")
Back to Journals » Journal of Inflammation Research » Volume 17
Restore Intestinal Barrier Integrity: An Approach for Inflammatory Bowel Disease Therapy
Authors Kong C, Yang M, Yue N, Zhang Y , Tian C, Wei D, Shi R, Yao J , Wang L , Li D
Received 27 March 2024
Accepted for publication 2 July 2024
Published 14 August 2024 Volume 2024:17 Pages 5389—5413
DOI https://doi.org/10.2147/JIR.S470520
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Tara Strutt
Chen Kong,1,* Meifeng Yang,2,* Ningning Yue,3,* Yuan Zhang,4 Chengmei Tian,5 Daoru Wei,6 Ruiyue Shi,1 Jun Yao,1 Lisheng Wang,1 Defeng Li1
1The Second Clinical Medical College, Jinan University; Shenzhen, Guangdong, People’s Republic of China; 2Department of Hematology, Yantian District People’s Hospital, Shenzhen, Guangdong, People’s Republic of China; 3Department of Gastroenterology, Shenzhen People’s Hospital (the Second Clinical Medical College, Jinan University), Shenzhen, Guangdong, People’s Republic of China; 4Department of Medical Administration, Huizhou Institute of Occupational Diseases Control and Prevention, Huizhou, Guangdong, People’s Republic of China; 5Department of Emergency, Shenzhen People’s Hospital (the Second Clinical Medical College, Jinan University; the First Affiliated Hospital, Southern University of Science and Technology), Shenzhen, Guangdong, People’s Republic of China; 6Department of Rehabilitation, Shenzhen People’s Hospital (the Second Clinical Medical College, Jinan University; the First Affiliated Hospital, Southern University of Science and Technology), Shenzhen, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lisheng Wang; Defeng Li, Department of Gastroenterology, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University; the First Affiliated Hospital, Southern University of Science and Technology), No. 1017, Dongmen North Road, Luohu District, Shenzhen, 518020, People’s Republic of China, Tel +86 755 25533018, Email [email protected]; [email protected]
Abstract: The intestinal barrier maintained by various types of columnar epithelial cells, plays a crucial role in regulating the interactions between the intestinal contents (such as the intestinal microbiota), the immune system, and other components. Dysfunction of the intestinal mucosa is a significant pathophysiological mechanism and clinical manifestation of inflammatory bowel disease (IBD). However, current therapies for IBD primarily focus on suppressing inflammation, and no disease-modifying treatments specifically target the epithelial barrier. Given the side effects associated with chronic immunotherapy, effective alternative therapies that promote mucosal healing are highly attractive. In this review, we examined the function of intestinal epithelial barrier function and the mechanisms of behind its disruption in IBD. We illustrated the complex process of intestinal mucosal healing and proposed therapeutic approaches to promote mucosal healing strategies in IBD. These included the application of stem cell transplantation and organ-like tissue engineering approaches to generate new intestinal tissue. Finally, we discussed potential strategies to restore the function of the intestinal barrier as a treatment for IBD.
Keywords: inflammatory bowel disease, intestinal barrier, pathophysiology, treatment
Graphical Abstract:

Introduction
Inflammatory bowel disease (IBD) represents a chronic, non-specific inflammation stemming from autoimmune disorders that predominantly impact the gastrointestinal (GI) tract. Typically, IBD manifests in two principal forms: Crohn’s disease (CD) and ulcerative colitis (UC). While the exact etiology of IBD remains elusive, several factors have been confirmed to be associated with the disease processes, including environmental factors, genetic susceptibility, intestinal microbiota, and mental health problems.1,2
Significant distinctions exist between CD and UC, spanning systematic manifestations to molecular levels. For instance, UC primarily targets the colon, causing superficial mucosal inflammation that extends proximally in a continuous pattern. Conversely, CD can non-contiguously affect different segments of the digestive tract, characterized by transmural inflammation.3 However, both CD and UC share dysfunctional or premature death of epithelial cells, a diminishing mucus layer, and dysregulated underlying intestinal immunity, resulting in compromised barrier protection.4 The compromise of barrier integrity not only manifests clinically in IBD but also contributes to various alimentary diseases. This underscores a potential therapeutic avenue: the restoration of intestinal barrier integrity.5
The components of the intestinal barrier typically include three sections: the mucus layer, the mucosa, and the underlying mucosal immune system.6 The intestinal epithelium comprises a single layer of epithelial cells interconnected with tight junctions (TJs), playing a crucial role in regulating interactions among intestinal contents (eg, microbiota), the immune system, and other components.3,7 The diverse intestinal epithelial cells, each with unique functions, maintain intestinal health, and any reduction in their quantity or diversity weakens the epithelium’s protective and digestive capacity.
The mucus barrier functions as a crucial protective layer in the GI tract, serving as the initial defense against bacterial invasion.8,9 Goblet cells are the principal producers of mucus.10 Beneath the epithelial layer, the intestinal immune system provides defensive capabilities. A multitude of immune cells resides within the GI tract, either in organized anatomical structures such as Peyer’s patches and mesenteric lymph nodes or dispersed throughout the intestinal epithelium and lamina propria.11 Unlike the immune systems of other organs, the intestinal immune system autonomously down-regulates immune reactions to sustain an anti-inflammatory environment in a healthy state.3 In the unique environment of the intestine, filled with exogenous antigens (eg food antigens), the intestinal immune system must defend against pathogen invasion while preventing excessive immune damage to its tissues.12 Consequently, intestinal immunity maintains a dynamic balance.3 Typically, effector immune cells produce inflammatory factors to prevent infection, while regulatory cells inhibit inflammation.13 In a healthy state, intestinal macrophages produce large amounts of the anti-inflammatory interleukin (IL)-10 when exposed to bacteria, and dendritic cells (DCs) produce retinoic acid and transforming growth factor β (TGF-β) to promote the generation of regulatory T cells (Tregs).3
To uphold the integrity of the barrier, heightened attention must be directed towards the molecular mechanisms governing barrier function in IBD, particularly focusing on epithelial cell junctions. Cell junctions establish connections between adjacent epithelial cells and the extracellular matrix (ECM), unifying the epithelium. They also regulate the selective permeability of the epithelium, enabling it to control paracellular transportation.6 Among various types of junctions, TJs play a pivotal role in determining cell junction function.6,14 Consequently, the absence of TJs significantly compromises defense, a common characteristic observed in IBD (Figure 4).
Furthermore, the process of intestinal mucosal healing is intricate, involving the migration and proliferation of intestinal epithelial cells (IECs) and the regulation of intestinal microbial peptides, such as mucins, growth factors, and defensins, along with the involvement of various molecules.15,16 Simultaneously, the complex mechanisms underlying mucosal repair offer diverse therapeutic targets for restoring intestinal integrity. However, current IBD therapies predominantly focus on inflammation suppression, with no disease-modifying therapies available targeting the restoration of epithelial barrier integrity.17 Consequently, there is promising potential for further investigation into the intestinal barrier to propose more effective alternative therapies for IBD.
This review aims to elucidate the components of the intestinal barrier and their respective roles, along with delineating the changes occurring in the barrier within the context of IBD. Subsequently, we delve into the self-repair process of the intestinal barrier and the involved mechanisms. In the course of this exploration, we pinpoint therapeutic targets unearthed in the repair program, thereby prompting the contemplation of potential strategies aimed at reinstating both the function and integrity of the intestinal barrier in the treatment of IBD.
Components and Mechanisms of the Intestinal Barrier
Epithelium
The intestinal epithelium, constituting the innermost layer of the intestinal barrier, forms the lining of the GI tract. Comprising six specialized cell lineages, enterocytes (absorptive cells), microfold cells (M cells), goblet cells, Paneth cells, hormone-producing enteroendocrine cells (EECs), and stem cells (Figure 1).17 The epithelium plays a crucial role in maintaining barrier function. Enterocytes and M cells primarily facilitate absorption, while goblet cells, Paneth cells, and EECs are in charged of secretory mission.18 Enterocytes, the most abundant cells in the intestinal epithelium, significantly expand the surface area to enhance nutrient absorption.19 Another differentiated cell type, tuft cells, resemble enterocytes and are primarily responsible for sensing and responding to various signals involved in immune responses, particularly type 2 mucosal immunity.20,21
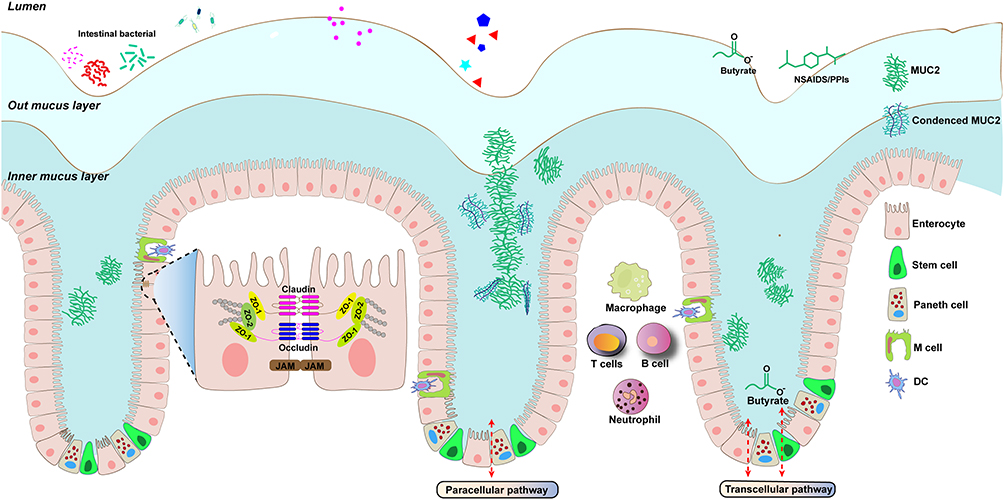 |
Figure 1 Illustration of the structure and functions of intestinal barrier. The intestinal barrier consists of a chemical barrier and a physical barrier. The epithelial cells form a physical barrier consisting of tight junction-associated proteins, including occludin, claudin; JAM: junctional adhesion molecule. These tight junction protein components close off the paracellular pathways and function as gates and fences. The mucus layer is a chemical barrier consisting of a dense inner layer and a lax outer layer that is essential for prevent invading microorganisms and the host. The skeleton that makes up the mucus layer is primarily MUC2. Immune cells are also involved in the immune response and host tolerance to external substances. Damage to the intestinal barrier leads to infiltration of intestinal microorganisms from the lumen into the lamina propria, inducing immune stress in host immune cells. |
M cells play a pivotal role in sampling gut contents and facilitating the transportation of luminal antigens to immune cells located in Peyer’s patches.22 Goblet cells, crucial in producing a protective mucus layer and secreting IgA to resist pathogens, also serve as antigen couriers conveying signals from the intestinal lumen to the underlying immune system.10,23 This initiates time-specific and location-specific processes to induce adaptive immune responses.10,24 Paneth cells contribute to the production of antimicrobial peptides (AMPs), including alpha-defensins, lysozyme, and secretory phospholipase A.3 Notably, the higher concentration of negatively charged phospholipids in bacterial cell membranes enables defensins to selectively and preferentially bind to them25. EECs synthesize and secrete different hormones to regulate digestive movement.26 Intestinal stem cells (ISCs), residing at the base of the crypts, give rise to all absorptive and secretory cells (Table 1).27,28 In the context of IBD, both the functionality and quantity of epithelial cells are compromised, resulting in a weakened protective function of the epithelium. This impairment highlights a significant aspect of IBD pathology.
 |
Table 1 Cell Types and Their Functions in the Composition of Intestinal Epithelium |
Cell Junction
The epithelial barrier’s functionality hinges on both a contiguous layer of cells and the existence of paracellular space between these cells. Cell junctions play a crucial role by binding epithelial cells into a cohesive unit and regulating selective paracellular transportation.6 In an inflammatory state, the contiguous layer breaks down, accompanied by an upregulation in paracellular conductance, indicating the involvement of TJ dysfunction. Within the intestinal epithelium, intercellular junctions include TJs, adherens junctions (AJs), and desmosomes, collectively referred to as the apical junctional complex (Figure 1).29
TJs form an apical belt-like structure around IECs and are integral in maintaining the integrity of the epithelium and restricting material exchange.29 Destruction of TJs has been identified in IBD, leading to barrier leakage that permits the passage of bacteria.25 Therefore, restoring the quantity and function of TJs has become a promising therapeutic strategy for IBD.
