")
Back to Journals » Journal of Inflammation Research » Volume 18
Recent Advances of Type I Interferon on the Regulation of Immune Cells and the Treatment of Systemic Lupus Erythematosus
Authors Wang X, Wen B, Duan X, Zhang Y, Hu Y, Li H, Shang H, Jing Y
Received 26 January 2025
Accepted for publication 18 March 2025
Published 30 March 2025 Volume 2025:18 Pages 4533—4549
DOI https://doi.org/10.2147/JIR.S516195
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Xiaoyu Liu
Xiaocui Wang,1,* Bin Wen,2,* Xuemei Duan,1 Yunfei Zhang,1 Ying Hu,1 Haonan Li,1 Huifeng Shang,2 Yukai Jing1
1Department of Clinical Laboratory, Third Hospital of Shanxi Medical University, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Tongji Shanxi Hospital, Taiyuan, 030032, People’s Republic of China; 2Department of Clinical Laboratory, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Tongji Shanxi Hospital, Third Hospital of Shanxi Medical University, Taiyuan, 030032, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yukai Jing, Department of Clinical Laboratory, Third Hospital of Shanxi Medical University, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Tongji Shanxi Hospital, Taiyuan, 030032, People’s Republic of China, Tel +86-13834234594, Email [email protected] Huifeng Shang, Department of Clinical Laboratory, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Third Hospital of Shanxi Medical University, Tongji Shanxi Hospital, Taiyuan, 030032, People’s Republic of China, Email [email protected]
Abstract: Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with multiple organ damage. Several studies have found that, in addition to significant production of autoantibodies, the majority of SLE patients exhibit increased expression of type I interferon (IFN-I) regulated genes (also known as IFN-I traits), and that IFN-I plays a crucial role in the pathogenesis of SLE. In SLE, virtually all immune cells are dysregulated, and most of these aberrant dysregulations are directly or indirectly affected by IFN-I. The mechanism of action of IFN-I in these immune cells is multifaceted. In this review, we focus on the immune cell types that produce IFN-I and are affected by IFN-I in SLE. Importantly, we explore the research progress of related drugs in terms of IFN-I production, itself, and downstream. Here we provide the most up-to-date information on the mechanisms that lead to the pathogenesis of SLE, providing the basis for the development of innovative future therapies and future research directions.
Keywords: systemic lupus erythematosus, type I interferon, autoimmune, B cell, T cell
Introduction
Systemic lupus erythematosus (SLE) is a persistent, incapacitating, diverse autoimmune condition distinguished by type I interferons (IFN-I), circulating antinuclear antibodies (such as dsDNA and snRNP), and immune complex deposition, leading to multiple organ damage.1 In addition to multiple organ involvement, SLE manifests three distinct patterns: chronic persistent disease, periodic remissions and relapses, and prolonged quiescence. Symptoms include rashes (71%), arthritis (85%), nephritis (21%), hematological disorders (35%), and neurological disorders (18%).2 An abnormal immune response plays a key role in SLE pathogenesis, characterized by impaired clearance of apoptotic cells, loss of self-tolerance to autoantigens, alteration of T-cell and B-cell functions, and altered cytokine profiles.3
 |
Table 1 Classification of IFN |
Interferons (IFN) are released by host cells in response to invasion by pathogens. Depending on the specific receptor type, human interferons can be categorized into three distinct groups: interferon I, interferon II, and interferon III (Table 1). IFN-I include IFN-α, β, ε, κ, and ω.4 Almost all nucleated cells in the body produce IFN-α in response to infection. Particularly notable in this regard are plasmacytoid dendritic cells (pDCs), which produce greater quantities of IFN-I than any other cell.4 IFN-β is the high-affinity short-lived priming IFN and the major IFN-I produced by fibroblasts.5 IFN-II is mainly secreted by T lymphocytes, natural killer cells (NKs), and antigen-presenting cells (APCs) such as monocytes, macrophages, and dendritic cells (DCs).6 IFN-III is produced by APCs and epithelial cells.4 The innate and adaptive immunity would initiate and produce IFN-I while viruses infected. The genetic association between IFN-I pathway and SLE susceptibility has been extensively investigated. Genome-wide association studies (GWAS) studies have shown single nucleotide polymorphisms (SNPs) in IFN regulatory factor (IRF)5, IRF7, IRF8, signal transducers and activators of transcription 4 (STAT), and tyrosine kinase 2 (TYK2) are associated with higher risks of developing SLE.7,8 Epigenome wide association study (EWAS) found that the most pronounced differences in methylation between SLE patients and healthy controls (HC) were in the interferon-stimulated genes (ISGs), including IRF5 and IRF7.9 Meanwhile, droplet-based microfluidics and other techniques to further define the relationship between IFN-I and SLE.10 Several studies have found a negative correlation was observed between serum IFN-α levels and Toll-like receptor (TLR)-7 expression, as well as C3 and C4 levels in SLE patients.11 Recent studies have shown that an elevated baseline level of IFN-α in SLE is associated with poor renal outcomes, including the development of two or more renal flares and a significant decline in kidney function, and could serve as a biomarker for upfront identification of patients at high risk of poor renal outcomes.12 The physiological importance of IFN-I in SLE patients remains unclear. Next, we will summarize the additional description of IFN-I and SLE.
IFN-I Signaling
IFN-I can be produced by a variety of nucleated cell types through the activation of multiple pattern recognition receptors (PRRs), including TLRs (TLR3 and TLR7/8/9) by pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), including cyclic GMP-AMP synthase (cGAS), melanoma differentiation-associated protein 5 (MDA5) and retinoic acid-inducible gene I (RIG-I).13,14 TLRs recognize different PAMPs to induce IFN-I production. TLR3 is a double-stranded RNA (dsRNA) sensor localized in the endosomes of many cell types but not plasmacytoid DCs (pDCs), the most potent producers of IFN-I.15 TLR7/8 recognises single-stranded RNA and TLR9 recognises DNA.15 cGAS is a DNA sensor that produces cGMP-AMP (cGAMP) by recognizing cytoplasmic nucleic acids including dsDNA.16 cGAMP induces IFN-I production and activates the nuclear factor kappa-B (NF-κB) pathway via a stimulator of interferon genes (STING).17–19 The most common RNA sensors are members of the RIG-I like (RLR) family, including RIG-I, MDA5, and laboratory of genetics and physiology 2 (LGP2).14,20 The helicase domains and C-terminal domains of the RLR bind to RNA ligands and the N-terminus binds to mitochondria antiviral signaling (MAVS) to induce IFN production.21–24 Tumor necrosis factor (TNF), receptor activator of nuclear factor kappa-B ligand (RANKL), and macrophage colony-stimulating factor (M-CSF) are also physiological inducers of IFN-I. It induces an antiviral state by activating ISG in a paracrine and autocrine mechanism upon induced IFN-I.25
Research has recently highlighted IRFs, a group of transcription factors involved in the IFN signaling cascade that modulate immune responses.26 The IRF family specifically includes IRF1, IRF3, IRF5, and IRF7,27 and IRF1 is the first member to activate the IFN-I gene promoter.28 Studies have recently implicated IRF3 and IRF7 as negative regulators of IFN responses in association with NF-κB pathway.29 Significantly, targeting IRF3 and IRF7 signaling has only shown partial effects on IFN-I, indicating the potential involvement of other mediators. Methyltransferase 3 (METTL3) promoted plasma cell infiltration and kidney damage by increasing IRF4 expression in MRL/lpr mice.30 While there is some indication that IRFs may influence IFN characteristics, current evidence is limited.
The binding of IFN-I to the IFN-α/β receptor (IFNAR) phosphorylates STAT1/STAT2, forming a trimer that moves into the nucleus and activates signaling downstream of ISG transcription initiation.31 RhoA siRNA reduces STAT1 phosphorylation but not STAT2, leading to downregulation of the IFN-I response, and has been demonstrated in patients with SLE.32 In response to cytokine binding, janus kinase (JAK) activation facilitates the phosphorylation of STAT and dimerization, resulting in nuclear translocation and regulation of target gene expression. A critical role for TYK2 is to mediate the signaling pathways of IFN-I and interleukin (IL)-12/23.33 IFN-β stimulation induced an increase in STAT2 phosphorylation for a longer duration in cells with higher HC. This activation induced higher MxA mRNA levels (an ISG), which may be due to insufficient upregulation of the regulatory suppressor of cytokine signaling 1 (SOCS1) protein.34,35 Individuals with SLE produce T follicular helper (Tfh)- T helper (Th)1-like cells as a result of the IL-12-mediated coactivation of STAT1 and STAT4. Tfh-Th1 type cells may expand through this process, providing a potential mechanism for targeted SLE therapy.36
Overall, inborn errors in the production of, or responses to, IFN-I, as well as their autoimmune phenocopies, underlie the threat of SLE pathogenesis (Figure 1). However, current studies have focused on IFN-I causing SLE disease by regulating its downstream targets. We believe that future studies could focus on how IFN-I production is regulated by SLE disease development.
 |
Figure 1 Pathways of action of IFN-I and related drugs. (A) IFN-I can be produced by a variety of nucleated cell types through the activation of multiple PRRs, including TLRs (TLR3 and TLR7/8/9) by PAMPs and DAMPs, including cGAS, MDA5, and RIG-I. (B) IFN-I binds to IFNAR on target cells and activates the JAK-STAT pathway. This image was created by Figdraw. Abbreviations: IFN-I, Interferon; PRRs, pattern recognition receptors; TLRs, Toll-like receptors; PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; cGAS, cyclic GMP-AMP synthase; MDA5, melanoma differentiation-associated protein 5; RIG-I, retinoic acid-inducible gene I; IFNAR, IFN-α/β receptor; JAK, janus kinase; STAT, signal transducers and activators of transcription. |
Mechanisms of IFN-I in Innate Immunity in SLE
SLE patients have a systemic immune stimulation marked by hyperactivated and senescent lymphocytes as well as altered myeloid subpopulations, including monocytes, DCs, and neutrophils.37 The earliest stages of inflammation in SLE involve the activation of the innate immune system, triggered by cellular and nuclear debris. Patients with SLE have been found to have elevated serum IFN-I levels.38 Multiple lines of evidence indicate that the interaction between different immune cells plays a crucial role in systemic inflammation and organ damage associated with SLE, nearly all cells can respond to IFN-I.
pDCs are recognized as the main producers of IFN-I, with their output being modulated by a variety of other immune cells that are key to the development of SLE.39 There is a marked dysregulation across diverse immune cell types, commonly thought to result from both direct and indirect interactions with IFN in the process of SLE. Additionally, innate immune players, including monocytes and conventional DCs (cDCs), significantly contribute to the IFN-I milieu by chiefly generating IFN-β.40,41 Ongoing studies aim to reveal the role played by IFN-I in SLE disease in the context of innate immune overactivation.
