-2B cells, and its expression increased with the increase in PM2.5 concentration. Similarly, we observed that miRNA-155-5p expression upregulated with PM2.5 concentration in BEAS-2B-derived exosomes. These data imply that PM2.5 exposure can change BEAS-2B-derived miRNA-155-5p, which may be transmitted by exosomes to Mφ to induce inflammatory changes.
Exosome plays a vital role in cell communication by transporting functional proteins, metabolites, and nucleic acids to recipient cells. Recent research has illustrated that exosomes are responsible for the development of lung diseases. Wang et al36 indicated that exosomes obtained from cigarette smoke extract-treated mouse AECs induce M1 macrophage polarization by inducing receptors in myeloid cells-1 in COPD. The expression of type I collagen, αSMA, BIP, XBP1s, and P-eIF2α were upregulated in macrophage-derived exosomes (SiO2-Exos) exposed to silicon and co-cultured with fibroblasts, suggesting that SiO2-Exos promoted pulmonary fibrosis.37 CSE-exposed BEAS-2B cells alleviated M2 macrophage polarization by modulating exosomes and inhibiting BEAS-2B cell EMT in COPD.34 We found that the exosomes derived from PM2.5-stimulated BEAS-2B cells can be internalized by Mφ, and Mφ-derived IL-6, IL-1β, and TNF-α were obviously increased after co-culturing. The exosome inhibitor GW4869 reduced Mφ-mediated inflammatory response after co-culturing with PM2.5-stimulated BEAS-2B cells. These data imply that BEAS-2B cells transmit signals to Mφ via exosomes to induce inflammatory response.
miRNA is diminutive, single-stranded, non-coding RNA molecule implicated in various cellular activities. miRNAs can be loaded onto exosomes, stably transported to target cells, and expressed to exert biological effects. In sepsis, miR-125b-5p in adipose-stem cell-derived exosomes may regulate the expression of Kelch-like cycloassociated protein 1 (Keap1)/nuclear transcription factor (Nrf2)/glutathione peroxidase 4 (GPX4), reduce iron-mediated death of pulmonary microvascular endothelial cells.38 miR-377-3p in HUCMSC-derived exosomes improved lipopolysaccharide-induced acute respiratory failure by targeting a regulatory associated protein of mTOR (RPTOR) and inducing autophagy.39 Wang et al40 investigated the role of pulmonary exosomes in PM2.5-stimulated cardiomyocyte apoptosis and impaired heart function and showed that pulmonary exosomes can transfer miR-421 and target cardiac ACE2, thereby mediating PM2.5-induced cardiac muscle cell death and impaired heart function. Similarly, our study revealed the role of exosomal miRNA in PM2.5-stimulated Mφ inflammatory responses. We exposed BEAS-2B cells to PM2.5, extracted exosomes from cell supernatant, and observed that exosomal miRNA-155-5P was upregulated with the increase in PM2.5 concentration. Extracted exosomes were co-cultured with Mφ, and the findings demonstrated that miRNA-155-5P expression increased in Mφ with the increase of PM2.5 concentration. Similarly, the expression of Mφ inflammatory factors also increased with changes in PM2.5 concentration.
miR-155 regulates inflammation in various pathological conditions. Using bioinformatics tools, SOCS1, a negative modulator of NF-κB, was found to be a promising target of miRNA-155. Inhibition of miRNA-155 significantly altered the inflammatory response after stroke.41 In staphylococcal enterotoxin B-induced acute respiratory failure, miRNA-155 promoted lung inflammation by targeting SOCS1.42 The total saponins from Panax japonicus reduced adipocyte inflammation by inhibiting miRNA-155/SOCS1/NF-κB signaling pathway.43 In addition, other signaling pathways besides NF-κB signaling pathway also play an important role in PM-induced pulmonary inflammation. TLR3 activation in macrophages has been observed in PM2.5-containing cigarette smoke.44 PM2.5 can cause lung inflammation in mice via the TLR4/NF-κB/IκBα signaling pathway.45 The NF-κB signaling pathway was studied in the study. This study also demonstrated that miRNA-155-5P was upregulated, SOCS1 expression was downregulated, PP65 expression was increased, and IL-6, IL-1β, and TNF-α were upregulated in Mφ after co-culturing PM2.5-stimulated BEAS-2B cells with Mφ. Comparable findings were achieved after co-culturing PM2.5-stimulated BEAS-2B cell-derived exosomes with Mφ. These results suggest that the miRNA-155-5P/SOCS1/NF-κB axis may be responsible for PM2.5-induced Mφ inflammation.