TJs consist of a complex network of specialized proteins, broadly categorized into transmembrane proteins, cytosolic plaque (scaffolding) proteins, and regulatory proteins.6,30 Transmembrane proteins include claudin, occludin, and junctional adhesion molecule (JAM), with claudin being particularly crucial.31,32 Claudins form pores by connecting with corresponding claudins on adjacent cells, regulating the selectivity of TJs.5 For instance, studies have revealed that claudin-2 constructs pores with ion selectivity, and its absence increases paracellular ion conductance without a concomitant increase in the flux of larger molecules.33–35 Gates of these pores exist in a dynamic state of opening and closing, similar to transmembrane ion channels, presenting a potential target for regulating barrier function.34 Occludins, tetraspanning proteins located at the cell-cell surface membrane, are phosphorylated by myosin light chain kinase (MLCK)-1, triggering their endocytosis and increasing TJ permeability.30,36 JAMs facilitate TJ assembly and leukocyte transmigration, and their absence increases susceptibility to inflammation.6,37 Cytosolic plaque proteins like ZO proteins directly interact with transmembrane TJ proteins and the actin cytoskeleton, regulating junction assembly.38
AJs comprise cadherin adhesion receptors and associated cytoplasmic proteins, such as actin filaments.39 E-cadherins form adhesive dimers at cell-cell boundaries, and their interaction with p120 catenin and β‑catenin on the cytoplasmic side, followed by α‑catenin binding to β‑catenin, establishes the cadherin–catenin core complex.39–41 AJs connect adjacent cells and the actin cytoskeleton, establishing a robust cell-cell connection. In vitro studies suggest that AJs facilitate TJ assembly through E‑cadherin and α-catenin.42,43 Furthermore, AJs are dynamic structures regulated by the cytoskeleton, enabling tissues remodeling in response to damage.39
Three pericellular pathways exist in TJs: pore, leak, and unrestricted pathways.44 Pore pathways, regulated by claudin proteins, are high-capacity, charge- and size-selective routes.45–47 Leak pathways are size-selective and regulated by occludin, tricellulin, ZO-1, and perijunctional actomyosin.44 In healthy gut conditions, these pathways reflect TJ permeability.44 However, in severe UC, particularly at sites of extensive epithelial damage, the unrestricted pathway allows the passage of massive ions, macromolecules, and even whole bacteria through the intestinal barrier.5 Complex mechanisms regulate these pathways, offering potential therapeutic targets to reduce barrier permeability. For instance, casein-kinase-2 can induce the phosphorylation of occludins, and inhibiting this process accelerates the assembly of the TJ complex while restricting passage through claudin-2 pores.48 As a result, cell junctions hold the potential to identify pericellular permeability-regulating mechanisms, representing future therapeutic targets for IBD.
Mucus Layer
Mucus, a crucial component of the intestinal barrier, is composed of materials originating from the epithelium, predominantly produced by goblet cells. Key constituents include mucin-2 (MUC2), Fcγ binding protein (FCGBP), calcium-activated chloride channel regulator1 (CLCA1), and others (Figure 1).10,49,50 The mucus core is primarily comprised of MUC2, a densely glycosylated protein that serves as a skeleton, forming a multimeric and crosslinked network.10,51,52 Upon secretion into the intestinal lumen, the alkaline environment and the removal of calcium ions cause the unfolding of MUC2, leading to its expansion and the formation of a net of stratified mucus gel.53,54 Na+/H+ exchanger 3 (NHE3), a reverse transport protein expressed on the apical surface of IECs, constructs an acidic environment beneath the outer mucus layer. This acidic environment may contribute to the formation of the compact inner mucus layer by maintaining the tight structure of MUC2.55,56
Electron microscopy has revealed the predominantly reticular three-dimensional structure of mucus, featuring minute pores.57 This unique structure, along with its electrostatic properties, selectively blocks the movement of substances.10,58 The outer mucus layer is loose, allowing bacteria to enter and colonize, and is easily cleared out. In contrast, the inner layer is tightly structured, preventing bacterial penetration.23,52,53 Nevertheless, the permeability of the inner layer is not uniform across all regions, influenced by two types of goblet cells that produce distinct mucus. Goblet cells residing in the intestinal crypts secrete a dense mucus effective against bacteria, whereas inter-crypt goblet cells on the colonic epithelial layer produce a relatively penetrable mucus.58 The distinct properties of different mucus types enable the inner layer to prevent bacterial invasion while facilitating substance exchange.58
Mucus is secreted in response to damage factors, such as mechanical stress, elevated levels of microbial ligands, or ischemia.49,59–62 The mucus layer can execute rapid self-recovery after a certain level of injury. However, when the extent of mucosal injury surpasses a threshold or when damage factors persist, the self-repair capacity becomes insufficient to compensate. This situation is comparable to mucus dysfunction observed in IBD.55,63
The mucus layer serves as a multifunctional protective barrier in the intestine, forming a physical shield that covers the intestinal wall and encapsulates fecal matter. This barrier prevents direct contact between harmful substances and the epithelium.10,64,65 Additionally, mucus provides lubrication, facilitating the smooth passage of gut content and reducing the risk of mechanical damage.9,10,66 With its sticky texture and multi-layered net structure, mucus can effectively trap pathogens and undergo constant renewal through coordinated muscular contractions, thereby expelling trapped pathogens from the gut.67–69
Vitally, mucus plays a crucial role in maintaining gut microbiota homeostasis.6,8 The mucus layer includes specific carbohydrates that serve as an energy source for certain commensal bacteria, fostering the growth of beneficial symbiotic bacteria and hindering the colonization of invasive pathogens.70,71 Consequently, strategies focused on restoring mucus and its function have the potential to alleviate IBD. This emphasizes the significance of regarding mucus as a therapeutic target for interventions in IBD.
The Self-Repair Mechanism of a Healthy Intestinal Epithelium
Apart from the previously mentioned protective barrier, the self-renewal capability for restoring intestinal integrity plays a crucial role in defending against damaging factors (Figure 1). The healthy epithelium undergoes updates every four to 7 days.72,73 When the epithelium is damaged, adjacent epithelial cells gradually lose their columnar morphology, migrate toward the damaged area, and subsequently cover the wound in a process known as “epithelial restitution”.27 In the regeneration process, LGR5+ stem cells serve as the “origin” of the repair process. LGR5+ stem cells, a subtype of ISCs, express the LGR5 protein and possess high regenerative and differentiation potential.28,74–76 Primarily located at the crypt base of the small intestine and colon, these cells undergo programmatic activation, migrating upwards along the crypt-villus axis. During this process, ISCs undergo proliferation and differentiation and ultimately fill the gaps in epithelial damage.77 The intricate molecular mechanisms regulating this process highlight its complexity.
The repair process relies on signaling originating from the microenvironment, which is constructed by various cells, ECM, and more.77 Mesenchymal cells residing below the epithelium in the lamina propria secrete various factors into the microenvironment, playing essential roles in the repair process.3,78 For instance, FOXL1+ telocytes and GLI1-expressing mesenchymal cells, acting as important sources of Wnts, activate the Wnt/β-Catenin signaling pathway, promoting the proliferation of ISCs and transit amplifying cells.79,80 Fibroblasts produce IL-33, which directly acts on LGR5+ cells and promotes their differentiation towards the secretory lineage by suppressing Notch signaling.81 CD34+ Gp38+ fibroblasts upregulate the expression of Grem1, Rspo1, Wnt2b, and several chemokines, cytokines, and epithelial growth factors, participating in immune response and wound repair.82–84 Enteric glial cells (EGCs) enhance wound repair by promoting the spreading of IECs through pro-epidermal growth factor (EGF) secretion.85 Paneth cells, located at the base of the niche adjacent to ISCs, provide essential signals such as Delta-like 1/4, EGF, and Wnt to support ISC homeostasis.86,87 Additionally, the ECM can activate mechanical sensors, such as Yes-associated protein (YAP) and Tafazzin (TAZ), suppressing adult stem cell markers and reprogramming them into a primitive state.88
Moreover, studies have demonstrated that the underlying immune system regulates barrier restoration, with immune cells secreting cytokines with distinct protective effects.77 IL-6 can activate STAT3 and YAP to promote epithelial regeneration.89 Group 3 innate lymphoid cells (ILCs) secrete IL-22 in response to damage, inducing the phosphorylation of STAT3 in CBCs, subsequently enhancing organoid growth without activating the Wnt or Notch pathway.90,91 IL-10, rapidly secreted by macrophages upon intestinal injury, promotes epithelial proliferation through epithelial cAMP response element-binding protein (CREB) signaling and subsequent WNT1-inducible signaling protein 1 (WISP-1) secretion.92 Additionally, the host’s nutritional state also affects the repair process, as calorie restriction and a high-fat diet can induce the expansion of the ISC pool.93 In summary, given the epithelial damage in IBD, studies focused on the mechanisms involved in the repair process can identify therapeutic targets with the potential to facilitate epithelium restoration.
Intestinal Barrier Dysfunction in IBD Disease
The integrity of the intestinal barrier is significantly compromised in the context of IBD. This impairment can be primarily attributed to abnormalities in five key elements: epithelial cells, cell junctions, the mucus layer, the intestinal immune system, and commensal microbiota. Due to the complex interactions among these defenders, their dysfunctions can mutually exacerbate each other, potentially establishing a vicious cycle. Consequently, interrupting this cycle at an intermediate stage becomes crucial for controlling the progression of the disease.
