")
Back to Journals » Journal of Inflammation Research » Volume 17
Preclinical Models of Atopic Dermatitis Suitable for Mechanistic and Therapeutic Investigations
Authors Maskey AR , Mo X, Li XM
Received 17 March 2024
Accepted for publication 7 August 2024
Published 2 October 2024 Volume 2024:17 Pages 6955—6970
DOI https://doi.org/10.2147/JIR.S467327
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Anish R Maskey,1 Xian Mo,1,2 Xiu-Min Li1,3,4
1Department of Pathology, Microbiology and Immunology, New York Medical College, Valhalla, NY, 10595, USA; 2The Department of Allergy and Clinical Immunology, Guangzhou Institute of Respiratory Health, Guangzhou, People’s Republic of China; 3Department of Otolaryngology, New York Medical College, Valhalla, NY, 10595, USA; 4Department of Dermatology, New York Medical College, Valhalla, NY, 10595, USA
Correspondence: Xiu-Min Li, Department of Pathology, Microbiology, & Immunology, New York Medical College, Valhalla, New York, 10595, USA, Tel +1-914-595-1497, Email [email protected]
Abstract: Atopic dermatitis (AD) is a complex immune-mediated abnormality of the skin characterized by impaired barrier function, eczematous dermatitis, chronic pruritus and itch. The immunological response in AD is mediated by a Th2-dominated immune response in the early acute phase followed by a Th1/ Th2 mixed immune response in the chronic phase. AD is the first step of the “atopic march” that progresses into food allergy, allergic rhinitis, and asthma. Different models are indispensable for studying AD pathogenesis and for designing pre-clinical studies for therapeutic discovery. They reflect the characteristic morphological features of typical human AD with regard to epidermal thickening, hyperkeratosis, acanthosis, and spongiosis and help understand the immunopathogenesis of the disease with respect to IgE levels and cellular infiltration of eosinophils, mast cells, and lymphocytes. Although it is difficult to replicate all human AD clinical features in a model, several AD in vivo models comprising spontaneous, induced, transgenic, and humanized and in vitro models, including 2D, co-culture, and 3D, have been described previously. However, several questions remain regarding whether these models satisfactorily reflect the complexity of human AD. Therefore, this review comprehensively highlights the diversity of currently available models and provides insights into the selection of suitable models based on research questions. It also summarizes the diverse mechanisms associated with each model, which may be valuable for better study design to test new therapeutic options.
Keywords: atopic dermatitis, eczema, in vitro models, in vivo models, Th2 cytokine, microbiome
Introduction
Atopic dermatitis (AD) is an inflammatory condition that causes dry skin, rash, scaly patches, blisters, and inflammation. It is a non-contagious condition but is often painful and irritating, leading to recurrent eczematous lesions and changes in skin color. AD lesions are commonly distributed on the face, trunk, and limbs, particularly in the popliteal and cubital fossa. The prevalence of AD is high in many Western countries, and its incidence is rising in developing countries.1 The disease is common in 50–60% of individuals within the first year of life and 90% in patients up to five years.2 It can also begin during adolescence or adulthood. The clinical symptoms include erythema, papules, exudative lesions, and varying degrees of dryness as well as itch mediated by neuroinflammation. Drying and constant scratching also lead to skin thickening and lichenification. The pathogenesis of eczema is mediated by a complex interplay between multiple factors, such as altered skin barrier function, host genetics, immune cell abnormalities, and environmental factors, including exposure to specific pathogens such as Staphylococcus aureus. Patients diagnosed with eczema often show an increased incidence of other allergies such as food allergies, asthma, and allergic rhinitis.3 Furthermore, the majority of patients in the severe disease category show IgE-mediated sensitization to common allergens.4 This sequence of diseases is often called atopic or allergic diseases.
Pathological Mechanisms
The exact mechanism underlying the complex progression of AD is not yet well understood. However, considerable progress in understanding the disease mechanism has led to many findings related to genetic abnormalities, immunological dysfunction, and environmental factors as the prime drivers of AD exacerbation.5 Any factor contributing to an impaired skin barrier, including environmental factors, allergens, or genetic predispositions, leads to rupture of skin integrity, allowing allergen access to the epidermis. Activated skin cells release thymic stromal lymphopoietin (TSLP), interleukin (IL)-25, and IL-33 from keratinocytes, leading to the activation of the Th2 immune axis through type 2 innate lymphoid cells (ILC2) and Th2 cells. In the acute phase of AD, Th2 cytokines (IL-4, IL-5, and IL-13) play major roles in barrier dysfunction. Subsequently, these cytokines are responsible for the increased production of immunoglobulin E (IgE) and eosinophil recruitment. Clinically, IL-4 and IL-13 contribute to skin barrier defects, cutaneous infections, inflammation, thickening, and itching.6 Another cytokine, IL-31, accounts for the stimulation of itch receptors, ultimately leading to pruritus in AD (Figure 1). 7 Chronic AD is characterized by Th1/ Th2 mixed immune responses with intensification of Th2, Th1, and Th17 responses.8 Increased levels of Th1 cytokine interferon-gamma (IFN-γ), IL-12, and granulocyte-macrophage colony-stimulating factor (GM-CSF) are highly dominant in chronic AD. Furthermore, the increased activity of IL-36, which stimulates the maturation and function of DCs, drives T cell proliferation to amplify the skin immune response is observed.9 Additionally, dendritic cell subclass, Langerhans Cells (LC), and inflammatory dendritic epidermal cells (IDECs), which normally reside beneath the tight junction in the epidermis, penetrate the tight junction, and probably increase allergen uptake to prime T cells.10 Similarly, Th17 and Th22 (IL-17, IL-19, and IL-22) contribute to the formation of chronic skin lesions in AD. These cytokines are responsible for barrier function disruption, the elevation of anti-microbial proteins, and an increase in abnormal epidermal markers. Furthermore, IL-17 stimulates B cells to differentiate into IgE-producing plasma cells and promotes the release of IL-8, TNF-α, and TSLP, the combined actions of which trigger chronic AD (Figure 1).