IFN-I and Neutrophils
Neutrophils are the most abundant effector cells in the innate immune system and are considered a potential source of endogenous antigenic triggers in SLE.42 Neutrophils disrupt vascular integrity, show increased sensitivity to IFN-I, and contribute to the development of SLE. In SLE patients, neutrophils exhibit pronounced IFN-I signaling and an elevated tendency to form neutrophil extracellular traps (NETs).43 NETs have been posited to initiate the activation of IFN-I genes, leading to heightened autoantibody production.44 Empirical evidence from both in vivo and in vitro studies implicates the link between IFN-I levels and neutrophil behavior.45 The level of tRF-His-GTG-1 (a transfer RNA-derived small RNAs) in neutrophils was positively correlated with NETs formation in SLE patients and regulated IRF7 activation in neutrophils upon TLR8 binding, leading to upregulation of IFN-α.46 Additionally, tempering IFN-I could lead to the normalization of neutrophil counts by curtailing non-neutrophilic responses.47 Genetic markers related to IFN-α and IFN-ω inhibitors, such as JNJ-55920839, have been detected through transcriptomic profiling of whole blood from SLE studies.45 These markers, however, require further corroboration in broader experiments. The low-density granulocyte (LDG) subset of neutrophils is associated with vascular damage in SLE, characterized by high IFN-I production and inflammatory cytokines.48 Traditional neutrophils in SLE increased the production of a proliferation inducing ligand (APRIL), IL-21, and the IFN-associated chemokine interferon γ-induced protein 10 (IP-10).49 Overall, there is growing evidence that neutrophils can participate in SLE by acting as antigens to drive autoantibody production, enhancing IFN-I and cytokine release, and inducing organ damage.50
IFN-I and Macrophage
M1 and M2 belong to two types of macrophage (Mφ). M1 is pro-inflammatory, usually activated by Lipopolysaccharide (LPS) and IFN-γ while M2 is anti-inflammatory, depending mainly on IL-10 and transforming growth factor-β (TGF-β).51 Mφ mediates the activation of innate immunity by recognizing microorganisms via TLR. Mouse bone marrow-derived macrophages induce aconitate decarboxylase 1 (ACOD1) via IFN-I receptor signaling in response to TLR7 stimulation, which plays an important immunomodulatory and vasoprotective role in SLE.52 Research indicates that IRF4 interacts with the intermediate region of myeloid differentiation primary response protein (MyD88) and induces anti-inflammatory cytokine secretion,53 the MyD88 adapter-like (Mal) plays a role in the TLR9-driven gene expression of IFN-β and TNF-α, an activity that is mediated by the activation of extracellular regulated protein (ERK) 1/2 kinases in macrophages infected with herpes simplex virus-1 (HSV-1). This process notably relies on the noncanonical NF-κB pathways.54 In addition, M2 polarisation was significantly reduced in SLE mice, and IRF4 affects Mφ polarisation and is involved in the pathogenesis of SLE.55 Principal component analysis (PCA) and clustering analyses revealed that IFN-I may be involved in the development of macrophage activation syndrome (MAS) in SLE patients.56,57 Spermine concentration is reduced in peripheral blood mononuclear cells (PBMC) of individuals with SLE, and spermine binds directly to JAK, thereby inhibiting JAK1 phosphorylation triggered by cytokines IFN-I, IFN-II, IL-2, and IL-6.58 Mφ in the lupus nephritis (LN) undergo phenotypic changes from inflammation patrolling macrophages to phagocytic macrophages to antigen-presenting macrophages and secrete a variety of pro-inflammatory factors and complement components.59 Plasma 7α, 25-dihydroxycholesterol is increased in SLE patients and binds to Epstein–Barr virus‐induced gene 2 (EBI2) to inhibit the IFN-I response in Mφ.60 In summary, IFN-I alters the release of inflammatory factors in vivo, Mφ polarisation, and IRF4 are associated with the pathogenesis of SLE.
Monocytes were classified into “classical” Ly6Chi cells (CM) and “non-classical” Ly6Clo cells (NCM).61 Seunghee Cha performed transcriptome analysis and found that IFN-I signaling on monocytes is essential for SLE, identifying some potential therapeutic targets.62 In the pristane-induced lupus model, the source of IFN-I is thought to be CM specifically induced by pristane administration.63 The pathogenic features of the SLE NCM are disturbed DNA repair, enhanced cell cycle and IFN signaling, and cellular differentiation. This is associated with activated macrophage-like and enriched M1 pro-inflammatory responses.64 SLE monocytes exhibited a cellular senescence phenotype, as evidenced by the upregulation of cyclin dependent kinase inhibitor 2A (CDKN2A), which led to the expression of GATA binding protein 4 (GATA4) and enhanced IFN-α production through activation of the cGAS-STING pathway.65 IL-10 and IFN-γ induce CD64 expression on the surface of monocytes, which is associated with elevated SLE Disease Activity Index (SLEDAI), blood urea nitrogen levels, and anti-Sm antibodies in SLE patients.66 Monocytes in SLE patients exhibit a variety of pathological features, including key roles through the IFN-I signaling pathway, a cellular senescence phenotype, impaired DNA repair, enhanced cell cycle, and cellular differentiation associated with an M1-type pro-inflammatory response, all of which may be potential therapeutic targets.
IFN-I and Dendritic Cells
Research into the response of myeloid DCs (mDCs) to IFN-I in SLE remains underexplored. However, Kristen L. Chen’s67 investigation into dermatitis revealed that in patients with moderate to severe dermatomyositis, CD11c+ mDCs along with CD69+ cells constitute the main cell types present in the skin, underscoring the vital influence of mDCs on the skin’s IFN-I profile.
The continuous release of IFN-I is linked to the aberrant stimulation of pDCs by their genetic material. Among circulating leukocytes, pDCs represent a unique class notably responsible for the persistent release of IFN-I in those with SLE.68 The pDC expresses the Immunoglobulin (Ig)A1 receptor, the Fc α receptor (FcαR), and the IgA1-containing immune complex drives IFN-α production by these cells in vitro. ISG expression is positively correlated with the amount of FcαR in whole blood in patients with SLE.69 Current research points to exosome-transferred microRNAs as emerging contributors to the development of human autoimmune diseases that are characterized by excessive production of IFN-I.70 Such as microRNAs induce pDCs to secrete IFN-I more intensely in SLE patients. These insights mark microRNAs as new triggers for pDC activation, implying that more detailed studies are needed to determine their role in IFN-I-driven autoimmune conditions using animal models.71 Furthermore, EGA, a late endosome trafficking inhibitor, has demonstrated efficacy in reducing both the levels and release of IFN-α by TLR7-activated pDCs. EGA also appears to lessen the number of pDCs that express pro-TNF, ultimately leading to reduced production and release of TNF-α from imiquimod (R837)-encouraged pDC cultures.72 αvβ3, a receptor for apoptotic cells and cellular debris, regulates TLR signaling and prevents self-reactive B-cell activation in a lupus model providing important environmental cues for pDC and limiting responses to self-produced nucleic acids.73 pDCs are key drivers in the cellular activation and production of soluble factors seen in SLE.74
Effect of IFN-I on Adaptive Immunity in SLE
An elevated IFN-I profile in SLE is implicated in disruptions of both the innate and adaptive immune responses. The augmentation of IFN-I is associated with the development of DCs and B cells, as well as T cell activation, which may lead to the emergence of autoantibodies.75 Following this, a comprehensive discussion is presented on the role of IFN-I in the maturation of B and T cells, along with its contribution to the pathogenesis of SLE.
Effects of IFN-I on T Cells
Following positive and negative selection processes, T cells depart from the thymus as naïve T (TN) cells, capable of recognizing specific epitopes.76 Upon engaging with their corresponding antigens (Ag), Double-negative (DN) T cells proliferate and mature into effector cells. Predominantly, they migrate to the periphery and inflammation sites, contributing to the elimination of the offending pathogen. A subset of these T cells evolves into either regulatory or memory variants.77 Additionally, CD4+ T cells may branch into several subsets like Th1, Th2, Th9, Th17, Th22, regulatory T (Treg), Tfh, and peripheral helper T (Tph) cells, contingent on their surrounding milieu. Clinical studies78 and large-scale single-cell RNA sequencing analysis79 confirmed the inverse association between IFN-I activity and the abundance of circulating lymphocytes or TN cells in SLE patients. Conversely, CD8+ T cells primarily transform into cytotoxic T lymphocytes (CTL). (Figure 2)
 |
Figure 2 IFN exerts significant impacts on various T-cell subsets in SLE pathology. (I) IFN-I enhances RSAD2 expression in naïve CD4+ T cells, contributing to their differentiation into Th17 and Tfh cells. (II) IFN-I enhances STAT4 phosphorylation and mTOR hyperactivation in Tfh cells. IFN-α amplifies IL-2-STAT5 pathway activation, impairs STAT1, and promotes the transformation of Tfh cells from Tfh cells into Tph cells. (III) AHR and JUN synergistically regulate the balance between Tph/Tfh and Th22, whereas IFN-I is an endogenous inhibitor of AHR and also disrupts the gene binding site of JUN. (IV) IFN-β directly promotes Treg cell induction through STAT1- and P300-dependent Foxp3 acetylation. This image was created by Figdraw. Abbreviations: IFN, Interferon; SLE, Systemic lupus erythematosus; RSAD2, Radical S-Adenosyl Methionine Domain Containing 2; Th17, T helper 17; Tfh, T follicular helper; STAT, signal transducers and activators of transcription; mTOR, mammalian target of rapamycin; IL-2, Interleukin 2; Tph, peripheral helper T; AHR, aryl hydrocarbon receptor. |
Effects of IFN-I on Th1
Increased levels of IL-12 and IL-18 initiate the activation of CD4+ T cells, leading to the subsequent activation of the STAT4 transcription factor. This activates specific transcription factors, namely STAT1 and T-bet within Th1 cells, prompting the transformation of CD4+ T cells into Th1 cells and later producing IFN-γ, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-2, and TNF-α consequently.80–83 A study showed that Lipocalin-2 (LCN2)-deficient mice, exhibited decreased glomerular IgG deposition, and reduced macrophage and neutrophil infiltration, alongside a diminished presence and quantity of Th1 cells in the LN model.84 This indicates that the exacerbation of lupus is influenced by the enhanced differentiation of Th1 cells. Reactivation of T cell immunoglobulin and ITIM domain (TIGIT) reversed Th1 polarization of Tregs by suppressing protein kinase B (AKT)/mammalian target of rapamycin (mTOR) and STAT4 signaling, a potential therapeutic target for SLE.85 Furthermore, immunization with a CD4αβ Th1 clone stimulated an anti-allogeneic T-cell reaction in individuals suffering from lupus. The Th1-specific transcription factor, T-bet, plays a crucial role in SLE, with recent findings suggesting that T-bet acts to obstruct Th17 cell differentiation and the synthesis of associated transcription factors and cytokines.86 Research into the role of IFN-α produced by Th1 cells in SLE has been sparse, primarily focusing on studies related to IFN-γ.