Interplay of IEC Death and Immune Dysregulation in Barrier Disruption
During the active phase of IBD, an upregulation of programmed cell death within the intestine significantly compromises the integrity of the intestinal barrier. The premature death of stem cells has particularly serious consequences, inherently weakening the regenerative capacity of the epithelium.84,94 Common forms of programmed cell death include anoikis, apoptosis, necroptosis, and pyroptosis.84 In a healthy state, IECs undergo regular self-renewal, with cells shedding through apoptosis without triggering an inflammatory response.95
In individuals with IBD, there is an excessive apoptosis of IECs attributed to the synergistic effects of genetic susceptibility and environmental damaging factors.96 Macrophages, unfortunately, are unable to promptly clear these cells, resulting in secondary necrosis and pyroptosis, thereby triggering inflammation. The sustained and excessive release of inflammatory factors can fuel an overactive immune response, thereby contributing to the progression of autoimmune diseases such as IBD.97 In a healthy intestine, apoptotic IECs are engulfed by macrophages. Subsequently, macrophages undergo M2 polarization and metabolic switching, generating eicosanoids such as prostaglandins (PGs) and resolvins. These substances promote tissue repair and angiogenesis and suppress the immune system.84 Simultaneously, DCs sense cellular debris, transition to a tolerogenic state, and migrate to the mesenteric lymph nodes. There, regulatory T cells (Tregs) are activated and proliferate, suppressing effector T cell responses.84
In contrast, necroptotic or pyroptotic cell death, followed by the release of damage-associated molecular patterns (DAMPs), induces M1 polarization of macrophages and a pro-inflammatory transition of DCs.98 M1 macrophages secrete pro-inflammatory cytokines (IL-1β, IL-36, etc.), activating effector T cells in the lymphoid node and fibroblasts.99 DCs migrate to the mesenteric lymphoid node and present antigens, activating naive T cells (Tn) while inhibiting the activation of Tregs.84
The therapeutic potential of macrophage state switching in IBD has gained significant attentions recently. A recent study has found that turmeric-derived nanovesicles (TNVs) alleviate colitis-related symptoms by restoring the intestinal epithelial barrier. TNVs can suppress the expression of CD16/32 and increase the expression of CD206 in the colonic lamina propria, promoting the conversion of M1 to M2 macrophages.100 Additionally, CS-IGF-1C hydrogel has been found to alleviates inflammatory responses through PGE2-mediated polarization of M2 macrophages.101 Moreover, it has found that the HA/CS/Dex nanoparticles (HCD NPs) could be internalized by macrophages, thereby regulating the polarization from M1 to M2 macrophages and exerting anti-inflammatory effects.102 This mechanism provides a safe, feasible, and effective approach for treating ulcerative colitis.100
In the context of IBD, the progression of inflammation encompasses several crucial steps. The initial stage involves the activation of the inflammatory cell death pathway by environmental signals, culminating in pyroptosis or necrosis of IECs and the subsequent release of DAMPs. Pattern recognition receptors (PRRs) on both epithelial and immune cells identify specific stimuli, such as signals of tissue damage or infection.103–106 Once recognized, these receptors form inflammasomes with adaptor proteins and pro-caspases, activating caspases.107 Activated caspases cleave and translocate gasdermin D (GSDMD) to the cell membrane, creating pores that lead to the leakage of DAMPs into neighboring tissues.104,108 Simultaneously, inflammasomes mature pro-inflammatory cytokines (such as pro-IL-1β and pro-IL-18) and release them into surrounding tissues through cell membrane pores.95,99,109
As cell death advances, GSDMD molecules undergo cleavage, subsequently forming pores. This process leads to membrane rupture and the release of cellular contents into the surrounding tissue.110 DAMPs represent a class of molecular motifs typically released as endogenous molecules when cells are damaged or dead.111 Recognized by the immune system, these molecules can induce inflammatory responses.111 Under normal circumstances, these molecules are selectively expressed and sequestered within the nucleus, mitochondria, and cytoplasm, avoiding direct interaction with the immune system.105,112 Examples include double-stranded DNA (usually confined to the cell nucleus), ATP (commonly found in the cytoplasm), and various molecules existing at high concentrations exclusively within the mitochondria.105 In addition to releasing molecules typically sequestered inside cells, DAMPs may also encompass modified components of the ECM, such as hyaluronic acid (HA).105
The second step involves the activation of immune cells and the release of pro-inflammatory factors. DAMPs are recognized by PRRs on sentinel cells, including macrophages, DCs, and ILCs. This recognition prompts these sentinel cells to produce pro-inflammatory cytokines, including tumor necrosis factor (TNF)-α, IL-1, histamine, nitric oxide (NO), reactive oxygen species (ROS), PGs, and leukotrienes.105,106,113 Ultimately, the uncontrolled release of pro-inflammatory cytokines triggers adaptive immune dysregulation, resulting in dysfunction of the immune system and loss of tolerance to normal tissues and organs.96 In this scenario, autoantibodies and self-reactive T cells may erroneously attack the intestinal tissues, contributing to the progression of IBD inflammation.12,97,114–116
As research progresses, numerous therapeutic targets have been identified in this process. In patients with IBD, particularly those with UC, an increased expression of GSDMD has been observed in the intestinal mucosa.99 This heightened expression can stimulate IECs to release IL-1β, thereby enhancing the inflammatory response. Experimental evidence also indicates that mice with GSDMD defects exhibit reduced inflammation in response to dextran sulfate sodium (DSS) induction.99
Studies have found that SETDB, a histone methyltransferase, safeguards genome stability. In patients with IBD, its decreased expression leads to genome instability of ISCs and induces Z-DNA binding protein 1 (ZBP1)-mediated necroptosis.117,118 Therefore, interfering with the sensing of endogenous retrovirus (ERV) RNA by ZBP1 may hold potential therapeutic benefits for patients with IBD.117 Additionally, upregulated TNF-α has been found to promote necroptosis of ISCs, especially active LGR5+ ISCs sensitive to acute injury. Prostaglandin E2 (PGE2) has a potential targeted therapeutic effect, as it can facilitate the expansion of LGR5+ ISCs via EP2-mediated signaling.119 Furthermore, it has been discovered that XBP1 deletion triggers endoplasmic reticulum (ER) stress, heightening the sensitivity of IECs to cell death and subsequently contributing to the spontaneous apoptosis of differentiated Paneth and goblet cells.120
Mitochondrial metabolism dysbiosis has also been identified as relevant to IBD. Prohibitin 1 (PHB1), a major component of the inner mitochondrial membrane, is absent in ISCs, leading to spontaneous ileal inflammation, which can be reversed with a mitochondrial-targeted antioxidant.121 Studies have also discovered that deletion of heat shock protein 60 (Hsp60) in ISCs induces mitochondrial dysfunction, resulting in reduced LGR5+ expression and then inhibiting the differentiation of LGR5+ cells into Paneth cells.122 Dichloroacetate (DCA) regulates mitochondrial disorders by diminishing glycolysis and improving mitochondrial respiration, presenting the potential to prevent IBD recurrence.122 Thus, therapies aimed at enhancing epithelial renewal ability or inhibiting cell death provide therapeutic modalities for IBD.
Increased Barrier Permeability
In the context of IBD, an increase in barrier permeability and abnormalities in cell junctions have been observed.25,123 These manifestations in IBD are associated with the upregulation of inflammatory factors, such as TNF-α, IFN-γ, and interleukin.124,125 TNF-α, for instance, facilitates the phosphorylation of the myosin light chain (MLC), leading to a decrease in occludins, and an increase in conductance through the leak pathway.36,126–130 Simultaneously, the Th2 cytokine IL-13 induces the expression of claudin-2, resulting in increased pore pathway flux.6,94,128 IFN and TNF synergistically affect TJs by upregulating each other’s receptors.131,132 High doses of TNF and IFN can reduce zonula occludens (ZO), further compromising the protective function of TJs.133,134 In CD, there is a predominant increase in IFN and TNF levels, while in UC, the primary elevation is observed in IL-13 and TNF, leading to a significant increase in claudin-2 (Figure 2).94,128,135
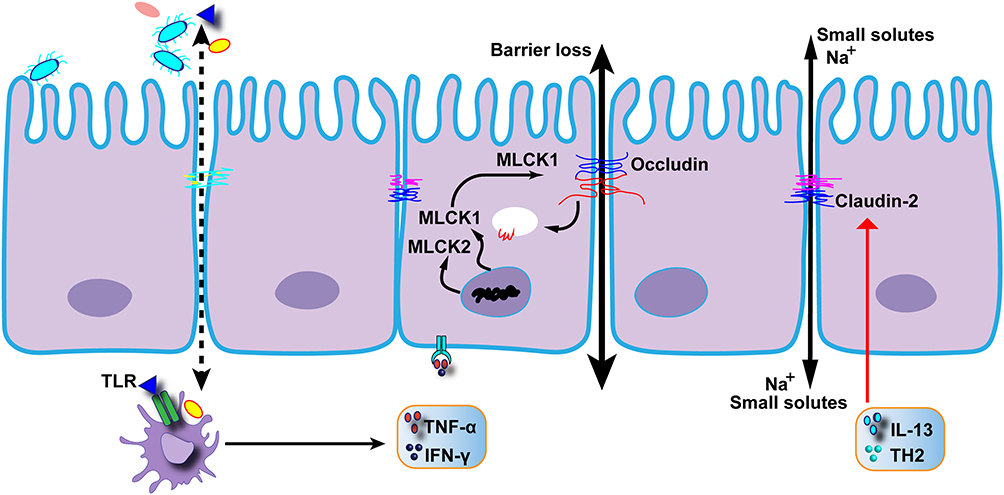 |
Figure 2 The molecule diverts a key signaling molecule away from the intercellular junctions that regulate the intestinal barrier, preventing the immune system from disrupting the barrier. |
Active CD patients exhibit distinctly impaired intestinal barrier function. Despite the presence of localized epithelial lesions, freeze-fracture electron microscopy reveals that discontinuous TJ strands contribute to the dysfunction of the intestinal barrier.94 In the active state of IBD, there is a significant decline in the expression of claudin 5 and claudin 8, which inhibits cation permeability, whereas there is a notable upregulation of claudin 2, which forms pores.94
During the remission phase of CD, TJ proteins are almost restored, and cell necrosis is basically under control, leading to normalized barrier function.94 Unexpectedly, some research has indicated that certain patients still exhibit distinct increased intestinal permeability.136,137 Further studies have subsequently demonstrated that the upregulated permeability is caused by increased transcellular transport, primarily mediated by immune-epithelial interactions, rather than ascending pericellular conductance.138 For example, CD23, a low-affinity receptor for IgE, is overexpressed at the apical surface of intestinal cells in CD, leading to increased transcellular transport of IgE-allergen immune complexes from the intestinal lumen to the lamina propria, subsequently triggering mast cell degranulation and allergic inflammatory responses in the subepithelial immune system.139 IIn general, in the inflamed region, the transportations through both pericellular and transcellular pathways increase, resulting in excessive stimulation to the local intestinal immune system, which, in turn, promotes inflammation.25,139 This process ultimately forms a vicious spiral, inducing the deterioration of barrier function.
Mucus Layer Alterations
The protective capability of the mucus layer is weakened in patients with IBD, contributing to increased bacterial penetrability.49,140,141 Tissue biopsies of UC patients reveal that the mucus layer becomes thinner and even absent in certain areas, and this change is independent of local inflammation.49,142 However, this phenomenon is not absolute. In IL-10 knockout mice, an unexpected increase in mucus layer thickness has been observed, possibly as a compensatory response to the weakened function of the mucus barrier.141 However, the quality of the mucus secreted in compensation is often inferior.52 The mechanisms underlying the thinner and poor-quality mucus layer are complex.