PM2.5 is involved in the occurrence and development of many respiratory diseases such as COPD, bronchial asthma and lung cancer. Chronic airway inflammation is one of the important mechanisms of COPD and bronchial asthma. The results of this study suggest that PM2.5 can promote Mφ inflammatory response by up-regulating the BEAS-2B cell-derived exosome miR-155-5P. This will provide a new possible theoretical basis for the pathogenesis of chronic pulmonary inflammatory diseases such as COPD and bronchial asthma.
In this study, we studied in vitro the promotion of macrophage-mediated inflammatory response by PM2.5 via the exosome miR-155-5p derived from airway epithelium cells, and the concentration of PM2.5 used in vitro may be somewhat different from that in vivo. The specificity of miRNA-155-5p also requires further study. In the follow-up experiment, we will further conducte in vitro experiments and study the specificity of miRNA-155-5p.
Conclusion
In summary, PM2.5 promoted Mφ inflammatory response by upregulating BEAS-2B cell-derived exosome miR-155-5P, which may be related to the miRNA-155-5P/SOCS1/NFκB axis. In addition, GW4869 improved Mφ inflammatory response. Together, our results suggest a new mechanism by which PM2.5-stimulated BEAS-2B cells promote Mφ inflammatory response. Exosomal miRNAs mediate cellular communication between BEAS-2B cells and Mφ, which may provide a new molecular target for treating pulmonary inflammation caused by PM2.5.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was financially supported by the National Natural Science Foundation of China (grant no. 82260010) and key research and development project of Gansu Province of China (grant no. 22YF7FA083).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wu M, Lai T, Jing D, et al. Epithelium-derived IL17A promotes cigarette smoke-induced inflammation and mucus hyperproduction. Am J Respir Cell Mol Biol. 2021;65:581–592.
2. Lugg ST, Scott A, Parekh D, et al. Cigarette smoke exposure and alveolar macrophages: mechanisms for lung disease. Thorax. 2022;77:94–101. doi:10.1136/thoraxjnl-2020-216296
3. Bu X, Xie Z, Liu J, et al. Global PM2.5-attributable health burden from 1990 to 2017: estimates from the global burden of disease study 2017. Environ Res. 2021;197:111123. doi:10.1016/j.envres.2021.111123
4. Landrigan PJ, Fuller R, Acosta NJR, et al. The lancet commission on pollution and health. Lancet. 2018;391:462–512.
5. Pun VC, Kazemiparkouhi F, Manjourides J, et al. Long-Term PM2.5 exposure and respiratory, cancer, and cardiovascular mortality in older US adults. Am J Epidemiol. 2017;186:961–969. doi:10.1093/aje/kwx166
6. Jia H, Liu Y, Guo D, et al. PM2.5-induced pulmonary inflammation via activating of theNLRP3/caspase-1 signaling pathway. Environ Toxicol. 2021;36:298–307. doi:10.1002/tox.23035
7. Zhao C, Wang Y, Su Z, et al. Respiratory exposure to PM2.5 soluble extract disrupts mucosal barrier function and promotes the development of experimental asthma. Sci Total Environ. 2020;730:139145. doi:10.1016/j.scitotenv.2020.139145
8. Shi J, Chen R, Yang C, et al. Association between fine particulate matter chemical constituents and airway inflammation: a panel study among healthy adults in China. Environ Res. 2016;150:264–268. doi:10.1016/j.envres.2016.06.022
9. Gao J, Lei T, Wang H, et al. Dimethylarginine dimethylaminohydrolase 1 protects PM2.5 exposure-induced lung injury in mice by repressing inflammation and oxidative stress. Part Fibre Toxicol. 2022;19. doi:10.1186/s12989-022-00460-3