 |
Figure 1 Mechanism of acute and chronic eczema. Genetic, environment and allergens lead to compromised skin barrier allowing allergen penetration along with impairment of skin microbiota (mostly Staphylococcus aureus) colonization. Activated keratinocytes release TSLP, IL-25 and IL-33 to activate skin LCs and IDECs to activate Th2 mediated immune response. In acute eczema, the Th2 immune response predominates with release of classical effector cytokines- IL-4, IL-5 and IL-13 causing further barrier dysfunction. B-cells differentiate to produce IgE which binds to basophils and mast cells. IL-31 binds to IL-31R and induces an itch response. In chronic eczema, Th1/Th17 immune response dominates with production of IL-17, IL-22. Activate Th1 cells produce IFN-γ and combined action of all these cytokines modulate local immune response leading to epidermal hyperplasia and skin lichenification. The colonization of Staphylococcus aureus is highly dominated in chronic eczema (created with BioRender). Abbreviations: LC, Langerhans cell; IDEC, Inflammatory dendritic epidermal cell; IL, Interleukin; ILC2, Innate lymphoid cell type 2; R, Receptor, TSLP, Thymic stromal lymphopoietin; IgE, Immunoglobulin E; S. aureus, Staphylococcus aureus. |
Skin Barrier Dysfunction
Skin barrier dysfunction is considered the first step in the development of atopic march.11 However, this remains controversial. The classical “atopic march” is but only one possibility with regards to progression of AD to atopic multimorbidity and that early life AD phenotypes, genotypes and environmental factors influence overall atopic disease trajectory and the relationships between and timing of onset of atopic comorbidities are more complex and many different patterns exist.12 A normal skin is comprised of a stratum corneum that acts as a permeable barrier to prevent transcutaneous water loss and provides an antimicrobial barrier favoring the colonization of nonpathogenic bacteria flora.13 Any factors contributing to the defect in this barrier, including environmental, allergens, or genetic predispositions, result in degradation of intracellular connections, higher protease activity, increased epidermal permeability, infiltration of antigens, and stimulation of proinflammatory cytokines.2
Genetic Susceptibility
Eczema has a strong heritability, as is evident from twin studies.14 Genome-wide association studies have identified 34 different loci that account for approximately 20% of eczema heritability.15 Most of which contain multiple genes with roles in immune response, T-cell activation, epidermal differentiation, and innate immunity.15–17 The most important risk gene identified in eczema is filaggrin (FLG), a major component of the stratum corneum (SC). It contributes to SC hydration and maintains the acidic pH of the skin.18 FLG mutation is associated with an increased risk of eczema, including early onset and persistent skin infection.19 Besides, other immune pathway genes associated with alterations in the Th2 signaling pathway have been identified. A gain of functional mutations in genes IL-4R and IL-13R is believed to lower FLG expression and contribute to eczema pathogenesis.20 Furthermore, other immune-related genes that have been reported in eczema include IL-31, IL-33, signal transducer and activator of transcription 6 (STAT-6), TSLP, toll-like receptor 2 (TLR-2), and FcRI-α.21,22 Recent studies have demonstrated that the vitamin D receptor and the cytochrome P450 variant (CYP27A1) are associated with eczema. Vitamin D plays a role in immune modulation and CYP27A1 is involved in metabolism.23
Th2 Skewed Immune Responses
Another important factor contributing to eczema pathogenesis is a dysregulated Th2 immune response. Excessive production of cytokines IL-4, IL-5, and IL-13 stimulates IgE antibodies and eosinophils in the tissue, leading to inflammation.24 These further damages the epidermal barrier and stimulates keratinocytes to produce proinflammatory cytokines such as TSLP, IL-25, and IL-33, which further activate the Th2-mediated immune response.25
Dysbiosis
The epidermis of the skin hosts various bacterial flora. These microorganisms participate in metabolic processes, maintain immune responses, protect against colonization by pathogenic microorganisms and support the epidermal barrier. Therefore, any disturbance by various microorganisms can lead to a number of diseases, including eczema. In eczema, a previous report indicated a reduction in commensal bacteria of the genera Streptococcus, Corynebacterium, Cutibacterium and Proteobacteria and an increase in the genus Staphylococcus (S. aureus in particular).26 Changes in qualitative and quantitative colonization by Staphylococcus aureus (S. aureus) precede the clinical manifestation of eczema and further exacerbate the disease.27 S. aureus has gained considerable attention for the development of eczema. It is a Gram-positive bacterium that appears on the skin of individuals with eczema. Skin lesion of 80–100% of patients with eczema are colonized by S. aureus. A correlation between eczema severity and colonization with S. aureus has been demonstrated, and it is believed that bacterial colonization is an important mechanism of aggravated skin lesions.28 Several factors associated with S. aureus infection contribute to eczema pathogenesis. First, the compromised epithelial barrier that leads to water loss provides a suitable platform for S. aureus colonization of the skin with S. aureus. Second, the adhesion factors located on the surface of the cell wall, such as fibronectin-binding proteins, clumping factors A and B, and iron-regulated surface determinants, facilitate binding on the epidermis of the skin.29 Through various virulence factors, S. aureus damages the epidermal layer and exacerbates the inflammation.30 Staphylococcal α-toxin causes membrane damage and lysis of keratinocytes, further enhancing biofilm formation, preventing bacterial clearance, damaging barrier components, and promoting the development of viral infection.31,32 Staphylococcal δ-toxin induces mast cell degranulation33 via PI3K and calcium influx, without IgE cross-linking.34 S. aureus isolates from the skin of eczema patients also released staphylococcal enterotoxins A, B, and C, as well as toxic shock syndrome toxin 1 (TSST-1), also known as a superantigen, and triggered inflammation by inducing uncontrolled activation of lymphocytes and macrophages.35 Superantigens are unconventional antigens that elicit a response by binding outside the complementarity-determining regions (CDRs) of their target immune receptor macromolecules (antibodies or T-cell receptors).36 These superantigens can also generate IgE responses and further enhance mast cell degranulation.37–39 Enterotoxin B increases the expression of IL-31, increasing itchiness, and inhibits filaggrin and AMP expression. Other virulence peptides, including phenol-soluble modulin (PSM-α), induce the release of keratinocyte IL-1α and IL-36α, which in turn induce IL-17 production and enhance neutrophil recruitment.40 Furthermore, S. aureus triggers Th2 skewing by initiating B cell activation via TLR2-dependent mechanisms.41 It also disrupts the proteolytic balance of the skin by producing proteases that disrupt the skin’s barrier.42 In addition to bacterial flora, fungi and viruses that colonize the skin are currently the focus of AD research. However, the role of fungi and viruses in patients with AD remains poorly understood. Shotgun metagenomic analysis could provide valuable insights into the species and subspecies levels in the whole picture, with the hope that the complex interplay between numerous microorganisms and allergic diseases, such as AD, can be delineated in a more accurate and precise manner.43,44
Murine Models to Study AD Pathophysiology
Mouse models have been valuable tools for studying and investigating mechanisms associated with complicated disease progression and optimizing the effect of drugs on multiple phenotypes and endotypes. These models are valuable for clinical interventional research involving novel therapeutics and preclinical platforms. There are a few challenges associated with selecting the right model to replicate human diseases in mice, as the majority of models documented to date cannot simulate the complex disease entity. It is clear that certain criteria need to be prioritized when selecting a murine model to replicate human AD. Although some of these defined models mimic the core clinical, immunological, histological, and neurophysiological characteristics of human AD, biomarkers that are of great therapeutic importance and can predict disease outcomes in human AD seem to be missing in these models.
To date, more than 20 AD mouse models have been reported. Although these models do not completely display all the characteristic features of human AD, several models that exhibit some AD-like symptoms but lack others have been developed and analyzed. These models can be broadly classified into four main categories: (1) mice induced by the application of exogenous agents, (2) genetically engineered mice in which certain genes of interest are overexpressed or deleted, (3) spontaneous mice, and (4) humanized mouse models. Based on the current understanding, selecting a system to answer specific questions that generate preclinical and clinical data as well as mechanistic and conceptual insights that are more clinically relevant to human AD using these mouse models is challenging. Therefore, it is necessary to have a deep understanding of how these models differ from one another, and which models best represent classical human AD. In this review, we discuss some of the existing models and provide a detailed comparison of how they differ from each other, and which best reflects human AD.
Induced Mice by Application of Exogenous Agents
AD induction using exogenous agents can be classified into different categories, depending on the agents used: haptens, antigens, and cytokines (Table 1). The advantage of using the induced model is that it allows the controlled application of different agents and provides the flexibility to monitor induction in a timely manner. This can be applied to many different strains of mice depending on what is intended to be achieved. The most common method of application of these agents to the skin is experimental disruption of the epidermal barrier and epicutaneous application. This can be achieved by tape stripping to disrupt the epidermal barrier, thereby allowing for increased allergen penetration.45 These models allow for the exploration of the roles of different allergens in AD.46 Moreover, studies have shown that these models contribute to a better understanding of mast cell activation and the role of IgE in AD47,48 and further understand the role of different chemokines, such as CCL20,49 TARC,50 and eosinophil chemoattractant protein eotaxin.51 One main disadvantage of this method is the standardization of some key agents used. Furthermore, there are many documented variable protocols, and each investigator has developed their own dosing, concentration, and duration using these agents. The immune response substantially differs between mouse strains. Finally, this process is labor-intensive, as it requires daily application of the agents for a longer period of time.
 |
Table 1 Induced Murine Model of AD |
Some widely used methods to induce AD in murine models using haptens have been documented. Haptens are small molecule irritants that elicit an immune response. Therefore, chronic skin surface application can lead to AD-like diseases in mice. Some of these include oxazolone, 2,4-dinitrochlorobenzene (DNCB), 2,4-dinitrofluorobenzene (DNFB), chloromethylisothiazolinone/methylisothiazolinone (CMIT/MIT), ovalbumin (OVA) and 2, 4, 6-trinitrochlorobenzene (TNCB). Oxazolone is the most commonly used hapten for AD induction.68 Repeated exposure to DNCB and DNFB often leads to hypersensitivity reactions in the skin and is not considered an AD model.69 Moreover, the majority of the immune responses induced by these haptens are Th1-dominant and often lead to keratinocyte necrosis.70 The majority of studies have used repeated application of these agents to the surface of the skin to develop AD-like lesions. Some of the strains widely used to achieve this phenotype include hairless mice, CBA/J, BALB/c, and NC/Nga mice.