The primary underlying factor is the malfunction of mucus producers in IBD. Examination of tissue biopsies from patients experiencing active IBD reveals a distinct reduction in mature goblet cells, likely stemming from a combination of factors such as excessive mucus release, diminished mucus storage, altered goblet cell differentiation, and increased apoptosis.49,143 Substantial support for this hypothesis is provided by the noticeable decline in sentinel cells.49,53 In the case of patients with remission in UC, biopsies indicate a decrease in mitochondrial β-oxidation, potentially disrupting goblet cell differentiation.144 Although the overall number of goblet cells remains relatively constant during the remission period, specific subtypes experience a reduction.141 For instance, inter-crypt goblet cells are prematurely shed during the remission phase, resulting in the absence of inter-crypt mucus. This suggests a potential link between defective inter-crypt mucus and the initiation of IBD.10,58
Moreover, the antibacterial protein WFDC2, secreted by goblet cells, is dysregulated in IBD. Given that WFDC2 inhibits the activity of serine and cysteine proteases, preventing the premature conversion of the inner mucus layer to the outer layer, its deficiency can lead to abnormal mucus layer formation, facilitating bacterial invasion.145 Additionally, electron microscopy has revealed the accumulation of incompletely synthesized or misfolded MUC2 mucin in the ER, indicating heightened ER stress in goblet cells.146 Recent studies suggest that elevated ER stress has the potential to inhibit the production of MUC2, thereby triggering the progression of IBD in the early stages.53,147–149
Mucus barrier dysfunction stems from structural irregularities and disrupted formation of the mucus layer. The aberrant mucus demonstrates reduced glycosylation, shortened oligosaccharide side chains, diminished sulfation, and a decrease in core mucus proteins.49,150,151 Additionally, early investigations have pinpointed the downregulation of the chloride anion exchanger SLC26A3 and the malfunction of sodium-hydrogen exchangers in IBD, both pivotal in mucus layer formation.56,152–154
Moreover, emerging research highlights the impact of dietary habits and the dysbiosis of the intestinal microbiota in the IBD process. Dietary fibers serve as the primary energy source for the microbiota in the lower GI tract.155 Certain microbial species, such as Bacteroides thetaiotaomicron, Bifidobacterium bifidum, Ruminococcus gnavus, Ruminococcus torques, and Akkermansia muciniphila, can utilize glycans for mucus barrier formation.53,156,157 Chronic and intermittent dietary fiber deficiency amplifies the survival competitiveness of bacteria dependent on mucins as a food source. As these bacteria proliferate, mucins are continually depleted, leading to a gradual thinning of the mucus layer.53 Consequently, the diminished mucus layer becomes more susceptible, facilitating bacterial penetration.
Dysbiosis in the GI Microbiomes
A noticeable alteration in the density and composition of intestinal bacteria has been identified, contributing to microbiota dysbiosis in individuals with IBD.53 In healthy individuals, the predominant bacterial phyla include Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria.158 The microbiota in the lower GI tract primarily utilize dietary fiber as their energy source.155 Fibrolytic bacteria break down polysaccharides into smaller carbohydrates, subsequently fermenting them into short-chain fatty acids (SCFAs) such as acetate, propionate, and butyrate.159 SCFAs exhibit immunomodulatory properties and stimulate the proliferation of Tregs in the intestine.160–164
Particularly noteworthy is butyrate’s ability to induce the expression of the actin-binding protein synaptopodin (SYNPO), promoting the repair of intestinal epithelium. Butyrate also serves as the primary energy source for colonocyte.39,163–165 Research has demonstrated a reduction in SCFA-producing bacteria in the context of IBD, leading to a gradual loss of the protective effects provided by SCFAs.25,162,166
Although these dysfunctions have been identified in the IBD process, the causality linking the onset of IBD to these dysfunctions, along with the intricate interplay among different dysfunctions, still requires clarification. Further research is essential to illuminate the pathogenesis of IBD, pinpoint suitable therapeutic targets, and subsequently devise effective treatment approaches aimed at restoring barrier integrity.
Current Therapeutic Strategies That Improve Barrier Dysfunction
Current therapies for IBD can be broadly categorized into two groups: untargeted treatments, including amino salicylates, glucocorticoids, and immunosuppression, and targeted biologic therapies, such as anti-TNF, anti-IL-12/IL-23, anti-α4β7 integrin, IL-10, and IL-22.15,167–170 Although biological therapies demonstrate efficacy for a substantial number of patients, around 30% do not respond to initial treatment, and up to 50% experience a loss of response over time.171 Notably, certain medications, especially steroids and immunomodulators, exert broad immunosuppressive effects, elevating the risk of infectious and neoplastic events.15,171 Consequently, there is a growing interest in therapies focused on restoring intestinal barrier integrity (Figure 3).
 |
Figure 3 Possible therapeutic strategies to improve the intestinal barrier function. |
 |
Figure 4 (a) Structural and functional modulation of the intestinal epithelial barrier under IBD will provide new ideas for the development of therapies for such chronic diseases. (b) The cell junctions of intestinal epithelium are disrupted in inflammatory bowel disease, providing potential targets for treatment. |
Stem Cells Transplantation
Promoting the proliferation and differentiation of stem cells emerges as a promising therapeutic strategy, with three noteworthy methods outlined below. Firstly, the engraftment of ISCs has been explored.172 Early research indicates that the therapeutic efficacy of enema-based transplantation of ISCs is notably limited and becomes apparent primarily in the presence of persistent damaging factors.173 However, transplantation of mesenchymal stem cells (MSCs) and hematopoietic stem cells (HSCs) has demonstrated effectiveness.174,175 The second approach involves the culture and transplantation of organoids.176 Organoids, possessing a structure and cellular composition akin to the intestinal epithelium in the body, prove beneficial.87 This property facilitates the engraftment of intestinal organoids at damaged mucosal sites, thereby accelerating the healing of inflamed mucosa.173,177 Additionally, through the in vitro expansion and transplantation of autologous ISCs, the risk of rejection can be effectively minimized.168,174 The third strategy aims to enhance epithelial expansion. EGF and R-spondin-1 play crucial roles in epithelial growth.6 Activation of the EGF receptor has a protective effect against TNF-induced apoptosis. Recent studies have indicated that R-spondin-1 can mitigate inflammation in colitis models characterized by epithelial damage.178,179
Inhibition of Intestinal Epithelium Permeability
Lowering intestinal epithelium permeability holds the potential for controlling the progression of inflammation. As mentioned earlier, the phosphorylation of MLC can induce an increase in pericellular permeability regulated by TJs.36 A novel oligopeptide, PIK, has been reported to inhibit MLCK, resulting in the reversible increase of transepithelial resistance and down-regulation of pericellular permeability.127 It is noteworthy that various subtypes of MLCK play unique and crucial roles. For instance, the smooth muscle isoform of MLCK is pivotal in intestinal contraction, airway constriction, and blood pressure regulation.180 However, all MLCK subtypes share the same catalytic domain. Therefore, unless drugs specifically targeting long MLCK are developed, the clinical application of MLCK inhibitors may not be feasible.6,126
Furthermore, PIK demonstrates significant efficacy in alleviating mild intestinal permeability defects but falls short in reversing severe damage induced by higher doses of IFN-γ and TNF-α.127 Additionally, occludin and claudin-2 present potentially druggable targets.6 For example, intestinal alkaline phosphatase (IAP) can down-regulate the expression of vascular endothelial growth factor and claudin-2, thereby reducing intestinal permeability.25
Mucus Layer Restoration
The restoration of the mucus layer holds the potential to enhance intestinal barrier function. In the remission phase of IBD, increasing dietary fiber intake may reduce microbial consumption of mucins, facilitating the restoration of the mucus layer and thereby extending the remission period.53 However, during the active phase of IBD, excessive secretion of MUC2 leads to increased ER stress in goblet cells.53 In such instances, the application of exogenous mucins can be considered to supplement the deficient mucus barrier function, alleviate ER stress, and promote the recovery of goblet cells.53
Gut Microbiota Homeostasis
Reestablishing microbiota homeostasis stands out as a promising alternative for IBD therapy, given the crucial role of the gut microbiota in maintaining the integrity of the intestinal barrier.7,181 SCFAs, protective factors produced by commensal microbiota, decrease during the course of IBD. Studies have indicated that SCFAs can stimulate the differentiation and proliferation of epithelial cells in vivo.182,183 Therefore, supplementing exogenous SCFAs holds the potential to promote epithelial repair in the treatment of IBD.182
Additionally, fecal microbiota transplantation (FMT) represents a straightforward yet effective method for modulating gut microbiota.53 Research has demonstrated that FMT can rapidly reduce mucosal permeability, improve intestinal barrier integrity, and decrease colonic inflammation. Consequently, FMT can be employed to inhibit the progression of IBD and prevent the recurrence of UC.184 However, the optimal treatment frequency of FMT and the identification of beneficial bacterial strains require further investigation in future studies.