10. Wu S, Ni Y, Li H, et al. Short-term exposure to high ambient air pollution increases airway inflammation and respiratory symptoms in chronic obstructive pulmonary disease patients in Beijing, China. Environ Int. 2016;94:76–82. doi:10.1016/j.envint.2016.05.004
11. Gao M, Liang C, Hong W, et al. Biomass-related PM2.5 induces mitochondrial fragmentation and dysfunction in human airway epithelial cells. Environ Pollut. 2022;292:118464. doi:10.1016/j.envpol.2021.118464
12. Cong LH, Li T, Wang H, et al. IL-17A-producing T cells exacerbate fine particulate matter-induced lung inflammation and fibrosis by inhibiting PI3K/Akt/mTOR-mediated autophagy. J Cell Mol Med. 2020;24:8532–8544. doi:10.1111/jcmm.15475
13. Fan X, Gao Y, Hua C, et al. Daphnetin ameliorates PM2.5-induced airway inflammation by inhibiting NLRP3 inflammasome-mediated pyroptosis in CS-exposed mice. Biomed Pharmacother. 2023;165:115047. doi:10.1016/j.biopha.2023.115047
14. Hsieh CC, Yu SH, Kuo HC, et al. Alleviation of PM2.5-induced alveolar macrophage inflammation using extract of fermented Chenopodium formosanum Koidz sprouts via regulation of NF-kappaB pathway. J Ethnopharmacol. 2024;318:116980. doi:10.1016/j.jep.2023.116980
15. Noonin C, Thongboonkerd V. Exosome-inflammasome crosstalk and their roles in inflammatory responses. Theranostics. 2021;11:4436–4451. doi:10.7150/thno.54004
16. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367. doi:10.1126/science.aau6977
17. Kadota T, Fujita Y, Yoshioka Y, et al. Extracellular vesicles in chronic obstructive pulmonary disease. Int J Mol Sci. 2016;17:1801. doi:10.3390/ijms17111801
18. Hough KP, Chanda D, Duncan SR, et al. Exosomes in immunoregulation of chronic lung diseases. Allergy. 2017;72:534–544. doi:10.1111/all.13086
19. Kubo H. Extracellular vesicles in lung disease. Chest. 2018;153:210–216. doi:10.1016/j.chest.2017.06.026
20. Zhang D, Lee H, Wang X, et al. A potential role of microvesicle-containing miR-223/142 in lung inflammation. Thorax. 2019;74:865–874. doi:10.1136/thoraxjnl-2018-212994
21. Liu F, Peng W, Chen J, et al. Exosomes derived from alveolar epithelial cells promote alveolar macrophage activation mediated by miR-92a-3p in sepsis-induced acute lung injury. Front Cell Infect Microbiol. 2021;11:646546.
22. Guiot J, Cambier M, Boeckx A, et al. Macrophage-derived exosomes attenuate fibrosis in airway epithelial cells through delivery of antifibrotic miR-142-3p. Thorax. 2020;75:870–881. doi:10.1136/thoraxjnl-2019-214077
23. Catalano M, O’Driscoll L. Inhibiting extracellular vesicles formation and release: a review of EV inhibitors. J Extracell Vesicles. 2020;9:1703244. doi:10.1080/20013078.2019.1703244
24. Mashima R. Physiological roles of miR-155. Immunology. 2015;145:323–333. doi:10.1111/imm.12468
25. Xu W, Feng S, Huang A. Role of miR-155 in inflammatory autoimmune diseases: a comprehensive review. Inflamm Res. 2022;71:1501–1517. doi:10.1007/s00011-022-01643-6
26. Jiang K, Yang J, Guo S, et al. Peripheral circulating exosome-mediated delivery of miR-155 as a novel mechanism for acute lung inflammation. Mol Ther. 2019;27:1758–1771. doi:10.1016/j.ymthe.2019.07.003
27. Li HF, Wu YL, Tseng TL, et al. Inhibition of miR-155 potentially protects against lipopolysaccharide-induced acute lung injury through the IRF2BP2-NFAT1 pathway. Am J Physiol Cell Physiol. 2020;319:C1070–C1081.