The use of antigens to induce AD in the mouse skin has also shown promising results in murine models. Antigens that have been used include ovalbumin, DNFB/DfE, crude extract of house dust mite (HDM), HDM in combination with Staphylococcus exotoxin B (SEB), and Staphylococcus exotoxin C (SEC). These antigens are then exposed to the external surface of the skin through repeated epicutaneous exposure. The three most common strains used in this study were BALB/c, NC/Nga, and DS-Nh. OVA, a protein allergen, is the most commonly used antigen to induce AD. This has led to murine models in where Th2 cytokines-IL-4, IL-5, and IL-13 predominate, followed by IgE synthesis.69 Furthermore, these models showed activation of IL-17 immune response.46 It is well known that S. aureus colonization is common in AD patients and more than 90% of the patients are colonized with S. aureus.71 The application of both HDM and staphylococcal enterotoxin B (SEB) in NC/Nga mice induced severe dermatitis and relatively mild dermatitis in BALB/c mice.60
Another approach involves the use of cytokines to induce AD. This approach involves the use of agents such as IL-23, vitamin D or its analog MC903 (calcipotriol), and particulate matter (PM). Some strains widely used for cytokine-based induction of AD include chemokine receptor 2 deficient (CCR2−/−), C57BL/6J, and hairless mice. The most common method of inducing AD using these agents is repeated application on the skin surface. MC903 is a well-characterized cytokine used to induce AD in mice. Atopic skin inflammation is dependent on TSLP and contributes to Th2 mediated cytokine expression via ILC2 mediated pathways.72,73 Furthermore, MC903 causes severe eosinophilic inflammation and skin irritation, which are the features of human AD. Th2 inflammation appeared to be dominant in these mouse models, although mixed endotypes were present. What is more special is the PM mice model. In this model, PM2.5, which induces TNF-α expression, may account for the AD phenotypes, with increased Th17 polarization linked to TNF-α exposure67 (Table 1).
Genetically Engineered Mice
The use of genetically engineered mice to study AD-related pathogenesis has been well-documented in the literature. These mice were genetically modified to alter the expression of AD-related genes in order to better understand the biological functions of each molecule. These genes were either overexpressed or ablated (Table 2). Some key genes that play important roles in AD pathogenesis and are overexpressed in murine models include IL-4, IL-13, IL-31, TSLP, IL-18, IL-33, Caspase-1, Cathepsin E, SCCE, and apolipoprotein C1. Similarly, the two key genes ablated in murine models are ADAM17 and CARMA-1. Genetically engineered mice provide detailed information on the gene-specific mechanisms of AD progression and can be valuable if these strains are crossed with other strains. In contrast, these mice are very expensive to maintain, as if required, for regular breeding, genotyping, and selecting the one with the desired alteration in the gene of interest. Furthermore, the generation of these mice is labor-intensive, and there is always a high chance of undesirable effects from unwanted or expected gene expression or alterations.
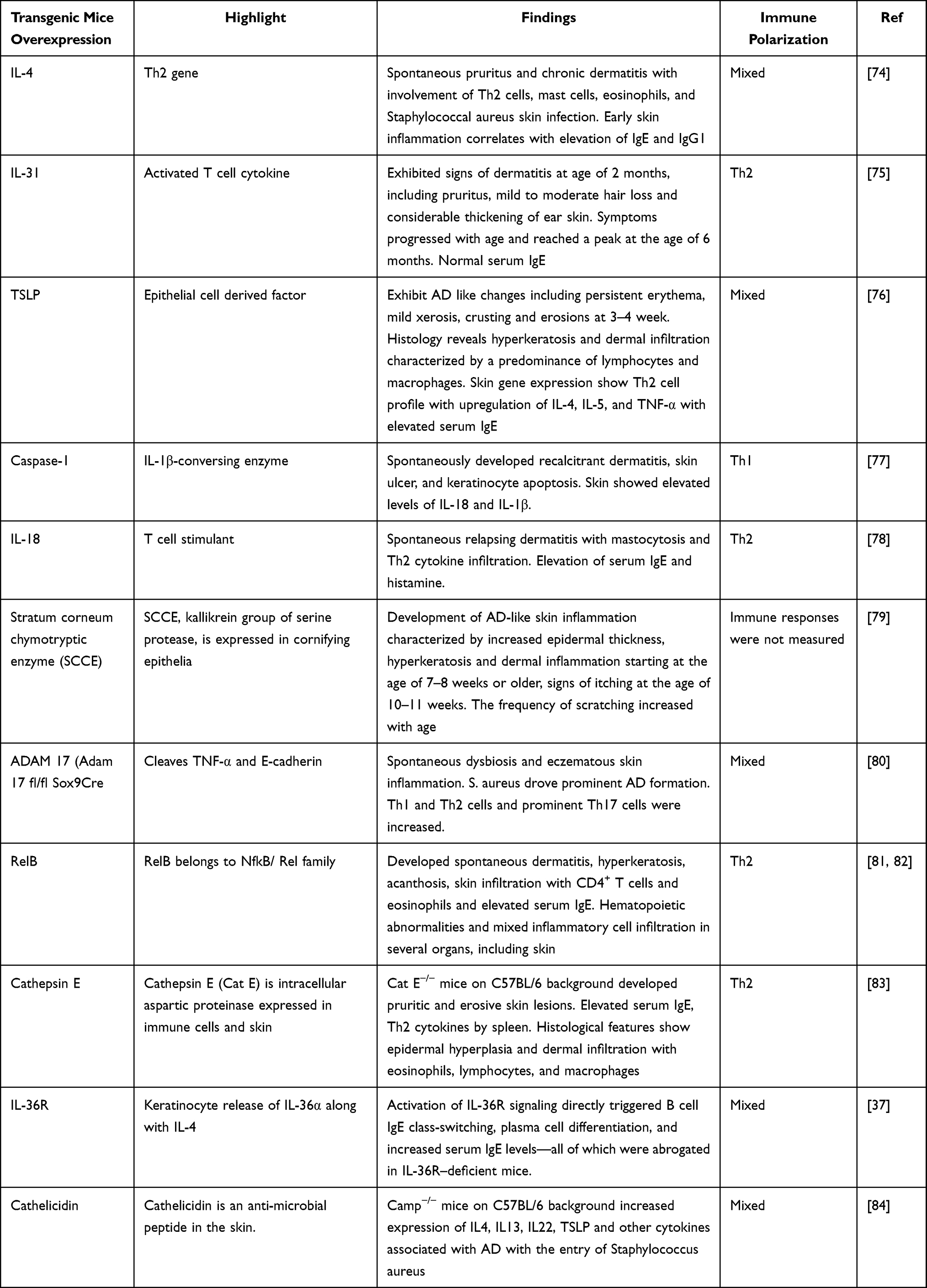 |
Table 2 Genetically Engineered Mice |
Spontaneous Mice
Spontaneous mice are often referred to as inbred mice. They show characteristics of AD-like lesions under normal housing conditions. The phenotypes observed in these mice arise from spontaneous genetic mutations caused by environmental factors or defects in DNA replication. Some of the well-defined stains used to study AD pathogenesis include Nc/Nga, Flaky tail, STAT-6 deficient, Otsuka, Sharpincpdm, Ds-Nh, and Naruto Research Institute (Table 3). The Nc/Ng inbred mouse is the first mouse model of AD reported.85 Flaky-tail mice have filaggrin gene defects and provide a better understanding of the role of filaggrin, as well as the role of Th2 related cytokines and chemokines in AD.86,87 This is the most widely studied spontaneous model and provides detailed information on the involvement of microbes and other environmental factors in the pathogenesis of AD. All mice showed multiple mixed immune responses (Table 3) and developed skin lesions beginning with itching, erythema, deep tissue erosion, excoriation, skin dryness, and hair loss.88–90
 |
Table 3 Spontaneous AD Mice |
The primary advantage of using these models is that they provide a natural course of human AD under normal conditions, without additional sensitization or manipulation. They can be enhanced by percutaneous sensitization to different haptens and allergens to exacerbate preexisting AD lesions. Similarly, these models allow the investigation of genetic abnormalities associated with AD. The major pitfalls associated with these strains are the lack of genetic information in some strains, development of disease only in an uncontrolled environment, disease progression in a very short period of time, and variable induction protocols when combined with other haptens or allergens. Other disadvantages of using this model include large variations among the groups, as some models do not spontaneously develop dermatitis under specific pathogen-free conditions.