Molecular Target
Regulating the molecular mechanisms within the cell emerges as a valuable therapeutic strategy worthy of attention. In chronic inflammation associated with IBD, there is a distinct decrease in BRG1 expression. BRG1 has been proposed to function as an autophagy checkpoint linked to colitis, playing a role in maintaining the homeostasis of IECs. However, the deficiency of BRG1 results in impaired autophagy, leading to the production of excessive ROS and compromising barrier integrity.185 Intriguingly, the antioxidant N-acetyl-L-cysteine (NAC) has been shown to reverse this process partially.185
Furthermore, mitochondria constitute another critical therapeutic target in IBD treatment.186 In IBD patients, there is a decreased expression of PHB1, an abundant inner mitochondrial membrane protein that stabilizes mitochondrial DNA-encoded proteins, regulates mitochondrial fusion, and maintains optimal electron transport chain activity.187,188 Consequently, PHB1 deficiency leads to loss of viability in the ISC niche and Paneth cell defects.189 Mitochondria-targeted antioxidants, such as Mito-Tempo or MitoQ, can alleviate this damage.190,191 During the active phase of UC, the release of mitochondrial DNA into cells serves as a trigger, prompting neutrophils in the peripheral circulation to secrete IL-8 and thereby exacerbating inflammation. Consequently, strategies to prevent the release of mitochondrial DNA present a promising therapeutic avenue for IBD.186 Treatments that specifically target mitochondria are particularly significant for Paneth cells, where mitochondrial dysfunction precipitates defects in these cells.186 Given that Paneth cell defects arise before the onset of severe inflammation, they serve as predictive markers for early relapse and molecular indicators of incipient inflammation.186
Others
In addition to the previously mentioned treatment strategies, several alternative therapeutic approaches show promise for restoring intestinal epithelial integrity. Early studies suggest that mannose treatment improves lysosomal integrity, reduces the release of cathepsin B, and prevents mitochondrial dysfunction, thereby inhibiting MLCK-induced disruption in colonic inflammation.192 Amino acids such as arginine, histidine, and glutamine can promote enterocyte proliferation and reduce mucosal permeability by regulating TJ proteins.193,194 Matrix metalloproteinases play a role in ECM remodeling, promoting cell migration, and driving the intestinal epithelial regeneration process.195
Moreover, the regulation of actin and microtubule cytoskeleton contraction holds the potential for promoting epithelial repair. For example, fidgetin-like 2 (FL2), a microtubule-severing enzyme, promotes successful wound repair in mice by regulating microtubule contraction and organizing the microtubule cytoskeleton.196 Various actin-remodeling proteins have also been identified as potential new targets for improving wound healing.165 Additionally, sacral nerve stimulation (SNS) shows promise as a specific therapeutic method by enhancing mucosal barrier function, with case reports supporting its use in treating IBD.197
The development of nanosystem targeting technology represents a significant advancement over traditional therapies, offering new perspectives for IBD treatment. The potential of nanozymes in treating IBD has garnered increasing attention. Recent studies have demonstrated that oxidized cerium nanozyme (D-CeO2), CeO2@PDA-PEG, and dopamine nanoparticles coupled with mCRAMP, can effectively clear ROS and alleviate colonic inflammation.198–201 Additionally, nano-medicines encapsulating antioxidant enzymes, such as superoxide dismutase (SOD) and catalase (CAT), have shown promise. Through the co-delivery of CaO2 (a CAT substrate) and dopamine, this nanosystem facilitates CAT-catalyzed oxygen generation and in-situ polymerization of dopamine to form a thin and integrated polydopamine (PDA) coating. Due to the high adhesive properties of PDA, it can cover the intestinal barrier, thereby limiting intestinal leakage and exhibiting a favorable anti-inflammatory effect.202 These nanoformulations exhibit high biocompatibility and demonstrate inflammatory targeting properties, anti-inflammatory effects, and positive modulation of gut microbiota, offering novel strategies for intervening in and treating colitis.198–201
In addition, the colon-targeted quercetin delivery system, COS-CaP-QT, targets the colon via pH-dependent release, regulating Th2 cells and ILC3s proliferation through the Notch pathway and reshaping the inflammatory microenvironment.203 HA-modified poly lactic-co-glycolic acid nanoparticles exert potent therapeutic effects in UC by modulating intestinal IEC/stem cell regeneration, angiogenesis, and inflammation improvement, leading to transcriptional reprogramming of immune response genes in colonic tissues.204 Ginger polysaccharides alleviate intestinal inflammation by inhibiting pro-inflammatory cytokine levels to regulate intestinal inflammation, up-regulating occludin-1 and ZO-1, and modulating gut microbiota.205 Nanocrystalline cellulose effectively treats UC by regulating the MAPK pathway of MUC2 and the relative abundance of Akkermansia and Odoribacter, without causing bodily harm as mucin substitutes.206 LBL-CO@MPDA specifically accumulates onto positively charged inflamed colon lesions through electrostatic interactions. CO@MPDA improves inflammatory conditions via MPDA-mediated ROS scavenging and CO-mediated immunomodulation. CO delivery activates heme oxygenase-1, leading to macrophage M2 polarization via the Notch/Hes1/Stat3 signaling pathway, while suppressing inflammation by downregulating the p38 MAPK and NF-κB (p50/p65) signaling pathways.207
HA holds potential in the treatment of IBD. Research indicates that HA enhances intestinal barrier integrity by increasing epithelial cell adhesion proteins and inhibiting the transcription of inflammatory factors, thereby reducing intestinal permeability.208 Specifically, HA enema plays a crucial role in suppressing intestinal inflammation, possibly by modulating cell growth and cytoskeletal rearrangement via epithelial cell adhesion signaling (eg, E-cadherin). Additionally, HA enema activates the LXR/RXR pathway, further inhibiting the NF-kB pathway of mucosal inflammation.208 However, limitations exist in its clinical application, including the lack of specific colitis models and testing in chronic or large animal models.208 Future investigations should focus on exploring HA’s mechanisms in intestinal inflammation and considering its combination with other therapeutic drugs for more effective treatment strategies.208
Dietary interventions also play a significant role in the treatment of IBD. Dietary therapy can alleviate IBD through the following four key steps: managing nutritional needs, influencing disease activity, controlling disease complications, and preventing disease in offspring.209 The Crohn’s Disease Exclusion Diet aims to limit harmful dietary components for gut microbiota, though concerns exist about potential long-term negative nutritional and psychosocial effects.209–211 In contrast, the Specific Carbohydrate Diet reduces inflammation by avoiding specific carbohydrates but may lead to nutritional deficiencies, particularly in calcium intake.212 The Paleo Auto-Immune Protocol eliminates potentially inflammatory foods, yet its safety is questioned due to lacking nutritional support and high meat consumption.209,213 On the other hand, the Plant-based diet emphasizes increased prebiotic intake for beneficial gut bacteria and is relatively safer, reducing the risk of nutritional inadequacies.209,214 The Mediterranean diet, with its anti-inflammatory properties from plant-based foods, aligns with healthy eating guidelines and is generally safer.212
While other approaches like CD-TREAT, low-sulfur diet, low-emulsifier or carrageenan diets, low-fat diet, low-meat diet, and Novel UC exclusion diet offer unique features, uncertainties remain regarding their safety and nutritional support.209,215–219 Therefore, selecting an appropriate dietary therapy should consider its characteristics, safety, and impact on nutritional intake to achieve the best treatment outcomes.
Finally, recent scientific studies have reported several alternative treatments for IBD, including ZINC40099027 (ZN27), phosphatidylcholine (LT-02), the PDE4 inhibitors Apremilast, Mongersen GED0301, STNM01, Alicaforsen, Momordica charantia (MCh), and others.220–223 While these approaches collectively propose numerous promising therapeutic strategies for IBD treatment, as of now, specific targeted drugs have not yet been discovered (Table 2).
 |
Table 2 Methods and Their Specific Principles for Restoring Intestinal Barrier |
Conclusion and Perspectives
This review initiates by scrutinizing the components and mechanisms of the intestinal barrier, covering the epithelium, cell junctions, mucus layer, and the self-repair capacity of the intestine. Next, we explore the vicious cycle created by dysfunction of these defenders in IBD. Finally, we provide an overview of therapeutic strategies and targets for restoring intestinal barrier integrity (Table 3. Comparison of Different Therapies).
 |
Table 3 Comparision of Different Therapies |
Previous studies have highlighted the negative side effects of non-targeted therapies, particularly immunosuppressive agents. However, technological advancements and a deeper understanding of IBD inflammation and intestinal self-repair have revealed new therapeutic targets for restoring epithelial integrity. Recent research has identified targets in the crosstalk between IEC death and the immune response, including ISC proliferation and differentiation, stem cell transplantation, and organoid culture. While growth factors are used for short bowel syndrome, their application in IBD remains unexplored.15 Restoring cell junction and mucus layer function, regulating the immune system, and rebuilding the intestinal microbiota are also promising approaches. Treatments targeting molecular mechanisms, such as blocking MLCK, show potential for IBD therapy.
While restoring the barrier integrity is an ideal option for IBD, its clinical application faces unresolved issues. Hematopoietic stem cell transplantation (HSCT) has shown certain efficacy in treating IBD in clinical trials. However, the occurrence of frequent severe adverse events has sparked a controversial debate on whether HSCT can be considered an alternative treatment for IBD.224 Screening for gene mutations associated with colorectal cancer is deemed necessary during the isolation of ISCs in IBD therapy due to an increased risk of post-transplantation malignant transformation.174 Injection of embryonic stem cell-derived MSCs derived from fetal sources can alleviate intestinal inflammation by increasing IGF-1 levels, upregulating IGF1R-PI3K-AKT expression, promoting cell proliferation, and reducing apoptosis.225 However, the therapeutic mechanism requires further clarification, particularly regarding its immunomodulatory properties. Additionally, MSCs derived from embryonic stem cells may face challenges in large-scale cultivation and production.225
Stem cells hold great promise in the treatment of IBD, particularly through the study of the intricate and complex interactions between stem cells and immune cells.174 Past research has shown that T cells and ILCs significantly impact the fate and function of stem cells. Additionally, cytokines can directly promote or restrict the proliferation, differentiation, and apoptosis of stem cells, making them crucial mediators in maintaining or disrupting the intestinal epithelial barrier.85,174 However, the interaction between stem cells and immune cells is highly complex, requiring further in-depth research.174 Considering the connections between stem cells and immune cells, the integration of biologic agents and stem cell transplantation may revolutionize the future treatment of IBD patients.174
Gene editing holds the potential to correct genetic defects in organoids, contributing to the high sensitivity of IBD.226 However, uncertainties persist regarding the precise identification of these defects and whether edited organoids can withstand future IBD-related damage.168,226 Evidence suggests that, despite significant plasticity, transplanted tissues may retain epigenetic features of their original tissue source.227 In vitro culture protocols for ISCs also require further refinement to maximize ISC yield.228 In the clinical realm, optimal indicators for evaluating the clinical outcomes of organoid transplantation remain uncertain, and it’s undetermined which types of IBD patients would derive the greatest benefits from these therapies.174 Additional clinical data collection is imperative.174 Notably, in some patients, the severity of the original damage to the epithelium may hinder the establishment of robust organoid cultures, necessitating alternative treatment approaches.168 Nonetheless, other therapeutic strategies also confront challenges preventing their clinical application. For instance, EGF and R-spondin-1 can reduce TNF-induced cell apoptosis by activating the Wnt pathway, though this may also contribute to cancer.6 Moreover, since a specific drug targeting MLCK has not yet emerged, MLCK inhibitors also cannot be used in clinical applications.
Recent research has highlighted the current challenges in repairing the intestinal barrier through various mechanisms and outlined promising future directions for development. Microbial tryptophan metabolites show great promise in treating IBD, though they come with challenges.229 Studies indicate that metabolites like IEt, IPyA, and I3A enhance gut barrier integrity by acting through the aryl hydrocarbon receptor (AhR), inhibiting actin-regulating proteins MyoIIA and ezrin, thereby reducing intestinal permeability.229 However, the exact concentrations and mechanisms of these metabolites in humans need further study. The variation in gut microbiota among individuals also affects metabolite production.229 While AhR’s role is crucial, the effects on different immune cells and pathways require more research. Future research should focus on understanding these metabolites’ mechanisms, personalizing treatments based on individual microbiota differences, and developing accurate human disease models, such as patient-derived organoids, to better study IBD.229
Polysaccharide nanoparticulate drug delivery systems show promise in treating IBD, overcoming the adverse effects associated with traditional targeted therapies.207 These nanoparticles can navigate through the mucus layer, adhere to inflamed areas as a protective layer, and release drugs in a controlled manner, enhancing therapeutic efficacy.230 However, several challenges persist in polysaccharide-based drug delivery. Polysaccharides are complex, varying in composition due to extraction conditions. Improved in vitro methods are needed to accurately simulate IBD conditions.230 Animal IBD models inadequately represent human IBD, necessitating more precise alternatives.230 Additionally, issues such as low encapsulation efficiency and premature drug release need addressing through strategies like smart-responsive drug-polysaccharide conjugates.230 Future research should focus on refining production methods, exploring new drug targets, and developing smart-responsive systems for targeted drug delivery in IBD treatment.230
The advancement of artificial intelligence (AI) offers a new perspective for developing IBD therapies. By employing computational algorithms to analyze vast datasets such as transcriptomics, AI constructs gene co-expression networks (GCNs) to unravel the complexity of human diseases, aiding in the prioritization and validation of treatment targets.231 These networks simplify intricate multi-cellular processes, identifying patterns beyond human analysis and thereby enhancing prediction accuracy. AI can simulate the progression of complex diseases like IBD, improving the precision of drug development.231 Looking ahead, With continuous advancements in AI technology and the ongoing integration of big data analytics, researchers can anticipate more accurate predictions, new target discoveries, and improved treatment outcomes.231
Therefore, despite the appeal of restoring barrier integrity as a therapy, implementing these treatments in clinical practice requires further research into the underlying pathogenesis of IBD, particularly the causative relationships among different dysfunctions. Additionally, a comprehensive understanding of the mechanisms behind these therapeutic strategies is necessary to identify and minimize potential side effects.