Effects of IFN-I on Th17
TGF-β, IL-6, and IL-23 initiate the activation of Th17 cell-specific transcription factors RAR Related Orphan Receptor C (RORC) and STAT3, facilitating their differentiation into Th17 cells that secrete IL-17, IL-22, and IL-23.87 In SLE, Tregs are reduced in comparison to the increased presence of Th17 cells in inflammatory tissues.88 Additionally, IFN-I elevates Radical S-Adenosyl Methionine Domain Containing 2 (RSAD2) expression in TN cells, which aids in their transformation into Th17 and Tfh cells, leading to the secretion of inflammatory cytokines and the development of SLE.89 Moreover, research into the dynamics between IFN-γ and Th17 in SLE is ongoing.90–92
Effects of IFN-I on Th22
Recent studies have demonstrated that the TNF-α and IL-6 can activate the aryl hydrocarbon receptor (AHR), which in turn encourages the conversion of CD4+ T cells into Th22 cells, known for their IL-22 production.93 In cases of SLE, a significant increase in both Th22 and IL-22 levels has been linked to the severity of disease symptoms. The percentage of Th22 cells in SLE patients with renal damage was positively correlated with erythrocyte sedimentation rate (ESR).94 In the SLE environment, AHR acts synergistically with AP-1 family member JUN to regulate the balance between Tph/Tfh and Th22, and IFN-I is an endogenous inhibitor of AHR that also disrupts the gene-binding site of JUN.95 However, research focusing on the influence of IFN on Th22 cells in SLE has been notably scarce in recent years.
Effects of IFN-I on Follicular Helper T Cells
Among the CD4+ T cells, Tfh cells, which can directly interact with B cells, have attracted attention as key factors in the pathogenesis of SLE. Tfh cells were identified using several markers including C-X-C Motif Chemokine Receptor (CXCR)5, programmed cell death protein 1 (PD-1), B-cell Lymphoma 6 (BCL6), B And T Lymphocyte Associated 4 (BTLA4), Inducible T Cell Costimulator (ICOS), IL-21 and SH2 Domain Containing 1A (SH2D1A).96 Tfh cells are correlated with SLE disease activity and organ damage.97 Tfh cells are implicated in the pathogenesis of autoimmune diseases such as SLE and rheumatoid arthritis (RA), where they drive aberrant germinal center (GC) reactions, aid B cell differentiation and proliferation, and contribute to increased autoantibody production.98 IFN-I has been found to intensify STAT4 phosphorylation in Tfh cells, leading to aberrant IL-21 and IFN-γ production.99 Overactivation of mTOR in Tfh cells by elevated IFN-I is another contributing factor to the lymphocytopenia phenotype in lupus.100 IFN-α was noted to upregulate CD25 on Tfh cells, amplifying IL-2-STAT5 pathway activation in SLE patients, further, STAT5 effectively competed at the BCL6 gene site by repressing the regulatory marker H3K4me3, to the detriment of STAT1, downregulating BCL6 and facilitating a shift from PD-1+CXCR5+ Tfh-like cells to a PD-1+CXCR5− Tph-like phenotype, thus disrupting Tfh cell stability and growth.101 Additionally, IFN-I offers protection for Tfh cells against NK cell-mediated cytotoxicity.102 IFN-I promotes aberrant activation and differentiation of Tfh in autoimmune diseases such as SLE through multiple mechanisms, leading to increased auto-antibody production and aberrant B-cell differentiation, and thus blocking IFN-α signaling may be a potential strategy for treating SLE.
Effects of IFN-I on Peripheral Helper T Cell
Tph cells, initially identified in RA, are a subset of T cells that localize to tertiary lymphoid follicles and contribute to SLE pathology.103 Tph cells share several phenotypic markers with Tfh cells, including C-X-C Motif Chemokine Ligand 13 (CXCL13), IL-21, ICOS, SH2D1A, Maf, TIGIT, CD38 and CD57. However, they express distinct chemokine receptors, such as CCR2, CX3CR1, and CCR5, which facilitate their migration to inflammatory sites.104 Studies have shown that the abundance of Tph cells increases in patients with high disease activity and LN.105 IFN-α-induced Tph-like cells promoted B cell differentiation into CD38hi CD27hi plasmablasts.101 Therefore, blocking IFN-α signaling could reduce Tph-like cell differentiation thus affecting the generation of CD38hi CD27hi plasmablasts in SLE patients.106 Tph cells promote B cell differentiation through IFN-α signaling, making them a potential therapeutic target for SLE.
Effects of IFN-I on Treg
Upon receiving signals from TGF-β, CD4+ T cells initiate the activation of Foxp3, a transcription factor essential for Treg cells. Francisco Fueyo-González et al found that IFN-β directly promotes Treg cell induction through STAT1 and P300-dependent Foxp3 acetylation.5 In the context of SLE, the compromised functionality or scarcity of Treg cells diminishes immunosuppressive action.107 Treg cells also impact bone metabolism via IFN-γ activity. The dysregulation of Treg operations and the disturbed equilibrium between Tregs and Th17 cells in SLE hinder Tregs’ capacity to control osteoclast functionality and uphold a negative feedback mechanism.108 Presently, the focus of numerous studies remains on IFN-γ.109–111
Effects of IFN-I on CD8+T Cells
The primary cytokine produced by CD8+ T cells is IFN-γ.112 In patients with LN, accumulation of CD8+ T cells in the periglomerular region correlates with disease severity. IFN-I signaling affects CD8+ T cell differentiation by overexpressing ISG and promoting CD8+ T cell cytotoxicity and migration.113 In IFNAR-cKO mice, Nba2-driven accumulation of CD44hiCD62Llo effector CD8+ T cells was reversed, whereas Foxp3+ CD8+ regulatory cells were up-regulated.92 Coziana Ciurtin114 also demonstrated that the transcriptome of SLE CD8+ T cells is distinguished from HC by a dysregulation in pathways associated with both ISG and mitochondrial biology, with IFN-α specifically increasing SLE effector memory (EM) CD8+ T cell apoptosis in vitro. IFN-I is associated with dysregulation of mitochondrial metabolism in CD8+ T cells, and modulation of the nicotinamide adenine dinucleotide (NAD+) pathway is a major mechanism by which it affects SLE.115
IFN-I and B Cells
IFN-I enhances the sensitivity of naïve B cells to RNA-associated antigens by up-regulating RNA-binding TLR7.116 Additionally, IFN-I up-regulates co-stimulatory molecules, thereby improving the ability of B cells to present antigens, receive T-cell help, and engage in GC reactions.117 Moreover, IFN-I supports the differentiation of B cells into plasma cells (PCs) and boosts antibody production. In SLE B cells, the transcriptional profile of IFN-I is up-regulated (Figure 3). Human B cells express the IFN-I gene in response to IFN-λ.118 In SLE, an increase in B cell subpopulations is commonly observed.