Humanized AD Mouse Models
In addition to these three models, the skin explant mouse model may provide a new and promising approach. For example, human skin xenotransplants on severe combined immunodeficiency (SCID) mice (humanized AD mouse models) offer a particularly promising preclinical research alternative to the currently available “AD” mouse models. These include the generation of entirely genetically engineered human skin on the back of NOC/SCID mice,96 bioengineering based humanized mouse models using samples derived from patients or healthy donors.97 And recent model where skin from health donors xenotransplanted on SCID mice followed by in-vivo intradermal injection of pre-activated, autologous human lymphocytes, CD4+ and CD8+ T cells was sufficient to induce AD, indistinguishable from spontaneous AD.98 These newer humanized models may be valuable for studying AD, as they mimic many spontaneous AD features with respect to lipid metabolism abnormalities, response to clinical AD treatment, and relapse upon exposure to insults. Furthermore, these SCID skin mouse models allow analysis of various steps in the migration of human T cells into the skin in response to selective chemokines.29
In vitro Models to Study AD Pathogenesis
Despite animal models serving as valuable tools for studying AD pathogenesis, strict implementation of ethical grounds limits the use of animals. Therefore, many alternative in vitro models that recapitulate AD pathogenesis of AD have been developed. These include 2D monolayers and co-culture models, 3D-skin models, and skin-on-a-chip systems. Monolayers are easily generated by culturing HaCaT cells or primary human keratinocytes (KC) with cytokines IL-4, IL-1399 and TSLP.100 Co-culture modalities have been widely used to understand the roles of cytokines and chemokines in AD. These models are generated by co-culturing fibroblasts (FB) and KC with basophils, eosinophils, and activated T-cells to study the expression and release of chemokines (CXCL8, CLL2, CCL5, CXCL8, and CCL4).101,102 Similarly, different 3D organoid models have been implemented to study the pathomechanisms and uncover therapeutic approaches. The two most common 3D models used to study epidermal properties are reconstructed human epidermis (RHE) and full-thickness human skin equivalents (HSE). Both models closely mimicked the in vivo characteristics of the human epidermis in terms of barrier function, differentiation, and morphology, as depicted by histology. Most RHE models rely on external challenges involving a variety of cytokines including IL-4, IL-13, TNF-α, IL-22, IL-25, and poly I:C.103–108 Lastly, adding to the complexity of AD, a relatively new technology that combines the concept of 2D and 3D cultures is developed as an alternative to current AD in vitro models. These models have used robust tissue engineering to generate a range of models that increase the movement of signaling molecules and cell-cell communication,109,110 increase vascularization for long-term survival, functionality, viable engraftment,111 and cell transmigration.112
Discussion
The progression of AD is very complex and consists of several different stages ranging from acute to chronic, with other phenotypes that are prominently associated with ichthyosis, keratosis pilaris, distinct variants such as nummular eczema and atopic prurigo.113 Furthermore, AD exhibits both racial and ethnic variation.114 It is evident that no single in vitro or murine model can stimulate all the phenotypes and endotypes that exist in human AD. Furthermore, the claims that these existing models emulate based on the data generated do not precisely define which phenotype or endotype of AD they are referring to, leading to a generalized claim that only a universal AD phenotype exists. It is very evident that researchers should consider minimal criteria to best reflect human AD to replicate more convincing, clinically relevant, and therapeutically equipped mouse models of AD.
Based on information extracted from the literature, it appears that their needs are a set of defined basic parameters considered to replicate human AD in mice. Impairment of skin barrier integrity is considered the most important factor contributing to AD; therefore, genetic abnormalities in the structural proteins filaggrin,5 TSLP,115 and claudin-1116 seem to be of utmost importance. These factors are associated with epidermal hyperplasia and hyperproliferation with increase in expression of keratin-16 ultimately leading to excessive trans epidermal water loss (TEWL) and increased levels PAR2 in keratinocytes driving barrier dysfunction, inflammation and sensitize skin to drive several levels of neuro-epidermal communication to drive itch response.117 Recent studies show IL-31 mediated signaling pathway is involved in inducing the itch response and orchestrating the release of a series of chemokines and cytokines from the skin.118 It is still not well established whether stress can trigger AD without previous sensitization and requires further investigation. Therefore, monitoring the itch response by scratching behavior is an important consideration in murine models. These findings suggest that neurogenic factors are highly relevant to the worsening of AD, and their interaction with immune pathways should be considered in AD studies.119,120 Other important factor contributing to AD and needs to be considered include dysbiosis of the skin microbiome particularly the overgrowth of Staphylococcus aureus and progressive increase in terminally differentiated proteins and antimicrobial peptides S100A7, S100A8, and S100A.121 Likewise, T-cell infiltration, Th2 cells, Th17, Th22, and type 2 innate lymphoid cells equally contribute to clinical manifestations and further favor epidermal barrier disruption.121,122 In addition, the presence of other inflammatory cells, including mast cells and eosinophils, should be considered in AD pathogenesis.5 A larger number of biomarkers have been identified in AD that correlate with disease severity and need attention when considering murine models of AD. Biomarkers include TARC, CCL17, and CCL27.123,124
Besides these findings, responses to the use of well-standardized clinical care for human patients that are available for the treatment of AD, such as glucocorticoids, calcineurin inhibitors, specific Th2 inhibitors against IL-4, IL-13 mediated signaling must be included to determine the effectiveness of these models in the replication of human AD.125 Furthermore, heterogeneity across different age groups and races/ethnicities, specifically focusing on different endotypes on how AD varies in these backgrounds with respect to their IgE levels, epidermal barrier genes, cytokines, and inflammatory pathways, may provide important frameworks for selecting murine models for more robust targeted therapy design in this era of personalized medicine.126
Considering these important factors, no single model can fully recapitulate the pathogenesis of AD. Moreover, none of the existing models reproduce a sufficient number of characteristics observed in human AD. This is because the anatomy of human and mouse skin is very different. Moreover, transcriptome-profiling studies have demonstrated that a full-blown AD mouse model shares approximately 4%–37% homology with human AD.53,127 Therefore, the selection of a murine model while considering AD studies should rely on outcome measures and should be restricted to a specific phenotype to make a credible claim. There needs to be a general consortium on minimal criteria that must be considered in any murine model, which is well defined in any endotype of AD, before selecting these murine models to make a big claim or design any therapeutic approaches. Furthermore, transplantation of human lesional skin in SCID mice may be an alternative approach to study mechanisms and therapeutic approaches and has shown fully developed human skin grafts expressing human adhesion molecules and selectins.29 However, a human skin biopsy is a painful process that is not readily available. To overcome these challenges, intradermally injected stimulated PBMCs in vitro with appropriate cytokines that can robustly induce skin lesions have been developed.69 Furthermore, the advancement in 3D models has significantly helped overcome some of the limitations, and it seems to be a reliable tool for studying AD at the cellular level. These in vitro models display various AD features, such as abnormal differentiation, loss of structural proteins, inflammation, hyperproliferation, disease-associated gene expression patterns, and apoptosis.128 Further upgrades in technologies are constantly needed to gain a closer understanding of the signaling processes and pathogenesis of skin diseases such as AD.
Conclusion
Overall, we now have a better understanding of AD pathogenesis. Therefore, the use of existing murine models, as well as exploring newer alternative models, especially the humanized mouse model and 3D in vitro models to replicate unique AD endotypes, might be a suitable approach to maximize human AD needs, understand immunological signatures, identify novel prevention strategies, and design appropriate therapeutic studies.
Abbreviations
AD, Atopic dermatitis; Th, T helper; ESAI, Eczema Area and Severity Index; SCORAD, Scoring Atopic Dermatitis; TSLP, Thymic stromal lymphopoietin; IL, Interleukin; IgE, Immunoglobulin E; IFN, Interferon; GM-CSF, Granulocyte-macrophage colony-stimulating factor; TNF, Tumor necrosis factor; FLG, Filaggrin; SC, Stratum corneum; STAT, Signal Transducer and Activator of Transcription); TLR, Toll like receptor; CYP, Cytochrome; S. aureus, Staphylococcus aureus; PI3K, Phosphoinositide 3-kinase; TSST, Toxic shock syndrome toxin; AMP, Antimicrobial peptide; PSM, Phenol-soluble modulin; FDA, Food and Drug Administration; CCL, Chemokine; TARC, Thymus and activation-regulated chemokine C; DNCB, Dinitrochlorobenzene; DNFB, Dinitrofluorobenzene; CMIT/ MIT, chloromethylisothiazolinone/methylisothiazolinone; OVA, Ovalbumin; TNCB, 2, 4, 6-trinitrochlorobenzene; SEC, Staphylococcus exotoxin C; HDM, House dust mite; SEB, Staphylococcus enterotoxin B; DfE, Dermatophagoides farina; PM, Particulate matter; CT, Cholera toxin; ADAM, A Disintegrin And Metalloprotease; CARMA, Caspase recruitment domain family; SCCE, Stratum corneum chymotryptic enzyme; TEWL, Trans epidermal water loss; PAR, Protease-activated receptor; SCID, Severe combined immunodeficiency; PBMC, Peripheral blood mononuclear cells.