Abbreviations
IBD, inflammatory bowel disease; CD, Crohn’s disease; UC, ulcerative colitis; IECs, intestinal epithelial cells; M cells, microfold cells; EECs, enteroendocrine cells; AMPs, antimicrobial peptides; TJ, tight junction; AJ, adherens junction; JAM, junctional adhesion molecule; MLC, myosin regulatory light chain; MLCK1, myosin light chain kinase 1; ZO, Zonula Occludens; E-cadherins, epithelial cadherins; MUC2, mucin-2; FCGBP, Fcγ binding protein; CLCA1, calcium-activated chloride channel regulator1; NHE3, Na+/H+ exchanger 3; ISCs, intestinal stem cells; IL, Interleukin; Grem1, Gremlin 1; Rspo1, R-spondin 1; Wnt2b, Recombinant Wingless Type MMTV Integration Site Family, Member 2B; EGCs, Enteric glial cells; proEGF, pro epidermal growth factor; EGF, epidermal growth factor; YAP, Yes-associated protein; TAZ, Tafazzin; STAT3, Signal Transducer and Activator of Transcription 3; ILCs, innate lymphoid cells; CBCs, Crypt Base Columnar Cells; CREB, cAMP response element-binding protein; WISP-1, WNT1-inducible signaling protein 1; DAMPs, Damage-Associated Molecular Patterns; SETDB, SET domain, bifurcated; ZBP1, Z-DNA binding protein 1; TNVs, turmeric-derived nanovesicles; PGE2, PGE2 prostaglandin E2; EP2, Prostaglandin E2 receptor 2; XBP1, Recombinant X-Box Binding Protein 1; PHB1, Prohibitin 1; Hsp60, Heat Shock Protein 60; DCA, Dichloroacetate; TNF-α, Tumor Necrosis Factor-α; IFN-γ, interferon-γ; Th2, T helper 2 cell; WFDC2, Recombinant WAP Four Disulfide Core Domain Protein 2; ER, endoplasmic reticulum; GI tract, Gastrointestinal tract; TGF-β, transforming growth factor β; Treg, regulatory T cell; SCFAs, short-chain fatty acids; SYNPO, actin-binding protein synaptopodin; MSCs, mesenchymal stem cells; HSCs, hematopoietic stem cells; PIK, membrane permeant inhibitor of MLC kinase; IAP, intestinal alkaline phosphatase; FMT, fecal microbiota transplantation; BRG1, Brahma-related gene 1; ROS, reactive oxygen species; NAC, N-acetyl-L-cysteine; MitoQ, Mitochondrial-targeted coenzyme Q10; FL2, fidgetin-like 2; SOD, Superoxide Dismutase; CAT, Catalase; PDA, polydopamine; ZN27, ZINC40099027; PDE4, Phosphodiesterase-4; SNS, sacral nerve stimulation; MCh, Momordica charantia; HSCT, Hematopoietic stem cell transplantation; HA, Hyaluronic acid; GCNs, gene co-expression networks; AI, Artificial intelligence.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Science and Technology Innovation Committee of Shenzhen (JCYJ20210324113802006, JCYJ 20210324113613035, and JCYJ2022053015180024).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hodson R. Inflammatory bowel disease. Nature. 2016;540(7634):S97. doi:10.1038/540S97a
2. Xu J, Xu HM, Yang MF, et al. New insights into the epigenetic regulation of inflammatory bowel disease. Front Pharmacol. 2022;13:813659. doi:10.3389/fphar.2022.813659
3. Chang JT, Longo DL. Pathophysiology of inflammatory bowel diseases. N Engl J Med. 2020;383(27):2652–2664. doi:10.1056/NEJMra2002697
4. Martini E, Krug SM, Siegmund B, et al. Mend your fences: the epithelial barrier and its relationship with mucosal immunity in Inflammatory Bowel Disease. Cell Mol Gastroenterol Hepatol. 2017;4(1):33–46. doi:10.1016/j.jcmgh.2017.03.007
5. Nalle SC, Turner JR. Intestinal barrier loss as a critical pathogenic link between inflammatory bowel disease and graft-versus-host disease. Mucosal Immunol. 2015;8(4):720–730. doi:10.1038/mi.2015.40
6. Odenwald MA, Turner JR. The intestinal epithelial barrier: a therapeutic target? Nat Rev Gastroenterol Hepatol. 2017;14(1):9–21. doi:10.1038/nrgastro.2016.169
7. Li DF, Yang MF, Xu J, et al. Extracellular vesicles: the next generation theranostic nanomedicine for Inflammatory Bowel Disease. Int J Nanomed. 2022;17:3893–3911. doi:10.2147/IJN.S370784
8. Yao Y, Kim G, Shafer S, et al. Mucus sialylation determines intestinal host-commensal homeostasis. Cell. 2022;185(7):1172–1188.e28. doi:10.1016/j.cell.2022.02.013
9. Paone P, Cani PD. Mucus barrier, mucins and gut microbiota: the expected slimy partners? Gut. 2020;69(12):2232–2243. doi:10.1136/gutjnl-2020-322260
10. Gustafsson JK, Johansson MEV. The role of goblet cells and mucus in intestinal homeostasis. Nat Rev Gastroenterol Hepatol. 2022;19(12):785–803. doi:10.1038/s41575-022-00675-x
11. Perez-Lopez A, Behnsen J, Nuccio SP, et al. Mucosal immunity to pathogenic intestinal bacteria. Nat Rev Immunol. 2016;16(3):135–148. doi:10.1038/nri.2015.17
12. Duchmann R, Kaiser I, Hermann E, et al. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin Exp Immunol. 1995;102(3):448–455. doi:10.1111/j.1365-2249.1995.tb03836.x
13. Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. 2014;15(12):1104–1115. doi:10.1038/ni.3031
14. Bertiaux-Vandaële N, Youmba SB, Belmonte L, et al. The expression and the cellular distribution of the tight junction proteins are altered in irritable bowel syndrome patients with differences according to the disease subtype. Am J Gastroenterol. 2011;106(12):2165–2173. doi:10.1038/ajg.2011.257
15. Villablanca EJ, Selin K, Hedin CRH. Mechanisms of mucosal healing: treating inflammatory bowel disease without immunosuppression? Nat Rev Gastroenterol Hepatol. 2022;19(8):493–507. doi:10.1038/s41575-022-00604-y
16. Jankowski JA, Goodlad RA, Wright NA. Maintenance of normal intestinal mucosa: function, structure, and adaptation. Gut. 1994;35(1 Suppl):S1–S4. doi:10.1136/gut.35.1_Suppl.S1
17. Kotla NG, Rochev Y. IBD disease-modifying therapies: insights from emerging therapeutics. Trends Mol Med. 2023;29(3):241–253. doi:10.1016/j.molmed.2023.01.001
18. Beumer J, Clevers H. Cell fate specification and differentiation in the adult mammalian intestine. Nat Rev Mol Cell Biol. 2021;22(1):39–53. doi:10.1038/s41580-020-0278-0
19. Snoeck V, Goddeeris B, Cox E. The role of enterocytes in the intestinal barrier function and antigen uptake. Microbes Infect. 2005;7(7–8):997–1004. doi:10.1016/j.micinf.2005.04.003
20. Gerbe F, Sidot E, Smyth DJ, et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature. 2016;529(7585):226–230. doi:10.1038/nature16527
21. Grencis RK, Worthington JJ. Tuft cells: a new flavor in innate epithelial immunity. Trends Parasitol. 2016;32(8):583–585. doi:10.1016/j.pt.2016.04.016
22. Kobayashi N, Takahashi D, Takano S, et al. The roles of Peyer’s Patches and microfold cells in the gut immune system: relevance to autoimmune diseases. Front Immunol. 2019;10:2345. doi:10.3389/fimmu.2019.02345
23. Pelaseyed T, Bergström JH, Gustafsson JK, et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol Rev. 2014;260(1):8–20. doi:10.1111/imr.12182
24. McDole JR, Wheeler LW, McDonald KG, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature. 2012;483(7389):345–349. doi:10.1038/nature10863
25. Miner-Williams WM, Moughan PJ. Intestinal barrier dysfunction: implications for chronic inflammatory conditions of the bowel. Nutr Res Rev. 2016;29(1):40–59. doi:10.1017/S0954422416000019
26. Gribble FM, Reimann F. Enteroendocrine Cells: chemosensors in the Intestinal Epithelium. Annu Rev Physiol. 2016;78(1):277–299. doi:10.1146/annurev-physiol-021115-105439
27. Neurath MF. New targets for mucosal healing and therapy in inflammatory bowel diseases. Mucosal Immunol. 2014;7(1):6–19. doi:10.1038/mi.2013.73
28. Gehart H, Clevers H. Tales from the crypt: new insights into intestinal stem cells. Nat Rev Gastroenterol Hepatol. 2019;16(1):19–34. doi:10.1038/s41575-018-0081-y
29. Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. 1963;17(2):375–412. doi:10.1083/jcb.17.2.375
30. Furuse M, Hirase T, Itoh M, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123(6):1777–1788. doi:10.1083/jcb.123.6.1777
31. Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol. 2006;68(1):403–429. doi:10.1146/annurev.physiol.68.040104.131404
32. Weber C, Fraemohs L, Dejana E. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol. 2007;7(6):467–477. doi:10.1038/nri2096
33. Amasheh S, Meiri N, Gitter AH, et al. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci. 2002;115(24):4969–4976. doi:10.1242/jcs.00165
34. Weber CR, Liang GH, Wang Y, et al. Claudin-2-dependent paracellular channels are dynamically gated. Elife. 2015;4:e09906. doi:10.7554/eLife.09906
35. Weber CR, Raleigh DR, Su L, et al. Epithelial myosin light chain kinase activation induces mucosal interleukin-13 expression to alter tight junction ion selectivity. J Biol Chem. 2010;285(16):12037–12046. doi:10.1074/jbc.M109.064808
36. Marchiando AM, Shen L, Graham WV, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. 2010;189(1):111–126. doi:10.1083/jcb.200902153
37. Vetrano S, Rescigno M, Cera MR, et al. Unique role of junctional adhesion molecule-A in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology. 2008;135(1):173–184. doi:10.1053/j.gastro.2008.04.002
38. Fanning AS, Anderson JM. Zonula occludens-1 and −2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann N Y Acad Sci. 2009;1165(1):113–120. doi:10.1111/j.1749-6632.2009.04440.x
39. Takeichi M. Dynamic contacts: rearranging adherens junctions to drive epithelial remodelling. Nat Rev Mol Cell Biol. 2014;15(6):397–410. doi:10.1038/nrm3802
40. Brasch J, Harrison OJ, Honig B, et al. Thinking outside the cell: how cadherins drive adhesion. Trends Cell Biol. 2012;22(6):299–310. doi:10.1016/j.tcb.2012.03.004
41. Rudini N, Dejana E. Adherens junctions. Curr Biol. 2008;18(23):R1080–R1082. doi:10.1016/j.cub.2008.09.018
42. Capaldo CT, Macara IG, Nusrat A. Depletion of E-cadherin disrupts establishment but not maintenance of cell junctions in Madin-Darby canine kidney epithelial cells. Mol Biol Cell. 2007;18(1):189–200. doi:10.1091/mbc.e06-05-0471
43. Maiers JL, Peng X, Fanning AS, et al. ZO-1 recruitment to α-catenin--A novel mechanism for coupling the assembly of tight junctions to adherens junctions. J Cell Sci. 2013;126(Pt 17):3904–3915. doi:10.1242/jcs.126565
44. Horowitz A, Chanez-Paredes SD, Haest X, et al. Paracellular permeability and tight junction regulation in gut health and disease. Nat Rev Gastroenterol Hepatol;2023. 1–16. doi:10.1038/s41575-022-00713-8