 |
Figure 3 Effects of IFN-I on B cells. IFN-I promotes the expansion of AFC and ICOShi ExFO−Th cells, triggering an autoantibody response. IFN-I triggers the hyperactivation of the ATR-Chk1 pathway in B cells. IFN-I activates FOXM1 by activating p38 and Akt in B cells. IFN-I drives the differentiation of B cells into PCs. This image was created by Figdraw. Abbreviations: IFN-I, Interferon-I; AFC, antibody-forming cell; ICOS, Inducible T Cell Costimulator; ATR, ataxia-telangiectasia mutated and Rad3 related; FOXM1, forkhead box M1; Akt, protein kinase B; PCs, plasma cells. |
IFN-I and B-Cell Differentiation
IFN-I is pivotal for the expansion of antibody-forming cells (AFCs) and ICOShi ExFO− Th cells, triggering the emergence of an autoantibody response in SLE. In vitro evidence has demonstrated that IFN-I is vital in driving the differentiation of B cells into CD138+ plasmablasts, a process that relies on B-cell receptor (BCR) and CD40 signaling.116 Autoantibody prevalence is associated with diminished IFN-I levels and clinical scores, suggesting the contribution of long-lived PCs to this antibody production.119 Studies have observed a reduction in double-negative (DN) B cells in SLE patients harboring neutralizing autoantibodies against IFN-I (anti-IFN-I-Abs).119 In SLE, the selection of self-reactive B cells is intensified within the mature B cell population, resulting in a higher proportion of APC cells within the mature naïve B cell compartment.120 There has been an observation of deficient B cells entering the GC and morphing into cells that produce autoantibodies in SLE. However, it remains unresolved whether these patterns are reliant on IFN.121 Furthermore, heightened levels of IFN-α augment the development of autoreactive B cells (ABC) in SLE mice by increasing their sensitivity to IL-21 and endosomal TLR ligands.121 Stimulation of CD19+ B cells by IFN-α induces the development of plasmacytoid cells and the production of antibodies, while also enhancing mitochondrial function.122 IL-4R and IFN-I receptor signaling plays a crucial role in regulating distinct pathways of memory B cell development, starting at the transitional stage of B cell development and progressing through differentiation to DN1 and classical memory (cMEM) B cells.123
IFN-I and B-Cell Function
IFN-α can alter B cell function in vitro, resulting in heightened BCR signaling, activation, and differentiation. High levels of IFN-α disrupt multiple B-cell tolerance checkpoints and lead to autoantibody production.121 IFN-α signaling enhances responses to T cell signals such as CD40 ligand (CD40L), thereby replacing B cell-activating factor (BAFF) as the differentiation signal for transitional 1 (T1)- transitional 2 (T2) transition.121 IFN-I has been demonstrated to enhance the production of serum BAFF as well as induce B cell proliferation and loss of tolerance.124 The pathogenic B cells contribute to SLE pathogenesis through autoantibody production, inflammatory cytokine secretion, and autoantigen presentation.125 Moreover, autoantibodies generated by these pathogenic B cells form immune complexes (ICs) in SLE. In particular, the IgE ICs further stimulate IFN-I in pDCs to participate in SLE disease activity.126 In SLE, specific DNA damage response (DDR) pathways are excessively activated in B cells, with IFN-I triggering the ataxia-telangiectasia mutated and Rad3 related (ATR)-Chk1 pathway. Targeted drugs that inhibit ATR activity have been found to suppress key aspects of SLE pathophysiology, such as B-cell activation, plasmacytoma formation, antibody production, and pro-inflammatory responses.127 Dysfunction of B cells plays a crucial role in SLE patients, while oxidative phosphorylation (OXPHOS) profiles show a correlation with IFN-I signaling-related genes (ISRGs) profiles.128
IFN-I and B Cell Receptor Signaling
IFN-I typically induces anti-proliferative and pro-apoptotic responses in target cells, with JAK/STAT activation being the primary signal transduction pathway triggered by IFN-α and other IFN-I upon binding to IFNAR. STAT3 and STAT5 become activated in a range of immune cell types, including monocytes, CD8+ T and CD4+ T cells, and B cells, in reaction to IFN-β.129 Using siRNAs or chemical inhibitors targeting enhancer of zeste homolog 2 (EZH2) has demonstrated a decrease in STAT1 phosphorylation and the induction of ISGs stimulated by IFN-I in vitro experiments. These findings suggest that an excess of EZH2 may be implicated in the heightened activity of the IFN-I signaling pathway in SLE.130 Elevated levels of ISGs prompt BCR activation in B-cells, which in turn more robustly activates Akt and p38, which would induce the upsurge in expression of forkhead box M1 (FOXM1).131 In MRL/lpr mice, NEAT1, the long non-coding RNA, is overexpressed in granulocytic-myeloid derived suppressor cells (G-MDSCs), which results in B cell activation of the IFN-I signaling through the secretion of the BAFF, triggered by G-MDSCs.132
Therapeutics Targeting IFN-I Pathway in SLE
Significant progress has been made with therapeutic agents targeting the IFN-I signaling pathway in SLE. This review will address three aspects, IFN-I production, IFN-I itself, and downstream of IFN-I. The monoclonal anti-blood dendritic cell antigen 2 (BDCA2) antibody Litifilimab inhibits IFN production in cutaneous lupus lesions by directly targeting pDC.133 CD123, the receptor for IL-3, is a cell surface marker for pDC and eosinophils. Anti-CD123 antibody reduces IFN-I production by targeting pDC.74 Cordycepin promotes STING autophagic degradation to alleviate autoimmunity upon DNA stimulation and might be a potential therapeutic candidate for alleviating aberrant IFN-I in autoimmune and autoinflammatory diseases.134 Clobenpropit binds effectively to CXCR4, significantly inhibiting IRF7 phosphorylation and reducing interferon production, and lowering levels of pro-inflammatory cytokines in a mouse model of lupus.135 HDAC10, which inhibits IRF3 phosphorylation, is a negative regulator of IFN-I production. HDAC10 is degraded by autophagy in SLE, so the autophagy inhibitors chloroquine and hydroxychloroquine are also commonly used in the treatment of SLE.136 The TLR7/8 inhibitors Enpatoran and Afimetoran are currently in Phase II SLE trials.137 The selective Sphingosine-1-phosphate (S1P) receptor modulator cenerimod reduces IFN-related biomarkers and has been shown in phase III clinical trials in patients with moderate to severe SLE.138 Methylprednisolone (MP) pulse therapy induces apoptosis in CD4+ T cells, promotes TGF-β production by monocytes, and further promotes Tregs differentiation. The newly differentiated Tregs inhibit CD4+ cell proliferation and IFN-γ production and contribute to the immunomodulatory environment after MP treatment for SLE.139
Drugs targeting IFN-α itself and IFNAR are currently used to treat SLE. Sifalimumab and rontalizumab are monoclonal antibodies targeting IFN-α. Using Sifalimumab reduces SLE disease activity,140 while rontalizumab is ineffective in the treatment of SLE141 and the reason for this has not been found. One of these drugs, Anifrolumab, is a therapeutic monoclonal antibody approved by the Food and Drug Administration (FDA) and the European Union for the treatment of moderate and severe SLE.142–144 QX006N monoclonal antibody that specifically targets IFNAR1, QX006N creates a spatial site-block on binding to IFNAR1-SD3 to prevent IFN binding and inhibit IFN/IFNAR1/IFNAR2 complex formation, has been used to treat SLE.145
A variety of small molecule inhibitors targeting JAK and TYK2 are currently in development, several of which are in clinical trials for autoimmune diseases. Phase II clinical trials have been completed for Baricitinib, which is more selective for JAK1 and JAK2 than JAK3, and is effective in relieving SLE rash and joint swelling.146 Another JAK inhibitor, Upadacitinib, has entered Phase III trials, and its specific effects are still being studied.147 Filgotinib (FIL) is a preferential inhibitor of JAK1 and improves cutaneous manifestations in SLE. Inhibition of JAK1 by FIL in SLE warrants further investigation.148 TYK2 inhibitors could represent a new class of drugs that inhibit interferon signatures and IL-12 signaling while preserving IL-2-mediated Treg cell differentiation, unlike other JAK inhibitors.149 Deurefacitinib is a potent, highly selective, small molecule inhibitor of TYK2 that strongly inhibits IFN-α-induced lymphopenia. This finding has important implications for the treatment of autoimmune diseases such as SLE.150 Tofacitinib was associated with inhibition of STAT1 phosphorylation in CD4+ T cells and downregulated ISG expression after treatment.151 Ruxolitinib reduced the level of ISG expression and inhibited JAK-STAT signaling for the treatment of SLE.152 IFN-I stimulated the production of BAFF, and the anti-BAFF antibody belimumab has been effective in treating SLE, especially in patients with active LN when added to therapy to improve remission rates and reduce relapse rates.153 The IFN signature score, defined based on the changes in the ISGs, can efficiently predict the SLE responder index (SRI) response after 12 months of belimumab treatment.154 Imatinib, an immune checkpoint molecule V-domain immunoglobulin suppressor of T-cell activation (VISTA) agonist, modulates VISTA through IFN-I and non-classical NF-κB pathways to alleviate symptoms in lupus-like mice.155
In summary, multiple drugs and cellular therapies targeting the IFN-I signaling pathway have made significant progress in the treatment of SLE, offering a variety of potential therapeutic strategies (Table 2). However, current studies are still mainly focused on IFN downstream targets, and more in-depth studies in the direction of upstream sources of IFN for the treatment of autoimmune diseases can be conducted in the future.
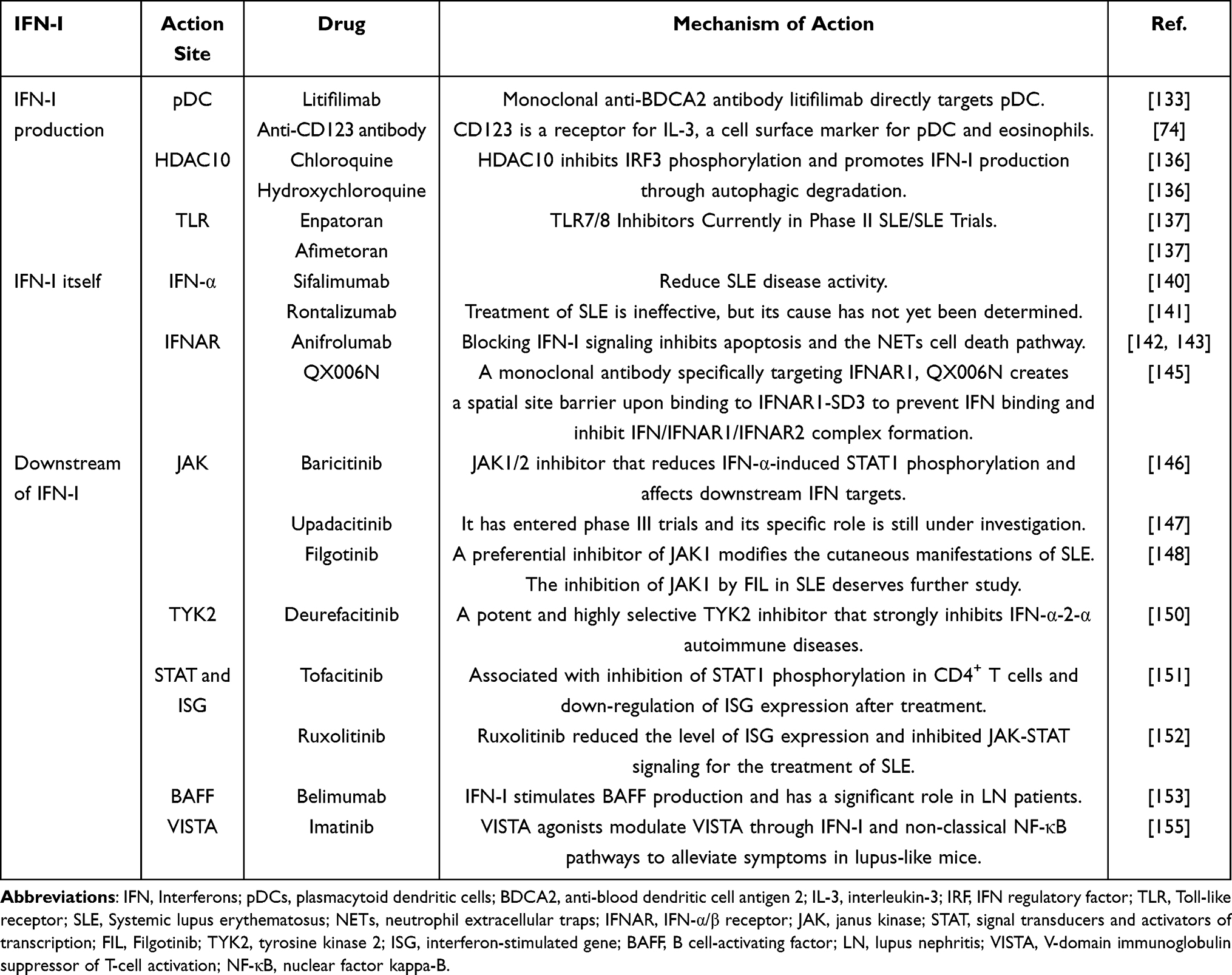 |
Table 2 Drugs That Treat SLE by Affecting the IFN-I |
Conclusion
Considerable evidence shows that IFN-I levels are significantly elevated in SLE patients, thereby contributing to the onset and progression of the disease. Research on the process of IFN production is well-established, and current studies focus on polymers that contribute to SLE disease by affecting IFN-I. The role of IFN-I in SLE pathogenesis has become a focal point in recent research, offering fresh insights into the disorder. Primarily produced by pDC, IFN-I significantly influences the pathophysiological progression of SLE by affecting various immune cells. This review clarifies the complex relationship between IFN-I and various immune cells, leading to a deeper understanding of SLE, advancing our comprehension of immune regulation, and opening up new possibilities for treatment (Figure 4). The limitations of this study include the lack of in-depth exploration of the differential effects of IFN-I on various tissues in SLE, such as the kidneys, skin, and hematopoietic system, as well as the absence of detailed analysis on the potential challenges of related drugs, including side effects and drug resistance. Nonetheless, more studies are needed to delve into IFN-I signaling transmission in B cells and its effects on Treg, as these aspects are not fully covered in existing studies.