Acknowledgment
We thank Henry Ehrlich for proofreading this manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by The Lie-Artati Family Fund (ORA LOG NO.:012756-101), and The Study of Integrative Medicine (ORA LOG NO.: 012874-101) to Xiu-Min Li at New York Medical College.
Disclosure
X-M Li received research support to her institution from the National Institutes of Health (NIH)/National Center for Complementary and Alternative Medicine (NCCAM) # 1P01 AT002644725-01“Center for Chinese Herbal Therapy 320 (CHT) for Asthma”, and grant #1R01AT001495-01A1 and 2R01 AT001495-05A2, NIH/NIAID R43AI148039, NIH/NIAID 1R21AI176061-01, NIH/NIAID 1R44AI177183-01, NIH/NIAID 1R41AI172572-01A1, Food Allergy Research and Education (FARE), Winston Wolkoff Integrative Medicine Fund for Allergies and Wellness, the Parker Foundation and Henan University of Chinese Medicine, the Study of Integrative Medicine, the Lie-Artati Family Fund; received consultancy fees from FARE and Johnson & Johnson Pharmaceutical Research & Development, L.L.C. Bayer Global Health LLC; received royalties from UpToDate; share US patent US7820175B2 (FAHF-2), US10500169B2 (XPP), US10406191B2 (S. Flavescens), US10028985B2 (WL); US11351157B2 (nanoBBR); take compensation from her practice at Center for Integrative Health and Acupuncture PC; US Times Technology Inc is managed by her related party; is a member of General Nutraceutical Technology LLC. The authors A Maskey, and X Mo declare that they have no conflict of interest.
References
1. Raimondo A, Lembo S. Atopic Dermatitis: epidemiology and Clinical Phenotypes. Dermatol Pract Concept. 2021;11(4):e2021146. doi:10.5826/dpc.1104a146
2. Sroka-Tomaszewska J, Trzeciak M. Molecular Mechanisms of Atopic Dermatitis Pathogenesis. Int J Mol Sci. 2021;22(8):4130. doi:10.3390/ijms22084130
3. Hill DA, Spergel JM. The atopic march: critical evidence and clinical relevance. Ann Allergy Asthma Immunol. 2018;120(2):131–137. doi:10.1016/j.anai.2017.10.037
4. Langan SM, Irvine AD, Weidinger S. Atopic dermatitis. Lancet. 2020;396(10247):345–360. doi:10.1016/s0140-6736(20)31286-1
5. Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers. 2018;4(1):1. doi:10.1038/s41572-018-0001-z
6. Bieber T. Interleukin-13: targeting an underestimated cytokine in atopic dermatitis. Allergy. 2020;75(1):54–62. doi:10.1111/all.13954
7. Sonkoly E, Muller A, Lauerma AI, et al. IL-31: a new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol. 2006;117(2):411–417. doi:10.1016/j.jaci.2005.10.033
8. Tsoi LC, Rodriguez E, Stölzl D, et al. Progression of acute-to-chronic atopic dermatitis is associated with quantitative rather than qualitative changes in cytokine responses. J Allergy Clin Immunol. 2020;145(5):1406–1415. doi:10.1016/j.jaci.2019.11.047
9. Foster AM, Baliwag J, Chen CS, et al. IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J Immunol. 2014;192(12):6053–6061. doi:10.4049/jimmunol.1301481
10. Bäsler K, Brandner JM. Tight junctions in skin inflammation. Pflugers Arch. 2017;469(1):3–14. doi:10.1007/s00424-016-1903-9
11. Lowe AJ, Leung DYM, Tang MLK, Su JC, Allen KJ. The skin as a target for prevention of the atopic march. Ann Allergy Asthma Immunol. 2018;120(2):145–151. doi:10.1016/j.anai.2017.11.023
12. Paller AS, Spergel JM, Mina-Osorio P, Irvine AD. The atopic march and atopic multimorbidity: many trajectories, many pathways. J Allergy Clin Immunol. 2019;143(1):46–55. doi:10.1016/j.jaci.2018.11.006
13. Elias PM. Stratum corneum defensive functions: an integrated view. J Invest Dermatol. 2005;125(2):183–200. doi:10.1111/j.0022-202X.2005.23668.x
14. Thomsen SF, Ulrik CS, Kyvik KO, et al. Importance of genetic factors in the etiology of atopic dermatitis: a twin study. Allergy Asthma Proc. 2007;28(5):535–539. doi:10.2500/aap2007.28.3041
15. Paternoster L, Standl M, Waage J, et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet. 2015;47(12):1449–1456. doi:10.1038/ng.3424
16. Weidinger S, Willis-Owen SA, Kamatani Y, et al. A genome-wide association study of atopic dermatitis identifies loci with overlapping effects on asthma and psoriasis. Hum Mol Genet. 2013;22(23):4841–4856. doi:10.1093/hmg/ddt317
17. Martin MJ, Estravís M, García-Sánchez A, Dávila I, Isidoro-García M, Sanz C. Genetics and Epigenetics of Atopic Dermatitis: an Updated Systematic Review. Genes. 2020;11(4):442. doi:10.3390/genes11040442
18. Egawa G, Kabashima K. Barrier dysfunction in the skin allergy. Allergol Int. 2018;67(1):3–11. doi:10.1016/j.alit.2017.10.002
19. Irvine AD, McLean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med. 2011;365(14):1315–1327. doi:10.1056/NEJMra1011040
20. Hussein YM, Shalaby SM, Nassar A, Alzahrani SS, Alharbi AS, Nouh M. Association between genes encoding components of the IL-4/IL-4 receptor pathway and dermatitis in children. Gene. 2014;545(2):276–281. doi:10.1016/j.gene.2014.04.024
21. Nedoszytko B, Reszka E, Gutowska-Owsiak D, et al. Genetic and Epigenetic Aspects of Atopic Dermatitis. Int J Mol Sci. 2020;21(18):6484. doi:10.3390/ijms21186484
22. Kaufman BP, Guttman-Yassky E, Alexis AF. Atopic dermatitis in diverse racial and ethnic groups-Variations in epidemiology, genetics, clinical presentation and treatment. Exp Dermatol. 2018;27(4):340–357. doi:10.1111/exd.13514
23. Suzuki H, Makino Y, Nagata M, et al. A rare variant in CYP27A1 and its association with atopic dermatitis with high serum total IgE. Allergy. 2016;71(10):1486–1489. doi:10.1111/all.12950
24. Kim KH. Overview of atopic dermatitis. Asia Pac Allergy. 2013;3(2):79–87. doi:10.5415/apallergy.2013.3.2.79
25. Klonowska J, Gleń J, Nowicki RJ, Trzeciak M. New Cytokines in the pathogenesis of Atopic Dermatitis-New Therapeutic Targets. Int J Mol Sci. 2018;19(10):3086. doi:10.3390/ijms19103086
26. Paller AS, Kong HH, Seed P, et al. The microbiome in patients with atopic dermatitis. J Allergy Clin Immunol. 2019;143(1):26–35. doi:10.1016/j.jaci.2018.11.015
27. Meylan P, Lang C, Mermoud S, et al. Skin Colonization by Staphylococcus aureus Precedes the Clinical Diagnosis of Atopic Dermatitis in Infancy. J Invest Dermatol. 2017;137(12):2497–2504. doi:10.1016/j.jid.2017.07.834
28. Goh CL, Wong JS, Giam YC. Skin colonization of Staphylococcus aureus in atopic dermatitis patients seen at the National Skin Centre, Singapore. Int J Dermatol. 1997;36(9):653–657. doi:10.1046/j.1365-4362.1997.00290.x
29. Carballido JM, Biedermann T, Schwärzler C, de Vries JE. The SCID-hu Skin mouse as a model to investigate selective chemokine mediated homing of human T-lymphocytes to the skin in vivo. J Immunol Methods. 2003;273(1–2):125–135. doi:10.1016/s0022-1759(02)00422-2
30. Geoghegan JA, Irvine AD, Foster TJ. Staphylococcus aureus and Atopic Dermatitis: a Complex and Evolving Relationship. Trends Microbiol. 2018;26(6):484–497. doi:10.1016/j.tim.2017.11.008
31. Alexander H, Paller AS, Traidl-Hoffmann C, et al. The role of bacterial skin infections in atopic dermatitis: expert statement and review from the International Eczema Council Skin Infection Group. Br J Dermatol. 2020;182(6):1331–1342. doi:10.1111/bjd.18643