45. Günzel D, Fromm M. Claudins and other tight junction proteins. Compr Physiol. 2012;2:1819–1852.
46. Günzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiol Rev. 2013;93(2):525–569. doi:10.1152/physrev.00019.2012
47. Tsukita S, Tanaka H, Tamura A. The claudins: from tight junctions to biological systems. Trends Biochem Sci. 2019;44(2):141–152. doi:10.1016/j.tibs.2018.09.008
48. Raleigh DR, Boe DM, Yu D, et al. Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. J Cell Biol. 2011;193(3):565–582. doi:10.1083/jcb.201010065
49. van der Post S, Jabbar KS, Birchenough G, et al. Structural weakening of the colonic mucus barrier is an early event in ulcerative colitis pathogenesis. Gut. 2019;68(12):2142–2151. doi:10.1136/gutjnl-2018-317571
50. Ehrencrona E, van der Post S, Gallego P, et al. The IgGFc-binding protein FCGBP is secreted with all GDPH sequences cleaved but maintained by interfragment disulfide bonds. J Biol Chem. 2021;297(1):100871. doi:10.1016/j.jbc.2021.100871
51. Javitt G, Khmelnitsky L, Albert L, et al. Assembly mechanism of mucin and von Willebrand factor polymers. Cell. 2020;183(3):717–729.e16. doi:10.1016/j.cell.2020.09.021
52. Johansson ME, Ambort D, Pelaseyed T, et al. Composition and functional role of the mucus layers in the intestine. Cell Mol Life Sci. 2011;68(22):3635–3641. doi:10.1007/s00018-011-0822-3
53. Yao D, Dai W, Dong M, et al. MUC2 and related bacterial factors: therapeutic targets for ulcerative colitis. EBioMedicine. 2021;74:103751. doi:10.1016/j.ebiom.2021.103751
54. Ambort D, Johansson ME, Gustafsson JK, et al. Calcium and pH-dependent packing and release of the gel-forming MUC2 mucin. Proc Natl Acad Sci U S A. 2012;109(15):5645–5650. doi:10.1073/pnas.1120269109
55. Johansson ME, Phillipson M, Petersson J, et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 2008;105(39):15064–15069. doi:10.1073/pnas.0803124105
56. Harrison CA, Laubitz D, Ohland CL, et al. Microbial dysbiosis associated with impaired intestinal Na(+)/H(+) exchange accelerates and exacerbates colitis in ex-germ free mice. Mucosal Immunol. 2018;11(5):1329–1341. doi:10.1038/s41385-018-0035-2
57. Critchfield AS, Yao G, Jaishankar A, et al. Cervical mucus properties stratify risk for preterm birth. PLoS One. 2013;8(8):e69528. doi:10.1371/journal.pone.0069528
58. Nyström EEL, Martinez-Abad B, Arike L, et al. An intercrypt subpopulation of goblet cells is essential for colonic mucus barrier function. Science;2021. 372. doi:10.1126/science.373.6553.372
59. Grootjans J, Hundscheid IH, Lenaerts K, et al. Ischaemia-induced mucus barrier loss and bacterial penetration are rapidly counteracted by increased goblet cell secretory activity in human and rat colon. Gut. 2013;62(2):250–258. doi:10.1136/gutjnl-2011-301956
60. Johansson ME, Hansson GC. The goblet cell: a key player in ischaemia-reperfusion injury. Gut. 2013;62(2):188–189. doi:10.1136/gutjnl-2012-302582
61. Lai SK, Wang YY, Wirtz D, et al. Micro- and macrorheology of mucus. Adv Drug Deliv Rev. 2009;61(2):86–100. doi:10.1016/j.addr.2008.09.012
62. Birchenough GM, Nyström EE, Johansson ME, et al. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science. 2016;352(6293):1535–1542. doi:10.1126/science.aaf7419
63. Bakshani CR, Morales-Garcia AL, Althaus M, et al. Evolutionary conservation of the antimicrobial function of mucus: a first defence against infection. NPJ Biofilms Microbiomes. 2018;4(1):14. doi:10.1038/s41522-018-0057-2
64. Allen A, Flemström G. Gastroduodenal mucus bicarbonate barrier: protection against acid and pepsin. Am J Physiol Cell Physiol. 2005;288(1):C1–C19. doi:10.1152/ajpcell.00102.2004
65. Hansson GC. Mucus and mucins in diseases of the intestinal and respiratory tracts. J Intern Med. 2019;285(5):479–490. doi:10.1111/joim.12910
66. Johansson ME, Sjövall H, Hansson GC. The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol. 2013;10(6):352–361. doi:10.1038/nrgastro.2013.35
67. Schneider H, Pelaseyed T, Svensson F, et al. Study of mucin turnover in the small intestine by in vivo labeling. Sci Rep. 2018;8(1):5760. doi:10.1038/s41598-018-24148-x
68. Arike L, Seiman A, van der Post S, et al. Protein turnover in epithelial cells and mucus along the gastrointestinal tract is coordinated by the spatial location and microbiota. Cell Rep. 2020;30(4):1077–1087.e3. doi:10.1016/j.celrep.2019.12.068
69. Johansson ME, Hansson GC. Immunological aspects of intestinal mucus and mucins. Nat Rev Immunol. 2016;16(10):639–649. doi:10.1038/nri.2016.88
70. Luis AS, Jin C, Pereira GV, et al. A single sulfatase is required to access colonic mucin by a gut bacterium. Nature. 2021;598(7880):332–337. doi:10.1038/s41586-021-03967-5
71. Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008;4(5):447–457. doi:10.1016/j.chom.2008.09.007
72. Snippert HJ, van der Flier LG, Sato T, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143(1):134–144. doi:10.1016/j.cell.2010.09.016
73. van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71(1):241–260. doi:10.1146/annurev.physiol.010908.163145
74. Kinchen J, Chen HH, Parikh K, et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell. 2018;175(2):372–386.e17. doi:10.1016/j.cell.2018.08.067
75. Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–265. doi:10.1038/nature07935
76. Ishikawa K, Sugimoto S, Oda M, et al. Identification of quiescent LGR5(+) stem cells in the human colon. Gastroenterology. 2022;163(5):1391–1406.e24. doi:10.1053/j.gastro.2022.07.081
77. Weichselbaum L, Klein OD. The intestinal epithelial response to damage. Sci China Life Sci. 2018;61(10):1205–1211. doi:10.1007/s11427-018-9331-y
78. Beumer J, Clevers H. Regulation and plasticity of intestinal stem cells during homeostasis and regeneration. Development. 2016;143(20):3639–3649. doi:10.1242/dev.133132
79. Shoshkes-Carmel M, Wang YJ, Wangensteen KJ, et al. Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature. 2018;557(7704):242–246. doi:10.1038/s41586-018-0084-4
80. Degirmenci B, Valenta T, Dimitrieva S, et al. GLI1-expressing mesenchymal cells form the essential Wnt-secreting niche for colon stem cells. Nature. 2018;558(7710):449–453. doi:10.1038/s41586-018-0190-3
81. Mahapatro M, Foersch S, Hefele M, et al. Programming of intestinal epithelial differentiation by IL-33 derived from pericryptal fibroblasts in response to systemic infection. Cell Rep. 2016;15(8):1743–1756. doi:10.1016/j.celrep.2016.04.049
82. Stzepourginski I, Nigro G, Jacob JM, et al. CD34+ mesenchymal cells are a major component of the intestinal stem cells niche at homeostasis and after injury. Proc Natl Acad Sci U S A. 2017;114(4):E506–E513. doi:10.1073/pnas.1620059114
83. Zushi S, Shinomura Y, Kiyohara T, et al. Role of heparin-binding EGF-related peptides in proliferation and apoptosis of activated ras-stimulated intestinal epithelial cells. Int J Cancer. 1997;73:917–923.