 |
Figure 4 Major effects of IFN-I on multiple immune cells in the pathogenesis of SLE. pDC are the major IFN-I-producing cells. SLE neutrophils have strong IFN-I signaling and have a greater NET capacity. Monocyte macrophages are the main IFN-β producing cells. IFN-I differentiates B cells into plasmablasts. IFN-I can alter Th1/Th2 and Th17/Treg balance. This image was created by Figdraw. Abbreviations: IFN-I, Interferon-I; SLE, Systemic lupus erythematosus; pDC, plasmacytoid dendritic cell; NET, neutrophil extracellular trap; Treg, regulatory T. |
Acknowledgments
This study was supported by the National Natural Science Foundation of China (82271761 and 82001656), Fundamental Research Program of Shanxi Province (202303021224012), and Research and Innovation Team Project for Scientific Breakthroughs at Shanxi Bethune Hospital (2024ZHANCHI09).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no competing interests in this work.
References
1. Caielli S, Wan Z, Pascual V. Systemic Lupus Erythematosus Pathogenesis: interferon and Beyond. Annu Rev Immunol. 2023;41:533–560. doi:10.1146/annurev-immunol-101921-042422
2. Lazar S, Kahlenberg JM. Systemic Lupus Erythematosus: new Diagnostic and Therapeutic Approaches. Annual Review Med. 2023;74:339–352. doi:10.1146/annurev-med-043021-032611
3. Crow MK. Pathogenesis of systemic lupus erythematosus: risks, mechanisms and therapeutic targets. Ann Rheumatic Dis. 2023;82(8):999–1014. doi:10.1136/ard-2022-223741
4. Psarras A, Emery P, Vital EM. Type I interferon-mediated autoimmune diseases: pathogenesis, diagnosis and targeted therapy. Rheumatology. 2017;56(10):1662–1675. doi:10.1093/rheumatology/kew431
5. Fava A, Buyon J, Mohan C, et al. Integrated urine proteomics and renal single-cell genomics identify an IFN-γ response gradient in lupus nephritis. JCI Insight. 2020;5(12). doi:10.1172/jci.insight.138345.
6. Fueyo-González F, McGinty M, Ningoo M, et al. Interferon-β acts directly on T cells to prolong allograft survival by enhancing regulatory T cell induction through Foxp3 acetylation. Immunity. 2022;55(3):459–474.e457. doi:10.1016/j.immuni.2022.01.011
7. Nian Z, Mao Y, Xu Z, et al. Multi-omics analysis uncovered systemic lupus erythematosus and COVID-19 crosstalk. Molecular Med. 2024;30(1):81.
8. Ding X, Cai M, Wang S, et al. Gene-based association analysis identified a novel gene associated with systemic lupus erythematosus. Ann Human Genetics. 2021;85(6):213–220. doi:10.1111/ahg.12439
9. Gallucci S, Meka S, Gamero AM. Abnormalities of the type I interferon signaling pathway in lupus autoimmunity. Cytokine. 2021;146:155633. doi:10.1016/j.cyto.2021.155633
10. Van Eyndhoven LC, Chouri E, Matos CI, et al. Unraveling IFN-I response dynamics and TNF crosstalk in the pathophysiology of systemic lupus erythematosus. Front Immunol. 2024;15:1322814. doi:10.3389/fimmu.2024.1322814
11. Paradowska-Gorycka A, Wajda A, Stypinska B, et al. Variety of endosomal TLRs and Interferons (IFN-α, IFN-β, IFN-γ) expression profiles in patients with SLE, SSc and MCTD. Clin Exp Immunol. 2021;204(1):49–63. doi:10.1111/cei.13566
12. Whittall Garcia LP, Gladman DD, Urowitz M, et al. Interferon-α as a biomarker to predict renal outcomes in lupus nephritis. Lupus Sci Med. 2024;11(2):1.
13. Trinchieri G. Type I interferon: friend or foe? J Exp Med. 2010;207(10):2053–2063. doi:10.1084/jem.20101664
14. Liu HM. Intracellular innate immunity and mechanism of action of cytosolic nucleic acid receptor-mediated type I IFN against viruses. IUBMB Life. 2022;74(2):180–189. doi:10.1002/iub.2551
15. Sakaniwa K, Fujimura A, Shibata T, et al. TLR3 forms a laterally aligned multimeric complex along double-stranded RNA for efficient signal transduction. Nat Commun. 2023;14(1):164. doi:10.1038/s41467-023-35844-2
16. Zhou W, Whiteley AT, de Oliveira Mann CC, et al. Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell. 2018;174(2):300–311.e311. doi:10.1016/j.cell.2018.06.026
17. Kumar V. A STING to inflammation and autoimmunity. J Leukocyte Biol. 2019;106(1):171–185. doi:10.1002/JLB.4MIR1018-397RR
18. Ergun SL, Fernandez D, Weiss TM, Li L. STING Polymer Structure Reveals Mechanisms for Activation, Hyperactivation, and Inhibition. Cell. 2019;178(2):290–301.e210. doi:10.1016/j.cell.2019.05.036
19. Zhang K, Huang Q, Li X, et al. The cGAS-STING pathway in viral infections: a promising link between inflammation, oxidative stress and autophagy. Front Immunol. 2024;15:2.
20. Wang C, Zhou W, Liu Y, et al. Nuclear translocation of RIG-I promotes cellular apoptosis. J Autoimmun. 2022;130:102840. doi:10.1016/j.jaut.2022.102840
21. Dunker W, Ye X, Zhao Y, Liu L, Richardson A, Karijolich J. TDP-43 prevents endogenous RNAs from triggering a lethal RIG-I-dependent interferon response. Cell Rep. 2021;35(2):108976. doi:10.1016/j.celrep.2021.108976
22. Chang CY, Liu HM, Chang MF, Chang SC. Middle East Respiratory Syndrome Coronavirus Nucleocapsid Protein Suppresses Type I and Type III Interferon Induction by Targeting RIG-I Signaling. J Virol. 2020;94(13). doi:10.1128/JVI.00099-20
23. Liu G, Lee JH, Parker ZM, et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat Microbiol. 2021;6(4):467–478. doi:10.1038/s41564-021-00884-1
24. Zhu Z, Zhang M, Yuan L, et al. LGP2 Promotes Type I Interferon Production To Inhibit PRRSV Infection via Enhancing MDA5-Mediated Signaling. J Virol. 2023;97(1):e0184322. doi:10.1128/jvi.01843-22
25. Arimoto KI, Miyauchi S, Stoner SA, Fan JB, Zhang DE. Negative regulation of type I IFN signaling. J Leukocyte Biol. 2018. doi:10.1002/JLB.2MIR0817-342R
26. Zhang XJ, Jiang DS, Li H. The interferon regulatory factors as novel potential targets in the treatment of cardiovascular diseases. Br J Pharmacol. 2015;172(23):5457–5476. doi:10.1111/bph.12881
27. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25(3):349–360. doi:10.1016/j.immuni.2006.08.009
28. Miyamoto M, Fujita T, Kimura Y, et al. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements. Cell. 1988;54(6):903–913. doi:10.1016/S0092-8674(88)91307-4
29. Wang J, Basagoudanavar SH, Wang X, et al. NF-kappa B RelA subunit is crucial for early IFN-beta expression and resistance to RNA virus replication. J Immunol. 2010;185(3):1720–1729. doi:10.4049/jimmunol.1000114
30. Liu Y, Wang X, Huang M, et al. METTL3 facilitates kidney injury through promoting IRF4-mediated plasma cell infiltration via an m6A-dependent manner in systemic lupus erythematosus. BMC Med. 2024;22(1):511. doi:10.1186/s12916-024-03735-y
31. Ritter J, Szelinski F, Aue A, et al. Elevated unphosphorylated STAT1 and IRF9 in T and B cells of primary sjögren’s syndrome: novel biomarkers for disease activity and subsets. J Autoimmun. 2024;147:103243. doi:10.1016/j.jaut.2024.103243
32. Fan W, Wei B, Chen X, et al. Potential role of RhoA GTPase regulation in type interferon signaling in systemic lupus erythematosus. Arthritis Res Therapy. 2024;26(1):31. doi:10.1186/s13075-024-03263-3
33. Fang Z, Sun H, Wang Y, Sun Z, Yin M. Discovery of WD-890: a novel allosteric TYK2 inhibitor for the treatment of multiple autoimmune disease. Biomed Pharmacother. 2023;167:115611.