32. Brauweiler AM, Goleva E, Leung DYM. Th2 cytokines increase Staphylococcus aureus alpha toxin-induced keratinocyte death through the signal transducer and activator of transcription 6 (STAT6). J Invest Dermatol. 2014;134(8):2114–2121. doi:10.1038/jid.2014.43
33. Syed AK, Reed TJ, Clark KL, Boles BR, Kahlenberg JM. Staphylococcus aureus phenol-soluble modulins stimulate the release of proinflammatory cytokines from keratinocytes and are required for induction of skin inflammation. Infect Immun. 2015;83(9):3428–3437. doi:10.1128/iai.00401-15
34. Nakamura Y, Oscherwitz J, Cease KB, et al. Staphylococcus δ-toxin induces allergic skin disease by activating mast cells. Nature. 2013;503(7476):397–401. doi:10.1038/nature12655
35. Spaulding AR, Salgado-Pabón W, Kohler PL, Horswill AR, Leung DY, Schlievert PM. Staphylococcal and streptococcal superantigen exotoxins. Clin Microbiol Rev. 2013;26(3):422–447. doi:10.1128/cmr.00104-12
36. Deacy AM, Gan SK, Derrick JP. Superantigen Recognition and Interactions: functions, Mechanisms and Applications. Front Immunol. 2021;12:731845. doi:10.3389/fimmu.2021.731845
37. Patrick GJ, Liu H, Alphonse MP, et al. Epicutaneous Staphylococcus aureus induces IL-36 to enhance IgE production and ensuing allergic disease. J Clin Invest. 2021;131(5). doi:10.1172/jci143334
38. Ong PY, Patel M, Ferdman RM, Dunaway T, Church JA. Association of staphylococcal superantigen-specific immunoglobulin e with mild and moderate atopic dermatitis. J Pediatr. 2008;153(6):803–806. doi:10.1016/j.jpeds.2008.05.047
39. Kim J, Kim BE, Ahn K, Leung DYM. Interactions Between Atopic Dermatitis and Staphylococcus aureus Infection: clinical Implications. Allergy Asthma Immunol Res. 2019;11(5):593–603. doi:10.4168/aair.2019.11.5.593
40. Nakagawa S, Matsumoto M, Katayama Y, et al. Staphylococcus aureus Virulent PSMα Peptides Induce Keratinocyte Alarmin Release to Orchestrate IL-17-Dependent Skin Inflammation. Cell Host Microbe. 2017;22(5):667–677.e5. doi:10.1016/j.chom.2017.10.008
41. Bekeredjian-Ding I, Inamura S, Giese T, et al. Staphylococcus aureus protein A triggers T cell-independent B cell proliferation by sensitizing B cells for TLR2 ligands. J Immunol. 2007;178(5):2803–2812. doi:10.4049/jimmunol.178.5.2803
42. Williams MR, Nakatsuji T, Sanford JA, Vrbanac AF, Gallo RL. Staphylococcus aureus Induces Increased Serine Protease Activity in Keratinocytes. J Invest Dermatol. 2017;137(2):377–384. doi:10.1016/j.jid.2016.10.008
43. Guzzo GL, Andrews JM, Weyrich LS. The Neglected Gut Microbiome: fungi, Protozoa, and Bacteriophages in Inflammatory Bowel Disease. Inflamm Bowel Dis. 2022;28(7):1112–1122. doi:10.1093/ibd/izab343
44. Vemuri R, Shankar EM, Chieppa M, Eri R, Kavanagh K. Beyond Just Bacteria: functional Biomes in the Gut Ecosystem Including Virome, Mycobiome, Archaeome and Helminths. Microorganisms. 2020;8(4):483. doi:10.3390/microorganisms8040483
45. Galand C, Leyva-Castillo JM, Yoon J, et al. IL-33 promotes food anaphylaxis in epicutaneously sensitized mice by targeting mast cells. J Allergy Clin Immunol. 2016;138(5):1356–1366. doi:10.1016/j.jaci.2016.03.056
46. Shershakova N, Bashkatova E, Babakhin A, et al. Allergen-Specific Immunotherapy with Monomeric Allergoid in a Mouse Model of Atopic Dermatitis. PLoS One. 2015;10(8):e0135070. doi:10.1371/journal.pone.0135070
47. Abboud G, Staumont-Salle D, Kanda A, et al. Fc(epsilon)RI and FcgammaRIII/CD16 differentially regulate atopic dermatitis in mice. J Immunol. 2009;182(10):6517–6526. doi:10.4049/jimmunol.0801055
48. Schwartz C, Moran T, Saunders SP, et al. Spontaneous atopic dermatitis in mice with a defective skin barrier is independent of ILC2 and mediated by IL-1β. Allergy. 2019;74(10):1920–1933. doi:10.1111/all.13801
49. Oshio T, Sasaki Y, Funakoshi-Tago M, et al. Dermatophagoides farinae extract induces severe atopic dermatitis in NC/Nga mice, which is effectively suppressed by the administration of tacrolimus ointment. Int Immunopharmacol. 2009;9(4):403–411. doi:10.1016/j.intimp.2008.12.013
50. Ehling S, Roßbach K, Dunston SM, Stark H, Bäumer W. Allergic inflammation is augmented via histamine H4 receptor activation: the role of natural killer cells in vitro and in vivo. J Dermatol Sci. 2016;83(2):106–115. doi:10.1016/j.jdermsci.2016.04.011
51. Yoshihisa Y, Makino T, Matsunaga K, et al. Macrophage migration inhibitory factor is essential for eosinophil recruitment in allergen-induced skin inflammation. J Invest Dermatol. 2011;131(4):925–931. doi:10.1038/jid.2010.418
52. Man M-Q, Hatano Y, Lee SH, et al. Characterization of a hapten-induced, murine model with multiple features of atopic dermatitis: structural, immunologic, and biochemical changes following single versus multiple oxazolone challenges. J Invest Dermatol. 2008;128(1):79–86. doi:10.1038/sj.jid.5701011
53. Kim Y-W, Ko E-A, Jung S-C, et al. Transcriptomic insight into the translational value of two murine models in human atopic dermatitis. Sci Rep. 2021;11(1):6616. doi:10.1038/s41598-021-86049-w
54. Kitamura A, Takata R, Aizawa S, Watanabe H, Wada T. A murine model of atopic dermatitis can be generated by painting the dorsal skin with hapten twice 14 days apart. Sci Rep. 2018;8(1):5988. doi:10.1038/s41598-018-24363-6
55. Go HN, Lee SH, Cho HJ, et al. Effects of chloromethylisothiazolinone/methylisothiazolinone (CMIT/MIT) on Th2/Th17-related immune modulation in an atopic dermatitis mouse model. Sci Rep. 2020;10(1):4099. doi:10.1038/s41598-020-60966-8
56. Wang G, Savinko T, Wolff H, et al. Repeated epicutaneous exposures to ovalbumin progressively induce atopic dermatitis-like skin lesions in mice. Clin Exp Allergy. 2007;37(1):151–161. doi:10.1111/j.1365-2222.2006.02621.x
57. Feng S, Liu W, Deng S, et al. An Atopic Dermatitis-Like Mouse Model by Alternate Epicutaneous Application of Dinitrofluorobenzene and an Extract of Dermatophagoides Farinae. Front Med Lausanne. 2022;9:843230. doi:10.3389/fmed.2022.843230
58. Riedl R, Kühn A, Rietz D, et al. Establishment and Characterization of Mild Atopic Dermatitis in the DNCB-Induced Mouse Model. Int J Mol Sci. 2023;24(15):12325. doi:10.3390/ijms241512325
59. Matsuoka H, Maki N, Yoshida S, et al. A mouse model of the atopic eczema/dermatitis syndrome by repeated application of a crude extract of house-dust mite Dermatophagoides farinae. Allergy. 2003;58(2):139–145. doi:10.1034/j.1398-9995.2003.23790.x
60. Kawakami Y, Yumoto K, Kawakami T. An improved mouse model of atopic dermatitis and suppression of skin lesions by an inhibitor of Tec family kinases. Allergol Int. 2007;56(4):403–409. doi:10.2332/allergolint.O-07-486
61. Yoshioka T, Hikita I, Matsutani T, et al. DS-Nh as an experimental model of atopic dermatitis induced by Staphylococcus aureus producing staphylococcal enterotoxin C. Immunology. 2003;108(4):562–569. doi:10.1046/j.1365-2567.2003.01588.x