28. De Smet EG, Van Eeckhoutte HP, Avila CF, et al. The role of miR-155 in cigarette smoke-induced pulmonary inflammation and COPD. Mucosal Immunol. 2020;13:423–436. doi:10.1038/s41385-019-0241-6
29. Wang W, Bian H, Li F, et al. HBeAg induces the expression of macrophage miR-155 to accelerate liver injury via promoting production of inflammatory cytokines. Cell Mol Life Sci. 2018;75:2627–2641. doi:10.1007/s00018-018-2753-8
30. Zhang H, Zhao Z, Pang X, et al. Genistein protects against Ox-LDL-induced inflammation through MicroRNA-155/SOCS1-mediated repression of NF-ĸB signaling pathway in HUVECs. Inflammation. 2017;40:1450–1459. doi:10.1007/s10753-017-0588-3
31. Lee YJ, Han JY, Byun J, et al. Inhibiting Mer receptor tyrosine kinase suppresses STAT1, SOCS1/3, and NF-κB activation and enhances inflammatory responses in lipopolysaccharide-induced acute lung injury. J Leukoc Biol. 2012;91:921–932. doi:10.1189/jlb.0611289
32. Wang Y, Zhong Y, Sun K, et al. Identification of exosome miRNAs in bronchial epithelial cells after PM2.5 chronic exposure. Ecotoxicol Environ Saf. 2021;215:112127. doi:10.1016/j.ecoenv.2021.112127
33. Xu Z, Wang N, Xu Y, et al. Effects of chronic PM2.5 exposure on pulmonary epithelia: transcriptome analysis of mRNA-exosomal miRNA interactions. Toxicol Lett. 2019;316:49–59. doi:10.1016/j.toxlet.2019.09.010
34. He S, Chen D, Hu M, et al. Bronchial epithelial cell extracellular vesicles ameliorate epithelial–mesenchymal transition in COPD pathogenesis by alleviating M2 macrophage polarization. Nanomedicine. 2019;18:259–271. doi:10.1016/j.nano.2019.03.010
35. Zou W, Wang X, Hong W, et al. PM2.5 induces the expression of inflammatory cytokines via the Wnt5a/Ror2 pathway in human bronchial epithelial cells. Int J Chron Obstruct Pulmon Dis. 2020;15:2653–2662. doi:10.2147/COPD.S270762
36. Wang L, Chen Q, Yu Q, et al. Cigarette smoke extract-treated airway epithelial cells-derived exosomes promote M1 macrophage polarization in chronic obstructive pulmonary disease. Int Immunopharmacol. 2021;96:107700. doi:10.1016/j.intimp.2021.107700
37. Qin X, Lin X, Liu L, et al. Macrophage‐derived exosomes mediate silica‐induced pulmonary fibrosis by activating fibroblast in an endoplasmic reticulum stress‐dependent manner. J Cell Mol Med. 2021;25:4466–4477. doi:10.1111/jcmm.16524
38. Shen K, Wang X, Wang Y, et al. miR-125b-5p in adipose derived stem cells exosome alleviates pulmonary microvascular endothelial cells ferroptosis via Keap1/Nrf2/GPX4 in sepsis lung injury. Redox Biol. 2023;62:102655. doi:10.1016/j.redox.2023.102655
39. Wei X, Yi X, Lv H, et al. MicroRNA-377-3p released by mesenchymal stem cell exosomes ameliorates lipopolysaccharide-induced acute lung injury by targeting RPTOR to induce autophagy. Cell Death Dis. 2020;11. doi:10.1038/s41419-019-2196-7
40. Wang H, Wang T, Rui W, et al. Extracellular vesicles enclosed-miR-421 suppresses air pollution (PM(2.5))-induced cardiac dysfunction via ACE2 signalling. J Extracell Vesicles. 2022;11:e12222.
41. Pena-Philippides JC, Caballero-Garrido E, Lordkipanidze T, et al. In vivo inhibition of miR-155 significantly alters post-stroke inflammatory response. J Neuroinflammation. 2016;13:287. doi:10.1186/s12974-016-0753-x
42. Rao R, Rieder SA, Nagarkatti P, et al. Staphylococcal enterotoxin B-induced microRNA-155 targets SOCS1 to promote acute inflammatory lung injury. Infect Immun. 2014;82:2971–2979. doi:10.1128/IAI.01666-14
43. Gao Y, Wang R, Li L, et al. Total saponins from Panax japonicus reduce inflammation in adipocytes through the miR155/SOCS1/NFkappaB signaling pathway. Phytomedicine. 2023;115:154827. doi:10.1016/j.phymed.2023.154827
44. Koarai A, Yanagisawa S, Sugiura H, et al. Cigarette smoke augments the expression and responses of toll-like receptor 3 in human macrophages. Respirology. 2012;17(6):1018–1025. doi:10.1111/j.1440-1843.2012.02198.x
45. Zhang J, Chen X, Li H, et al. Selenium-enriched soybean peptides pretreatment attenuates lung injury in mice induced by fine particulate matters (PM2.5) through inhibition of TLR4/NF-κB/IκBα signaling pathway and inflammasome generation. Food Funct. 2022;13(18):9459–9469. doi:10.1039/D2FO01585D
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.