34. Ramírez-Vélez G, Medina F, Ramírez-Montaño L, et al. Constitutive phosphorylation of interferon receptor A-associated signaling proteins in systemic lupus erythematosus. PLoS One. 2012;7(7):e41414. doi:10.1371/journal.pone.0041414
35. Du Y, Brodeur KE, Hsu E, et al. In cis “benign” SOCS1 variants linked to enhanced interferon signaling and autoimmunity. J Autoimmun. 2023;140:103119. doi:10.1016/j.jaut.2023.103119
36. Ma X, Nakayamada S, Kubo S, et al. Expansion of T follicular helper-T helper 1 like cells through epigenetic regulation by signal transducer and activator of transcription factors. Ann Rheumatic Dis. 2018;77(9):1354–1361. doi:10.1136/annrheumdis-2017-212652
37. López P, Rodríguez-Carrio J, Caminal-Montero L, Suárez A. Relationship Between T-Cell Exosomes and Cellular Subsets in SLE According to Type I IFN-Signaling. Front Med. 2020;7:604098. doi:10.3389/fmed.2020.604098
38. Garcia-Romo GS, Caielli S, Vega B, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Trans Med. 2011;3(73):73ra20. doi:10.1126/scitranslmed.3001201
39. Davison LM, Jorgensen TN. New Treatments for Systemic Lupus Erythematosus on the Horizon: targeting Plasmacytoid Dendritic Cells to Inhibit Cytokine Production. J Clin Cell Immunol. 2017;8(6):1.
40. Han S, Zhuang H, Lee PY, et al. Differential Responsiveness of Monocyte and Macrophage Subsets to Interferon. Arthritis Rheumatol. 2020;72(1):100–113. doi:10.1002/art.41072
41. Khiewkamrop P, Kaewraemruaen C, Manipuntee C, et al. Immunosuppressive Polymeric Nanoparticles Targeting Dendritic Cells Alleviate Lupus Disease in Fcgr2b(-/-) Mice by Mediating Antigen-Specific Immune Tolerance. Int J mol Sci. 2023;24(9). doi:10.3390/ijms24098313.
42. Lande R, Ganguly D, Facchinetti V, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Trans Med. 2011;3(73):73ra19. doi:10.1126/scitranslmed.3001180
43. Wigerblad G, Kaplan MJ. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat Rev Immunol. 2023;23(5):274–288. doi:10.1038/s41577-022-00787-0
44. Matta B, Barnes BJ. Coordination between innate immune cells, type I IFNs and IRF5 drives SLE pathogenesis. Cytokine. 2020;132:154731. doi:10.1016/j.cyto.2019.05.018
45. Seridi L, Cesaroni M, Orillion A, et al. Novel signatures associated with systemic lupus erythematosus clinical response to IFN-α/-ω inhibition. Lupus. 2021;30(5):795–806. doi:10.1177/0961203321995576
46. Chen YM, Tang KT, Liu HJ, Huang ST, Liao TL. tRF-His-GTG-1 enhances NETs formation and interferon-α production in lupus by extracellular vesicle. CCS. 2024;22(1):354. doi:10.1186/s12964-024-01730-7
47. Casey KA, Guo X, Smith MA, et al. Type I interferon receptor blockade with anifrolumab corrects innate and adaptive immune perturbations of SLE. Lupus Sci Med. 2018;5(1):e000286. doi:10.1136/lupus-2018-000286
48. Denny MF, Yalavarthi S, Zhao W, et al. A Distinct Subset of Proinflammatory Neutrophils Isolated from Patients with Systemic Lupus Erythematosus Induces Vascular Damage and Synthesizes Type I IFNs. J Immunol. 2010;184(6):3284–3297. doi:10.4049/jimmunol.0902199
49. Jog NR, Wagner CA, Aberle T, et al. Neutrophils isolated from systemic lupus erythematosus patients exhibit a distinct functional phenotype. Front Immunol. 2024;15:1339250. doi:10.3389/fimmu.2024.1339250
50. Ma S, Jiang W, Zhang X, Liu W. Insights into the pathogenic role of neutrophils in systemic lupus erythematosus. Current Opin Rheumatol. 2023;35(2):82–88. doi:10.1097/BOR.0000000000000912
51. Leitinger N, Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arteriosclerosis Thrombosis Vasc Biol. 2013;33(6):1120–1126. doi:10.1161/ATVBAHA.112.300173
52. Patiño-Martinez E, Nakabo S, Jiang K, et al. The Aconitate Decarboxylase 1/Itaconate Pathway Modulates Immune Dysregulation and Associates with Cardiovascular Disease Markers and Disease Activity in Systemic Lupus Erythematosus. J Immunol. 2024;2024:1.
53. Zhu Y, Deng J, Nan ML, et al. The Interplay Between Pattern Recognition Receptors and Autophagy in Inflammation. Adv Exp Med Biol. 2019;1209:79–108.
54. Zyzak J, Mitkiewicz M, Leszczyńska E, Reniewicz P, Moynagh Paul N, Siednienko J. HSV-1/TLR9-Mediated IFNβ and TNFα Induction Is Mal-Dependent in Macrophages. J Innate Immunity. 2020;12(5):387–398. doi:10.1159/000504542
55. Xiao ZX, Liang R, Olsen N, Zheng SG. Roles of IRF4 in various immune cells in systemic lupus erythematosus. Int Immunopharmacol. 2024;133:112077. doi:10.1016/j.intimp.2024.112077
56. Hiyama T, Kurasawa K, Hasegawa A, et al. Differences and similarities in cytokine profiles of macrophage activation syndrome in systemic lupus erythematosus and adult-onset Still’s disease. Clin Exp Med. 2023;23(7):3407–3416. doi:10.1007/s10238-023-00988-4
57. Qian Y, Lu M, Zheng Q. IL-10 and IFN-γ as markers for early recognition of pediatric systemic lupus erythematosus complicated with macrophage activation syndrome. Rheumatology. 2023;2023:1.
58. Xu H, Zhang X, Wang X, et al. Cellular spermine targets JAK signaling to restrain cytokine-mediated autoimmunity. Immunity. 2024;2024:1.
59. Wei S, Shen H, Zhang Y, et al. Integrative analysis of single-cell and bulk transcriptome data reveal the significant role of macrophages in lupus nephritis. Arthritis Res Therapy. 2024;26(1):84.
60. Zhang F, Zhang B, Ding H, et al. The Oxysterol Receptor EBI2 Links Innate and Adaptive Immunity to Limit IFN Response and Systemic Lupus Erythematosus. Adv Sci. 2023;10(27):e2207108. doi:10.1002/advs.202207108
61. Guilliams M, Mildner A, Yona S. Developmental and Functional Heterogeneity of Monocytes. Immunity. 2018;49(4):595–613. doi:10.1016/j.immuni.2018.10.005
62. Lee KE, Mun S, Kim SM, et al. The inflammatory signature in monocytes of Sjögren’s syndrome and systemic lupus erythematosus, revealed by the integrated Reactome and drug target analysis. Genes Genomics. 2022;44(10):1215–1229. doi:10.1007/s13258-022-01308-y
63. Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends in Immunology. 2009;30(9):455–464. doi:10.1016/j.it.2009.06.003
64. Stergioti EM, Manolakou T, Sentis G, et al. Transcriptomic and proteomic profiling reveals distinct pathogenic features of peripheral non-classical monocytes in systemic lupus erythematosus. Clinical Immunology. 2023;255:109765. doi:10.1016/j.clim.2023.109765
65. Kuga T, Chiba A, Murayama G, et al. Enhanced GATA4 expression in senescent systemic lupus erythematosus monocytes promotes high levels of IFNα production. Front Immunol. 2024;15:1320444. doi:10.3389/fimmu.2024.1320444
66. Jiang L, Han X, Qiu W, et al. Amelioration of Lupus Serum-Induced Skin Inflammation in CD64-Deficient Mice. Front Immunol. 2022;13:824008. doi:10.3389/fimmu.2022.824008
67. Chen KL, Patel J, Zeidi M, et al. Myeloid Dendritic Cells Are Major Producers of IFN-β in Dermatomyositis and May Contribute to Hydroxychloroquine Refractoriness. J Invest Dermatol. 2021;141(8):1906–1914.e1902. doi:10.1016/j.jid.2020.12.032
68. Pilon C, Subang R, Lonina E, et al. Type I Interferon-Dependent and -Independent TRIF Signaling are Required for Autoantibody Generation in an Induced Model of Systemic Lupus Erythematosus. J Immunol. 2023;210(1_Supplement):78.
69. Waterman HR, Dufort MJ, Posso SE, et al. Lupus IgA1 autoantibodies synergize with IgG to enhance plasmacytoid dendritic cell responses to RNA-containing immune complexes. Sci Trans Med. 2024;16(754):eadl3848. doi:10.1126/scitranslmed.adl3848
70. Sozzani S, Del Prete A, Bosisio D. Dendritic cell recruitment and activation in autoimmunity. J Autoimmun. 2017;85:126–140. doi:10.1016/j.jaut.2017.07.012
71. Salvi V, Gianello V, Busatto S, et al. Exosome-delivered microRNAs promote IFN-α secretion by human plasmacytoid DCs via TLR7. JCI Insight. 2018;3(10). doi:10.1172/jci.insight.98204.
72. Wiest MJ, Baert L, Gu C, et al. Endosomal trafficking inhibitor EGA can control TLR7-mediated IFNα expression by human plasmacytoid dendritic cells. Front Immunol. 2023;14:1202197. doi:10.3389/fimmu.2023.1202197
73. Lorant AK, Yoshida AE, Gilbertson EA, et al. Integrin αvβ3 Limits Cytokine Production by Plasmacytoid Dendritic Cells and Restricts TLR-Driven Autoimmunity. J Immunol. 2024;212(11):1680–1692. doi:10.4049/jimmunol.2300290
74. Monaghan KA, Hoi A, Gamell C, et al. CSL362 potently and specifically depletes pDCs in vitro and ablates SLE-immune complex-induced IFN responses. iScience. 2023;26(7):107173. doi:10.1016/j.isci.2023.107173
75. Bashal F. Hematological disorders in patients with systemic lupus erythematosus. Open Rheumatol J. 2013;7(1):87–95. doi:10.2174/1874312901307010087
76. Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who’s who of T-cell differentiation: human memory T-cell subsets. European J Immunol. 2013;43(11):2797–2809.
77. Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol. 2013;13(5):309–320. doi:10.1038/nri3442
78. Oke V, Gunnarsson I, Dorschner J, et al. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res Therapy. 2019;21(1). doi:10.1186/s13075-019-1878-y.
79. Perez RK, Gordon MG, Subramaniam M, et al. Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science. 2022;376(6589):eabf1970. doi:10.1126/science.abf1970
80. Iwata S, Zhang M, Hao H, et al. Enhanced Fatty Acid Synthesis Leads to Subset Imbalance and IFN-γ Overproduction in T Helper 1 Cells. Front Immunol. 2020;11:593103. doi:10.3389/fimmu.2020.593103
81. Oster C, Wilde B, Specker C, et al. BTLA Expression on Th1, Th2 and Th17 Effector T-Cells of Patients with Systemic Lupus Erythematosus Is Associated with Active Disease. Int J mol Sci. 2019;20(18):4505. doi:10.3390/ijms20184505
82. Morimoto S, Tokano Y, Nakano S, et al. Chemoattractant mechanism of Th1 cells in class III and IV lupus nephritis. Autoimmunity. 2009;42(2):143–149. doi:10.1080/08916930802438790
83. Mesquita Jr D, Kirsztajn GM, Franco MF, et al. CD4(+) T helper cells and regulatory T cells in active lupus nephritis: an imbalance towards a predominant Th1 response? Clin Exp Immunol. 2018;191(1):50–59. doi:10.1111/cei.13050
84. Fujii T, Okada M, Fujita Y, et al. Vaccination with autoreactive CD4(+)Th1 clones in lupus-prone MRL/Mp-Fas(lpr/lpr) mice. J Autoimmun. 2009;33(2):125–134. doi:10.1016/j.jaut.2009.06.001
85. Yu S, Gu J, Wang R, et al. TIGIT reverses IFN-α-promoted Th1-like Tregs via in-sequence effects dependent on STAT4. Arthritis Res Therapy. 2023;25(1):221.
86. Shimizu M, Kondo Y, Tanimura R, et al. T-bet represses collagen-induced arthritis by suppressing Th17 lineage commitment through inhibition of RORγt expression and function. Sci Rep. 2021;11(1):17357. doi:10.1038/s41598-021-96699-5
87. Lee GR. The Balance of Th17 versus Treg Cells in Autoimmunity. Int J mol Sci. 2018;19(3). doi:10.3390/ijms19030730
88. Alunno A, Bartoloni E, Bistoni O, et al. Balance between regulatory T and Th17 cells in systemic lupus erythematosus: the old and the new. Clinic Develop Immunol. 2012;2012:823085. doi:10.1155/2012/823085
89. Xin Y, He Z, Mei Y, et al. Interferon-α regulates abnormally increased expression of RSAD2 in Th17 and Tfh cells in systemic lupus erythematosus patients. Eur J Immunol. 2023;53(8):e2350420. doi:10.1002/eji.202350420
90. Li M, Yang C, Wang Y, et al. The Expression of P2X7 Receptor on Th1, Th17, and Regulatory T Cells in Patients with Systemic Lupus Erythematosus or Rheumatoid Arthritis and Its Correlations with Active Disease. J Immunol. 2020;205(7):1752–1762. doi:10.4049/jimmunol.2000222
91. Lande R, Palazzo R, Gestermann N, et al. Native/citrullinated LL37-specific T-cells help autoantibody production in Systemic Lupus Erythematosus. Sci Rep. 2020;10(1):5851. doi:10.1038/s41598-020-62480-3
92. Keller EJ, Dvorina N, Jørgensen TN. Spontaneous CD4+ T Cell Activation and Differentiation in Lupus-Prone B6.Nba2 Mice Is IFNAR-Independent. Int J mol Sci. 2022;23(2):874. doi:10.3390/ijms23020874
93. Jiang Q, Yang G, Xiao F, et al. Role of Th22 Cells in the Pathogenesis of Autoimmune Diseases. Front Immunol. 2021;12:688066.
94. Zhong W, Jiang Y, Ma H, Wu J, Jiang Z, Zhao L. Elevated levels of CCR6(+) T helper 22 cells correlate with skin and renal impairment in systemic lupus erythematosus. Sci Rep. 2017;7(1):12962. doi:10.1038/s41598-017-13344-w
95. Law C, Wacleche VS, Cao Y, et al. Interferon subverts an AHR-JUN axis to promote CXCL13(+) T cells in lupus. Nature. 2024;631(8022):857–866. doi:10.1038/s41586-024-07627-2
96. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41(4):529–542. doi:10.1016/j.immuni.2014.10.004
97. Choi JY, Ho JH, Pasoto SG, et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol. 2015;67(4):988–999.
98. Huang X, Wu H, Qiu H, et al. The expression of Bcl-6 in circulating follicular helper-like T cells positively correlates with the disease activity in systemic lupus erythematosus. Clinical Immunology. 2016;173:161–170. doi:10.1016/j.clim.2016.10.017
99. Dong X, Antao OQ, Song W, et al. Type I Interferon-Activated STAT4 Regulation of Follicular Helper T Cell-Dependent Cytokine and Immunoglobulin Production in Lupus. Arthritis Rheumatol. 2021;73(3):478–489.
100. Zhou X, Qi H, Li M, et al. mTORC2 contributes to systemic autoimmunity. Immunology. 2023;168(3):554–568. doi:10.1111/imm.13594
101. Jiang Q, Wang J, Jiang H, et al. Competitive binding of transcription factors underlies flexibility of T peripheral helper cells and T follicular helper cells in SLE. Rheumatology. 2022;61(11):4547–4557. doi:10.1093/rheumatology/keac112
102. Klarquist J, Cantrell R, Lehn MA, et al. Type I IFN Drives Experimental Systemic Lupus Erythematosus by Distinct Mechanisms in CD4 T Cells and B Cells. ImmunoHorizons. 2020;4(3):140–152. doi:10.4049/immunohorizons.2000005
103. Rao DA, Gurish MF, Marshall JL, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017;542(7639):110–114. doi:10.1038/nature20810
104. Yoshitomi H. Peripheral helper T cells, mavericks of peripheral immune responses. Int Immunol. 2024;36(1):9–16. doi:10.1093/intimm/dxad041
105. Shan Y, Nakayamada S, Nawata A, et al. TGF-β3 in differentiation and function of Tph-like cells and its relevance to disease activity in patients with systemic lupus erythematosus. Rheumatology. 2023;62(7):2464–2474. doi:10.1093/rheumatology/keac646
106. Tanemura S, Seki N, Tsujimoto H, et al. Role of interferons (IFNs) in the differentiation of T peripheral helper (Tph) cells. Int Immunol. 2022;34(10):533–544. doi:10.1093/intimm/dxac032
107. Zhao X, Wang S, Wang S, Xie J, Cui D. mTOR signaling: a pivotal player in Treg cell dysfunction in systemic lupus erythematosus. Clinical Immunology. 2022;245:109153. doi:10.1016/j.clim.2022.109153
108. Li H, Hong S, Qian J, Zheng Y, Yang J, Yi Q. Cross talk between the bone and immune systems: osteoclasts function as antigen-presenting cells and activate CD4+ and CD8+ T cells. Blood. 2010;116(2):210–217. doi:10.1182/blood-2009-11-255026
109. Blanco LP, Patino-Martinez E, Nakabo S, et al. Modulation of the Itaconate Pathway Attenuates Murine Lupus. Arthritis Rheumatol. 2022;74(12):1971–1983. doi:10.1002/art.42284
110. Peixoto TV, Carrasco S, Botte DAC, et al. CD4(+)CD69(+) T cells and CD4(+)CD25(+)FoxP3(+) Treg cells imbalance in peripheral blood, spleen and peritoneal lavage from pristane-induced systemic lupus erythematosus (SLE) mice. Adv Rheumatol. 2019;59(1):30. doi:10.1186/s42358-019-0072-x
111. Kailashiya V, Singh U, Kailashiya J. CTLA4 Gene Polymorphism and its Association with Disease Occurrence, Clinical Manifestations, Serum Markers and Cytokine Levels in SLE Patients from North India. Ind J Dermatol. 2022;67(3):311. doi:10.4103/ijd.ijd_82_22
112. Jonsson AH, Zhang F, Dunlap G, et al. Granzyme K(+) CD8 T cells form a core population in inflamed human tissue. Sci Trans Med. 2022;14(649):eabo0686. doi:10.1126/scitranslmed.abo0686
113. Huang B, Li H, Jiang Q, et al. Elevated type I IFN signalling directly affects CD8(+) T-cell distribution and autoantigen recognition of the skeletal muscles in active JDM patients. J Autoimmun. 2024;146:103232. doi:10.1016/j.jaut.2024.103232
114. Radziszewska A, Peckham H, Restuadi R, et al. Type I interferon and mitochondrial dysfunction are associated with dysregulated cytotoxic CD8+ T cell responses in juvenile systemic lupus erythematosus. Clin Exp Immunol. 2024;2024:1.
115. Buang N, Tapeng L, Gray V, et al. Type I interferons affect the metabolic fitness of CD8(+) T cells from patients with systemic lupus erythematosus. Nat Commun. 2021;12(1):1980. doi:10.1038/s41467-021-22312-y
116. Soni C, Perez OA, Voss WN, et al. Plasmacytoid Dendritic Cells and Type I Interferon Promote Extrafollicular B Cell Responses to Extracellular Self-DNA. Immunity. 2020;52(6):1022–1038.e1027. doi:10.1016/j.immuni.2020.04.015
117. Lam JH, Smith FL, Baumgarth N. B Cell Activation and Response Regulation During Viral Infections. Viral Immunol. 2020;33(4):294–306. doi:10.1089/vim.2019.0207
118. Barnas JL, Albrecht J, Meednu N, et al. B Cell Activation and Plasma Cell Differentiation Are Promoted by IFN-λ in Systemic Lupus Erythematosus. J Immunol. 2021;207(11):2660–2672. doi:10.4049/jimmunol.2100339
119. Bradford HF, Haljasmägi L, Menon M, et al. Inactive disease in patients with lupus is linked to autoantibodies to type I interferons that normalize blood IFNα and B cell subsets. Cell Rep Med. 2023;4(1):100894. doi:10.1016/j.xcrm.2022.100894
120. Quách TD, Manjarrez-Orduño N, Adlowitz DG, et al. Anergic responses characterize a large fraction of human autoreactive naive B cells expressing low levels of surface IgM. J Immunol. 2011;186(8):4640–4648.