62. Cau L, Williams MR, Butcher AM, et al. Staphylococcus epidermidis protease EcpA can be a deleterious component of the skin microbiome in atopic dermatitis. J Allergy Clin Immunol. 2021;147(3):955–966.e16. doi:10.1016/j.jaci.2020.06.024
63. Byrd AL, Deming C, Cassidy SKB, et al. Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Sci Transl Med. 2017;9(397). doi:10.1126/scitranslmed.aal4651
64. Li XM, Kleiner G, Huang CK, et al. Murine model of atopic dermatitis associated with food hypersensitivity. J Allergy Clin Immunol. 2001;107(4):693–702. doi:10.1067/mai.2001.114110
65. Bromley SK, Larson RP, Ziegler SF, Luster AD. IL-23 induces atopic dermatitis-like inflammation instead of psoriasis-like inflammation in CCR2-deficient mice. PLoS One. 2013;8(3):e58196. doi:10.1371/journal.pone.0058196
66. Li M, Hener P, Zhang Z, Kato S, Metzger D, Chambon P. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc Natl Acad Sci U S A. 2006;103(31):11736–11741. doi:10.1073/pnas.0604575103
67. Kim BE, Kim J, Goleva E, et al. Particulate matter causes skin barrier dysfunction. JCI Insight. 2021;6(5). doi:10.1172/jci.insight.145185
68. Kake T, Imai M, Takahashi N. Effects of β-carotene on oxazolone-induced atopic dermatitis in hairless mice. Exp Dermatol. 2019;28(9):1044–1050. doi:10.1111/exd.14003
69. Gilhar A, Reich K, Keren A, Kabashima K, Steinhoff M, Paus R. Mouse models of atopic dermatitis: a critical reappraisal. Exp Dermatol. 2021;30(3):319–336. doi:10.1111/exd.14270
70. Peiser M. Role of Th17 cells in skin inflammation of allergic contact dermatitis. Clinic Develop Immunol. 2013;2013:261037. doi:10.1155/2013/261037
71. Park HY, Kim CR, Huh IS, et al. Staphylococcus aureus Colonization in Acute and Chronic Skin Lesions of Patients with Atopic Dermatitis. Ann Dermatol. 2013;25(4):410–416. doi:10.5021/ad.2013.25.4.410
72. Wang Q, Du J, Zhu J, Yang X, Zhou B. Thymic stromal lymphopoietin signaling in CD4(+) T cells is required for TH2 memory. J Allergy Clin Immunol. 2015;135(3):781–91.e3. doi:10.1016/j.jaci.2014.09.015
73. Kim BS, Siracusa MC, Saenz SA, et al. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci, trans med. 2013;5(170):170ra16. doi:10.1126/scitranslmed.3005374
74. Chan LS, Robinson N, Xu L. Expression of interleukin-4 in the epidermis of transgenic mice results in a pruritic inflammatory skin disease: an experimental animal model to study atopic dermatitis. J Invest Dermatol. 2001;117(4):977–983. doi:10.1046/j.0022-202x.2001.01484.x
75. Dillon SR, Sprecher C, Hammond A, et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. 2004;5(7):752–760. doi:10.1038/ni1084
76. Yoo J, Omori M, Gyarmati D, et al. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med. 2005;202(4):541–549. doi:10.1084/jem.20041503
77. Yamanaka K, Tanaka M, Tsutsui H, et al. Skin-specific caspase-1-transgenic mice show cutaneous apoptosis and pre-endotoxin shock condition with a high serum level of IL-18. J Immunol. 2000;165(2):997–1003. doi:10.4049/jimmunol.165.2.997
78. Konishi H, Tsutsui H, Murakami T, et al. IL-18 contributes to the spontaneous development of atopic dermatitis-like inflammatory skin lesion independently of IgE/stat6 under specific pathogen-free conditions. Proc Natl Acad Sci U S A. 2002;99(17):11340–11345. doi:10.1073/pnas.152337799
79. Hansson L, Bäckman A, Ny A, et al. Epidermal overexpression of stratum corneum chymotryptic enzyme in mice: a model for chronic itchy dermatitis. J Invest Dermatol. 2002;118(3):444–449. doi:10.1046/j.0022-202x.2001.01684.x
80. Kobayashi T, Glatz M, Horiuchi K, et al. Dysbiosis and Staphylococcus aureus Colonization Drives Inflammation in Atopic Dermatitis. Immunity. 2015;42(4):756–766. doi:10.1016/j.immuni.2015.03.014
81. Weih F, Durham SK, Barton DS, Sha WC, Baltimore D, Bravo R. p50-NF-kappaB complexes partially compensate for the absence of RelB: severely increased pathology in p50(-/-)relB(-/-) double-knockout mice. J Exp Med. 1997;185(7):1359–1370. doi:10.1084/jem.185.7.1359
82. Barton D, HogenEsch H, Weih F. Mice lacking the transcription factor RelB develop T cell-dependent skin lesions similar to human atopic dermatitis. Eur J Immunol. 2000;30(8):2323–2332. doi:10.1002/1521-4141(2000)30:8<2323::Aid-immu2323>3.0.Co;2-h
83. Tsukuba T, Yamamoto K. Atopic dermatitis and cathepsin E. Nihon Yakurigaku Zasshi. 2003;122(1):15–20. doi:10.1254/fpj.122.15
84. Nakatsuji T, Chen TH, Two AM, et al. Staphylococcus aureus Exploits Epidermal Barrier Defects in Atopic Dermatitis to Trigger Cytokine Expression. J Invest Dermatol. 2016;136(11):2192–2200. doi:10.1016/j.jid.2016.05.127
85. Matsuda H, Watanabe N, Geba GP, et al. Development of atopic dermatitis-like skin lesion with IgE hyperproduction in NC/Nga mice. Int Immunol. 1997;9(3):461–466. doi:10.1093/intimm/9.3.461
86. Moniaga CS, Kabashima K. Filaggrin in atopic dermatitis: flaky tail mice as a novel model for developing drug targets in atopic dermatitis. Inflammation Allergy Drug Targets. 2011;10(6):477–485. doi:10.2174/187152811798104881
87. Vandeghinste N, Klattig J, Jagerschmidt C, et al. Neutralization of IL-17C Reduces Skin Inflammation in Mouse Models of Psoriasis and Atopic Dermatitis. J Invest Dermatol. 2018;138(7):1555–1563. doi:10.1016/j.jid.2018.01.036
88. Jin H, He R, Oyoshi M, Geha RS. Animal models of atopic dermatitis. J Invest Dermatol. 2009;129(1):31–40. doi:10.1038/jid.2008.106
89. Yagi R, Nagai H, Iigo Y, Akimoto T, Arai T, Kubo M. Development of atopic dermatitis-like skin lesions in STAT6-deficient NC/Nga mice. J Immunol. 2002;168(4):2020–2027. doi:10.4049/jimmunol.168.4.2020
90. Hikita I, Yoshioka T, Mizoguchi T, et al. Characterization of dermatitis arising spontaneously in DS-Nh mice maintained under conventional conditions: another possible model for atopic dermatitis. J Dermatol Sci. 2002;30(2):142–153. doi:10.1016/s0923-1811(02)00070-1
91. Natori K, Tamari M, Watanabe O, et al. Mapping of a gene responsible for dermatitis in NOA (Naruto Research Institute Otsuka Atrichia) mice, an animal model of allergic dermatitis. J Hum Genet. 1999;44(6):372–376. doi:10.1007/s100380050181
92. Fallon PG, Sasaki T, Sandilands A, et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet. 2009;41(5):602–608. doi:10.1038/ng.358
93. Sasaki T, Shiohama A, Kubo A, et al. A homozygous nonsense mutation in the gene for Tmem79, a component for the lamellar granule secretory system, produces spontaneous eczema in an experimental model of atopic dermatitis. J Allergy Clin Immunol. 2013;132(5):1111–1120.e4. doi:10.1016/j.jaci.2013.08.027
94. Saunders SP, Goh CS, Brown SJ, et al. Tmem79/Matt is the matted mouse gene and is a predisposing gene for atopic dermatitis in human subjects. J Allergy Clin Immunol. 2013;132(5):1121–1129. doi:10.1016/j.jaci.2013.08.046
95. Potter CS, Wang Z, Silva KA, et al. Chronic proliferative dermatitis in Sharpin null mice: development of an autoinflammatory disease in the absence of B and T lymphocytes and IL4/IL13 signaling. PLoS One. 2014;9(1):e85666. doi:10.1371/journal.pone.0085666
96. Del Rio M, Larcher F, Serrano F, et al. A preclinical model for the analysis of genetically modified human skin in vivo. Hum Gene Ther. 2002;13(8):959–968. doi:10.1089/10430340252939069