121. Ferri DM, Nassar C, Manion KP, et al. Elevated Levels of Interferon-α Act Directly on B Cells to Breach Multiple Tolerance Mechanisms Promoting Autoantibody Production. Arthritis Rheumatol. 2023;75(9):1542–1555.
122. Sumikawa MH, Iwata S, Zhang M, et al. An enhanced mitochondrial function through glutamine metabolism in plasmablast differentiation in systemic lupus erythematosus. Rheumatology. 2022;61(7):3049–3059. doi:10.1093/rheumatology/keab824
123. Gao M, Liu S, Chatham WW, Mountz JD, Hsu HC. IL-4-Induced Quiescence of Resting Naive B Cells Is Disrupted in Systemic Lupus Erythematosus. J Immunol. 2022;209(8):1513–1522. doi:10.4049/jimmunol.2200409
124. Liu Z, Davidson A. IFNα Inducible Models of Murine SLE. Front Immunol. 2013;4:306. doi:10.3389/fimmu.2013.00306
125. Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12(12):716–730. doi:10.1038/nrrheum.2016.186
126. Henault J, Riggs JM, Karnell JL, et al. Self-reactive IgE exacerbates interferon responses associated with autoimmunity. Nat Immunol. 2016;17(2):196–203. doi:10.1038/ni.3326
127. Manolakou T, Nikolopoulos D, Gkikas D, et al. ATR-mediated DNA damage responses underlie aberrant B cell activity in systemic lupus erythematosus. Sci Adv. 2022;8(43):eabo5840. doi:10.1126/sciadv.abo5840
128. Takeshima Y, Iwasaki Y, Nakano M, et al. Immune cell multiomics analysis reveals contribution of oxidative phosphorylation to B-cell functions and organ damage of lupus. Ann Rheumatic Dis. 2022;81(6):845–853. doi:10.1136/annrheumdis-2021-221464
129. van Boxel-Dezaire AH, Zula JA, Xu Y, Ransohoff RM, Jacobberger JW, Stark GR. Major differences in the responses of primary human leukocyte subsets to IFN-beta. J Immunol. 2010;185(10):5888–5899. doi:10.4049/jimmunol.0902314
130. Wu L, Jiang X, Qi C, Zhang C, Qu B, Shen N. EZH2 Inhibition Interferes With the Activation of Type I Interferon Signaling Pathway and Ameliorates Lupus Nephritis in NZB/NZW F1 Mice. Front Immunol. 2021;12:653989. doi:10.3389/fimmu.2021.653989
131. Akita K, Yasaka K, Shirai T, Ishii T, Harigae H, Fujii H. Interferon α Enhances B Cell Activation Associated With FOXM1 Induction: potential Novel Therapeutic Strategy for Targeting the Plasmablasts of Systemic Lupus Erythematosus. Front Immunol. 2020;11:498703. doi:10.3389/fimmu.2020.498703
132. Dong G, Yang Y, Li X, et al. Granulocytic myeloid-derived suppressor cells contribute to IFN-I signaling activation of B cells and disease progression through the lncRNA NEAT1-BAFF axis in systemic lupus erythematosus. Biochim Biophys Acta. 2020;1866(1):165554. doi:10.1016/j.bbadis.2019.165554
133. Li X, Zhang Y, Li B, et al. An immunomodulatory antibody-drug conjugate targeting BDCA2 strongly suppresses plasmacytoid dendritic cell function and glucocorticoid responsive genes. Rheumatology. 2024;63(1):242–250. doi:10.1093/rheumatology/kead219
134. Yang D, Peng N, Zhang H, Qiu Z, Xu L, Pan M. Cordycepin ameliorates autoimmunity by promoting STING degradation via autophagy pathway. Br J Pharmacol. 2024;182(7):1546–1560. doi:10.1111/bph.17425
135. Bekaddour N, Smith N, Caspar B, et al. The histamine analogue clobenpropit modulates IRF7 phosphorylation and interferon production by targeting CXCR4 in systemic lupus erythematosus models. Front Immunol. 2024;15:1490593. doi:10.3389/fimmu.2024.1490593
136. Zhou W, Wang J, Wang X, et al. Degradation of HDAC10 by autophagy promotes IRF3-mediated antiviral innate immune responses. Sci Signaling. 2022;15(765):eabo4356. doi:10.1126/scisignal.abo4356
137. Deshmukh A, Pereira A, Geraci N, et al. Preclinical Evidence for the Glucocorticoid-Sparing Potential of a Dual Toll-Like Receptor 7/8 Inhibitor in Autoimmune Diseases. J Pharmacol Exp Ther. 2024;388(3):751–764. doi:10.1124/jpet.123.001744
138. Suffiotti M, Brazauskas P, Keller MP, et al. Pharmacodynamics of the S1P(1) receptor modulator cenerimod in a Phase 2b randomised clinical trial in patients with moderate to severe SLE. Ann Rheumatic Dis. 2024;2024:1.
139. Sun JL, Lyu TB, Chen ZL, et al. Methylprednisolone pulse therapy promotes the differentiation of regulatory T cells by inducing the apoptosis of CD4(+) T cells in patients with systemic lupus erythematosus. Clinical Immunology. 2022;241:109079. doi:10.1016/j.clim.2022.109079
140. Khamashta M, Merrill JT, Werth VP, et al. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheumatic Dis. 2016;75(11):1909–1916. doi:10.1136/annrheumdis-2015-208562
141. Kalunian KC, Merrill JT, Maciuca R, et al. A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-α) in patients with systemic lupus erythematosus (ROSE). Ann Rheumatic Dis. 2016;75(1):196–202. doi:10.1136/annrheumdis-2014-206090
142. Kalunian KC, Furie R, Morand EF, et al. A Randomized, Placebo-Controlled Phase III Extension Trial of the Long-Term Safety and Tolerability of Anifrolumab in Active Systemic Lupus Erythematosus. Arthritis Rheumatol. 2023;75(2):253–265. doi:10.1002/art.42392
143. Gensous N, Lazaro E, Blanco P, Richez C. Anifrolumab: first biologic approved in the EU not restricted to patients with a high degree of disease activity for the treatment of moderate to severe systemic lupus erythematosus. Expert Rev Clin Immunol. 2024;20(1):21–30. doi:10.1080/1744666X.2023.2268284
144. Mosca M, Emmas C, Nekeman-Nan C, et al. Anifrolumab Study for Treatment Effectiveness in the Real World (ASTER) among patients with systemic lupus erythematosus: protocol for an international observational effectiveness study. BMJ open. 2024;14(11):e086055. doi:10.1136/bmjopen-2024-086055
145. Chen X, Ke H, Li W, et al. Structural basis for the recognition of IFNAR1 by the humanized therapeutic monoclonal antibody QX006N for the treatment of systemic lupus erythematosus. Int J Biol Macromol. 2024;268(Pt 2):131721. doi:10.1016/j.ijbiomac.2024.131721
146. Zaidi SMM, Ahmed Fatmi SA, Ashraf MH, et al. Efficacy of baricitinib in the treatment of Systemic lupus erythematosus (SLE): a Systemic review and meta-analysis. Heliyon. 2023;9(12):e22643. doi:10.1016/j.heliyon.2023.e22643
147. Wang S, Ning W, Tang H, Mu C, Huang X. Efficacy and safety study of targeted small-molecule drugs in the treatment of systemic lupus erythematosus. Arthritis Res Therapy. 2024;26(1):98. doi:10.1186/s13075-024-03331-8
148. Werth VP, Fleischmann R, Robern M, et al. Filgotinib or lanraplenib in moderate to severe cutaneous lupus erythematosus: a phase 2, randomized, double-blind, placebo-controlled study. Rheumatology. 2022;61(6):2413–2423. doi:10.1093/rheumatology/keab685
149. Satoh-Kanda Y, Nakayamada S, Tanaka Y. Fine-tuning SLE treatment: the potential of selective TYK2 inhibition. RMD Open. 2024;10(4):e005072. doi:10.1136/rmdopen-2024-005072
150. Morand E, Pike M, Merrill JT, et al. Deucravacitinib, a Tyrosine Kinase 2 Inhibitor, in Systemic Lupus Erythematosus: a Phase II, Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2023;75(2):242–252. doi:10.1002/art.42391
151. Hasni SA, Gupta S, Davis M, et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat Commun. 2021;12(1):3391. doi:10.1038/s41467-021-23361-z
152. Wenzel J, van Holt N, Maier J, Vonnahme M, Bieber T, Wolf D. JAK1/2 Inhibitor Ruxolitinib Controls a Case of Chilblain Lupus Erythematosus. J Invest Dermatol. 2016;136(6):1281–1283. doi:10.1016/j.jid.2016.02.015
153. Itotagawa E, Tomofuji Y, Kato Y, et al. SLE stratification based on BAFF and IFN-I bioactivity for biologics and implications of BAFF produced by glomeruli in lupus nephritis. Rheumatology. 2023;62(5):1988–1997. doi:10.1093/rheumatology/keac528
154. Li H, Ju B, Luo J, et al. Type I interferon-stimulated genes predict clinical response to belimumab in systemic lupus erythematosus. Eur J Pharmacol. 2024;987:177204. doi:10.1016/j.ejphar.2024.177204
155. Yang L, Zhang T, Wang P, et al. Imatinib and M351-0056 enhance the function of Vista and ameliorate the development of SLE via IFN-I and noncanonical NF-κB pathway. Cell Biol Toxicol. 2023;39(6):3287–3304. doi:10.1007/s10565-023-09833-6
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
miR-31-5p Regulates Type I Interferon by Targeting SLC15A4 in Plasmacytoid Dendritic Cells of Systemic Lupus Erythematosus
Li S, Wu Q, Jiang Z, Wu Y, Li Y, Ni B, Xiao J, Zhai Z
Journal of Inflammation Research 2022, 15:6607-6616
Published Date: 6 December 2022