97. Carretero M, Guerrero-Aspizua S, Del Río M. Bioengineered skin humanized model of psoriasis. Methods Mol Biol. 2013;961:305–323. doi:10.1007/978-1-62703-227-8_20
98. Keren A, Reich K, Bertolini M, et al. Autologous Th2-polarized lymphocytes induce atopic dermatitis lesions in non-atopic human skin xenotransplants. Allergy. 2023;78(6):1538–1553. doi:10.1111/all.15635
99. Omori-Miyake M, Yamashita M, Tsunemi Y, Kawashima M, Yagi J. In vitro assessment of IL-4- or IL-13-mediated changes in the structural components of keratinocytes in mice and humans. J Invest Dermatol. 2014;134(5):1342–1350. doi:10.1038/jid.2013.503
100. Tatsuno K, Fujiyama T, Yamaguchi H, Waki M, Tokura Y. TSLP Directly Interacts with Skin-Homing Th2 Cells Highly Expressing its Receptor to Enhance IL-4 Production in Atopic Dermatitis. J Invest Dermatol. 2015;135(12):3017–3024. doi:10.1038/jid.2015.318
101. Jiao D, Wong CK, Qiu HN, et al. NOD2 and TLR2 ligands trigger the activation of basophils and eosinophils by interacting with dermal fibroblasts in atopic dermatitis-like skin inflammation. Cell Mol Immunol. 2016;13(4):535–550. doi:10.1038/cmi.2015.77
102. Engelhart K, El Hindi T, Biesalski HK, Pfitzner I. In vitro reproduction of clinical hallmarks of eczematous dermatitis in organotypic skin models. Arch Dermatol Res. 2005;297(1):1–9. doi:10.1007/s00403-005-0575-7
103. Bernard FX, Morel F, Camus M, et al. Keratinocytes under Fire of Proinflammatory Cytokines: bona Fide Innate Immune Cells Involved in the Physiopathology of Chronic Atopic Dermatitis and Psoriasis. J Allergy. 2012;2012:718725. doi:10.1155/2012/718725
104. Danso MO, van Drongelen V, Mulder A, et al. TNF-α and Th2 cytokines induce atopic dermatitis-like features on epidermal differentiation proteins and stratum corneum lipids in human skin equivalents. J Invest Dermatol. 2014;134(7):1941–1950. doi:10.1038/jid.2014.83
105. De Vuyst É, Giltaire S, de Rouvroit CL, et al. Methyl-β-cyclodextrin concurs with interleukin (IL)-4, IL-13 and IL-25 to induce alterations reminiscent of atopic dermatitis in reconstructed human epidermis. Exp Dermatol. 2018;27(4):435–437. doi:10.1111/exd.13113
106. Hsu CY, Lecland N, Pendaries V, et al. Stabilization of microtubules restores barrier function after cytokine-induced defects in reconstructed human epidermis. J Dermatol Sci. 2018;91(1):87–96. doi:10.1016/j.jdermsci.2018.04.008
107. Mitra A, Raychaudhuri SK, Raychaudhuri SP. IL-22 induced cell proliferation is regulated by PI3K/Akt/mTOR signaling cascade. Cytokine. 2012;60(1):38–42. doi:10.1016/j.cyto.2012.06.316
108. Rabeony H, Petit-Paris I, Garnier J, et al. Inhibition of keratinocyte differentiation by the synergistic effect of IL-17A, IL-22, IL-1α, TNFα and oncostatin M. PLoS One. 2014;9(7):e101937. doi:10.1371/journal.pone.0101937
109. Ataç B, Wagner I, Horland R, et al. Skin and hair on-a-chip: in vitro skin models versus ex vivo tissue maintenance with dynamic perfusion. Lab Chip. 2013;13(18):3555–3561. doi:10.1039/c3lc50227a
110. Wufuer M, Lee G, Hur W, et al. Skin-on-a-chip model simulating inflammation, edema and drug-based treatment. Sci Rep. 2016;6(1):37471. doi:10.1038/srep37471
111. Abaci HE, Guo Z, Coffman A, et al. Human Skin Constructs with Spatially Controlled Vasculature Using Primary and iPSC-Derived Endothelial Cells. Adv Healthc Mater. 2016;5(14):1800–1807. doi:10.1002/adhm.201500936
112. Ren X, Getschman AE, Hwang S, et al. Investigations on T cell transmigration in a human skin-on-chip (SoC) model. Lab Chip. 2021;21(8):1527–1539. doi:10.1039/d0lc01194k
113. Cabanillas B, Brehler AC, Novak N. Atopic dermatitis phenotypes and the need for personalized medicine. Curr Opin Allergy Clin Immunol. 2017;17(4):309–315. doi:10.1097/aci.0000000000000376
114. Peters EM, Michenko A, Kupfer J, et al. Mental stress in atopic dermatitis--neuronal plasticity and the cholinergic system are affected in atopic dermatitis and in response to acute experimental mental stress in a randomized controlled pilot study. PLoS One. 2014;9(12):e113552. doi:10.1371/journal.pone.0113552
115. Nakajima S, Kabata H, Kabashima K, Asano K. Anti-TSLP antibodies: targeting a master regulator of type 2 immune responses. Allergol Int. 2020;69(2):197–203. doi:10.1016/j.alit.2020.01.001
116. Bergmann S, von Buenau B, Vidal YSS, et al. Claudin-1 decrease impacts epidermal barrier function in atopic dermatitis lesions dose-dependently. Sci Rep. 2020;10(1):2024. doi:10.1038/s41598-020-58718-9
117. Buhl T, Ikoma A, Kempkes C, et al. Protease-Activated Receptor-2 Regulates Neuro-Epidermal Communication in Atopic Dermatitis. Front Immunol. 2020;11:1740. doi:10.3389/fimmu.2020.01740
118. Meng J, Moriyama M, Feld M, et al. New mechanism underlying IL-31-induced atopic dermatitis. J Allergy Clin Immunol. 2018;141(5):1677–1689.e8. doi:10.1016/j.jaci.2017.12.1002
119. Pavlovic S, Daniltchenko M, Tobin DJ, et al. Further exploring the brain-skin connection: stress worsens dermatitis via substance P-dependent neurogenic inflammation in mice. J Invest Dermatol. 2008;128(2):434–446. doi:10.1038/sj.jid.5701079
120. Yosipovitch G, Berger T, Fassett MS. Neuroimmune interactions in chronic itch of atopic dermatitis. J Eur Acad Dermatol Venereol. 2020;34(2):239–250. doi:10.1111/jdv.15973
121. Gittler JK, Shemer A, Suárez-Fariñas M, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. 2012;130(6):1344–1354. doi:10.1016/j.jaci.2012.07.012
122. Ahn K, Kim BE, Kim J, Leung DY. Recent advances in atopic dermatitis. Curr Opin Immunol. 2020;66:14–21. doi:10.1016/j.coi.2020.02.007
123. Thijs JL, Nierkens S, Herath A, et al. A panel of biomarkers for disease severity in atopic dermatitis. Clin Exp Allergy. 2015;45(3):698–701. doi:10.1111/cea.12486
124. Ariëns LFM, van der Schaft J, Bakker DS, et al. Dupilumab is very effective in a large cohort of difficult-to-treat adult atopic dermatitis patients: first clinical and biomarker results from the BioDay registry. Allergy. 2020;75(1):116–126. doi:10.1111/all.14080
125. Fishbein AB, Silverberg JI, Wilson EJ, Ong PY. Update on Atopic Dermatitis: diagnosis, Severity Assessment, and Treatment Selection. J Allergy Clin Immunol Pract. 2020;8(1):91–101. doi:10.1016/j.jaip.2019.06.044
126. Nomura T, Wu J, Kabashima K, Guttman-Yassky E. Endophenotypic Variations of Atopic Dermatitis by Age, Race, and Ethnicity. J Allergy Clin Immunol Pract. 2020;8(6):1840–1852. doi:10.1016/j.jaip.2020.02.022
127. Ewald DA, Noda S, Oliva M, et al. Major differences between human atopic dermatitis and murine models, as determined by using global transcriptomic profiling. J Allergy Clin Immunol. 2017;139(2):562–571. doi:10.1016/j.jaci.2016.08.029
128. Morgner B, Tittelbach J, Wiegand C. Induction of psoriasis- and atopic dermatitis-like phenotypes in 3D skin equivalents with a fibroblast-derived matrix. Sci Rep. 2023;13(1):1807. doi:10.1038/s41598-023-28822-7
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.