84. Patankar JV, Becker C. Cell death in the gut epithelium and implications for chronic inflammation. Nat Rev Gastroenterol Hepatol. 2020;17(9):543–556. doi:10.1038/s41575-020-0326-4
85. Van Landeghem L, Chevalier J, Mahé MM, et al. Enteric glia promote intestinal mucosal healing via activation of focal adhesion kinase and release of proEGF. Am J Physiol Gastrointest Liver Physiol. 2011;300(6):G976–G987. doi:10.1152/ajpgi.00427.2010
86. Clevers HC, Bevins CL. Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol. 2013;75(1):289–311. doi:10.1146/annurev-physiol-030212-183744
87. Sato T, van Es JH, Snippert HJ, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469(7330):415–418. doi:10.1038/nature09637
88. Yui S, Azzolin L, Maimets M, et al. YAP/TAZ-dependent reprogramming of colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem Cell. 2018;22(1):35–49.e7. doi:10.1016/j.stem.2017.11.001
89. Taniguchi K, Wu LW, Grivennikov SI, et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature. 2015;519(7541):57–62. doi:10.1038/nature14228
90. Spits H, Artis D, Colonna M, et al. Innate lymphoid cells--A proposal for uniform nomenclature. Nat Rev Immunol. 2013;13(2):145–149. doi:10.1038/nri3365
91. Lindemans CA, Calafiore M, Mertelsmann AM, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. 2015;528(7583):560–564. doi:10.1038/nature16460
92. Quiros M, Nishio H, Neumann PA, et al. Macrophage-derived IL-10 mediates mucosal repair by epithelial WISP-1 signaling. J Clin Invest. 2017;127(9):3510–3520. doi:10.1172/JCI90229
93. Igarashi M, Guarente L. mTORC1 and SIRT1 cooperate to foster expansion of gut adult stem cells during calorie restriction. Cell. 2016;166(2):436–450. doi:10.1016/j.cell.2016.05.044
94. Zeissig S, Bürgel N, Günzel D, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut. 2007;56(1):61–72. doi:10.1136/gut.2006.094375
95. Newton K, Dixit VM, Kayagaki N. Dying cells fan the flames of inflammation. Science. 2021;374(6571):1076–1080. doi:10.1126/science.abi5934
96. You R, He X, Zeng Z, et al. Pyroptosis and its role in autoimmune disease: a potential therapeutic target. Front Immunol. 2022;13:841732. doi:10.3389/fimmu.2022.841732
97. Deets KA, Vance RE. Inflammasomes and adaptive immune responses. Nat Immunol. 2021;22(4):412–422. doi:10.1038/s41590-021-00869-6
98. Krysko DV, Agostinis P, Krysko O, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011;32(4):157–164. doi:10.1016/j.it.2011.01.005
99. Bulek K, Zhao J, Liao Y, et al. Epithelial-derived gasdermin D mediates nonlytic IL-1β release during experimental colitis. J Clin Invest. 2020;130(8):4218–4234. doi:10.1172/JCI138103
100. Gao C, Zhou Y, Chen Z, et al. Turmeric-derived nanovesicles as novel nanobiologics for targeted therapy of ulcerative colitis. Theranostics. 2022;12(12):5596–5614. doi:10.7150/thno.73650
101. Cao X, Duan L, Hou H, et al. IGF-1C hydrogel improves the therapeutic effects of MSCs on colitis in mice through PGE(2)-mediated M2 macrophage polarization. Theranostics. 2020;10(17):7697–7709. doi:10.7150/thno.45434
102. Zhang Y, Ma R, You C, et al. Hyaluronic acid modified oral drug delivery system with mucoadhesiveness and macrophage-targeting for colitis treatment. Carbohydr Polym. 2023;313:120884. doi:10.1016/j.carbpol.2023.120884
103. Gupta KH, Goldufsky JW, Wood SJ, et al. Apoptosis and compensatory proliferation signaling are coupled by CrkI-containing microvesicles. Dev Cell. 2017;41(6):674–684.e5. doi:10.1016/j.devcel.2017.05.014
104. Dannappel M, Vlantis K, Kumari S, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513(7516):90–94. doi:10.1038/nature13608
105. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol. 2020;15(1):493–518. doi:10.1146/annurev-pathmechdis-012419-032847
106. Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48(1):322–335. doi:10.1002/hep.22306
107. Sellin ME, Müller AA, Felmy B, et al. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe. 2014;16(2):237–248. doi:10.1016/j.chom.2014.07.001
108. Dondelinger Y, Declercq W, Montessuit S, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7(4):971–981. doi:10.1016/j.celrep.2014.04.026
109. Nowarski R, Jackson R, Gagliani N, et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell. 2015;163(6):1444–1456. doi:10.1016/j.cell.2015.10.072
110. Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–158. doi:10.1038/nature18629
111. Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–435. doi:10.1038/nature07201
112. Yatim N, Cullen S, Albert ML. Dying cells actively regulate adaptive immune responses. Nat Rev Immunol. 2017;17(4):262–275. doi:10.1038/nri.2017.9
113. Andonegui G, Zhou H, Bullard D, et al. Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J Clin Invest. 2009;119(7):1921–1930. doi:10.1172/jci36411
114. Tindemans I, Joosse ME, Samsom JN. Dissecting the heterogeneity in T-cell mediated inflammation in IBD. Cells. 2020;9(1):110. doi:10.3390/cells9010110
115. Xue Y, Enosi Tuipulotu D, Tan WH, et al. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. 2019;40(11):1035–1052. doi:10.1016/j.it.2019.09.005
116. Zarrin AA, Bao K, Lupardus P, et al. Kinase inhibition in autoimmunity and inflammation. Nat Rev Drug Discov. 2021;20(1):39–63. doi:10.1038/s41573-020-0082-8
117. Wang R, Li H, Wu J, et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature. 2020;580(7803):386–390. doi:10.1038/s41586-020-2127-x
118. Cuellar TL, Herzner A-M, Zhang X, et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J Cell Biol. 2017;216(11):3535–3549. doi:10.1083/jcb.201612160
119. Lee C, An M, Joung JG, et al. TNFα induces LGR5+ stem cell dysfunction in patients with Crohn’s Disease. Cell Mol Gastroenterol Hepatol. 2022;13(3):789–808. doi:10.1016/j.jcmgh.2021.10.010
120. Glimcher LH. XBP1: the last two decades. Ann Rheum Dis. 2010;69(Suppl 1):i67–i71. doi:10.1136/ard.2009.119388
121. Jackson DN, Panopoulos M, Neumann WL, et al. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut. 2020;69(11):1928–1938. doi:10.1136/gutjnl-2019-319523
122. Ray K. Mitochondrial dysfunction in Crohn’s disease. Nat Rev Gastroenterol Hepatol. 2020;17(5):260. doi:10.1038/s41575-020-0291-y
123. Madsen KL, Malfair D, Gray D, et al. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm Bowel Dis. 1999;5(4):262–270. doi:10.1097/00054725-199911000-00004
124. Gassler N, Rohr C, Schneider A, et al. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am J Physiol Gastrointest Liver Physiol. 2001;281(1):G216–228. doi:10.1152/ajpgi.2001.281.1.G216
125. Kucharzik T, Walsh SV, Chen J, et al. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol. 2001;159(6):2001–2009. doi:10.1016/S0002-9440(10)63051-9
126. Su L, Nalle SC, Shen L, et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology. 2013;145(2):407–415. doi:10.1053/j.gastro.2013.04.011
127. Zolotarevsky Y, Hecht G, Koutsouris A, et al. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology. 2002;123(1):163–172. doi:10.1053/gast.2002.34235
128. Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129(2):550–564. doi:10.1016/j.gastro.2005.05.002
129. Graham WV, Wang F, Clayburgh DR, et al. Tumor necrosis factor-induced long myosin light chain kinase transcription is regulated by differentiation-dependent signaling events. Characterization of the human long myosin light chain kinase promoter. J Biol Chem. 2006;281(36):26205–26215. doi:10.1074/jbc.M602164200
130. Blair SA, Kane SV, Clayburgh DR, et al. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest. 2006;86(2):191–201. doi:10.1038/labinvest.3700373
131. Madara JL, Stafford J. Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Invest. 1989;83(2):724–727. doi:10.1172/JCI113938
132. Taylor CT, Dzus AL, Colgan SP. Autocrine regulation of epithelial permeability by hypoxia: role for polarized release of tumor necrosis factor alpha. Gastroenterology. 1998;114(4):657–668. doi:10.1016/S0016-5085(98)70579-7
133. Youakim A, Ahdieh M. Interferon-gamma decreases barrier function in T84 cells by reducing ZO-1 levels and disrupting apical actin. Am J Physiol. 1999;276(5):G1279–G1288. doi:10.1152/ajpgi.1999.276.5.G1279
134. Mankertz J, Tavalali S, Schmitz H, et al. Expression from the human occludin promoter is affected by tumor necrosis factor alpha and interferon gamma. J Cell Sci. 2000;113(Pt 11):2085–2090. doi:10.1242/jcs.113.11.2085
135. Mankertz J, Amasheh M, Krug SM, et al. TNFalpha up-regulates claudin-2 expression in epithelial HT-29/B6 cells via phosphatidylinositol-3-kinase signaling. Cell Tissue Res. 2009;336(1):67–77. doi:10.1007/s00441-009-0751-8
136. Wyatt J, Vogelsang H, Hübl W, et al. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet. 1993;341(8858):1437–1439. doi:10.1016/0140-6736(93)90882-H
137. Hilsden RJ, Meddings JB, Hardin J, et al. Intestinal permeability and postheparin plasma diamine oxidase activity in the prediction of Crohn’s disease relapse. Inflamm Bowel Dis. 1999;5(2):85–91. doi:10.1097/00054725-199905000-00003
138. Söderholm JD, Streutker C, Yang PC, et al. Increased epithelial uptake of protein antigens in the ileum of Crohn’s disease mediated by tumour necrosis factor alpha. Gut. 2004;53(12):1817–1824. doi:10.1136/gut.2004.041426
139. Ménard S, Cerf-Bensussan N, Heyman M. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal Immunol. 2010;3(3):247–259. doi:10.1038/mi.2010.5
140. Van der sluis M, De Koning BA, De Bruijn AC, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131(1):117–129. doi:10.1053/j.gastro.2006.04.020
141. Johansson ME, Gustafsson JK, Holmén-Larsson J, et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut. 2014;63(2):281–291. doi:10.1136/gutjnl-2012-303207
142. Swidsinski A, Loening-Baucke V, Theissig F, et al. Comparative study of the intestinal mucus barrier in normal and inflamed colon. Gut. 2007;56(3):343–350. doi:10.1136/gut.2006.098160
143. Gersemann M, Becker S, Kübler I, et al. Differences in goblet cell differentiation between Crohn’s disease and ulcerative colitis. Differentiation. 2009;77(1):84–94. doi:10.1016/j.diff.2008.09.008
144. Sünderhauf A, Hicken M, Schlichting H, et al. Loss of mucosal p32/gC1qR/HABP1 triggers energy deficiency and impairs goblet cell differentiation in ulcerative colitis. Cell Mol Gastroenterol Hepatol. 2021;12(1):229–250. doi:10.1016/j.jcmgh.2021.01.017
145. Parikh K, Antanaviciute A, Fawkner-Corbett D, et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature. 2019;567(7746):49–55. doi:10.1038/s41586-019-0992-y
146. Heazlewood CK, Cook MC, Eri R, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008;5(3):e54. doi:10.1371/journal.pmed.0050054
147. Cloots E, Simpson MS, De Nolf C, et al. Evolution and function of the epithelial cell-specific ER stress sensor IRE1β. Mucosal Immunol. 2021;14(6):1235–1246. doi:10.1038/s41385-021-00412-8
148. Das I, Png CW, Oancea I, et al. Glucocorticoids alleviate intestinal ER stress by enhancing protein folding and degradation of misfolded proteins. J Exp Med. 2013;210(6):1201–1216. doi:10.1084/jem.20121268
149. McGuckin MA, Eri RD, Das I, et al. ER stress and the unfolded protein response in intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2010;298(6):G820–G832. doi:10.1152/ajpgi.00063.2010
150. Larsson JM, Karlsson H, Crespo JG, et al. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm Bowel Dis. 2011;17(11):2299–2307. doi:10.1002/ibd.21625
151. Wibowo AA, Pardjianto B, Sumitro SB, et al. Decreased expression of MUC2 due to a decrease in the expression of lectins and apoptotic defects in colitis patients. Biochem Biophys Rep. 2019;19:100655. doi:10.1016/j.bbrep.2019.100655
152. Xiao F, Yu Q, Li J, et al. Slc26a3 deficiency is associated with loss of colonic HCO3 (-) secretion, absence of a firm mucus layer and barrier impairment in mice. Acta Physiol. 2014;211(1):161–175. doi:10.1111/apha.12220
153. Smillie CS, Biton M, Ordovas-Montanes J, et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell. 2019;178(3):714–730.e22. doi:10.1016/j.cell.2019.06.029
154. Gurney MA, Laubitz D, Ghishan FK, et al. Pathophysiology of Intestinal Na(+)/H(+) exchange. Cell Mol Gastroenterol Hepatol. 2017;3(1):27–40. doi:10.1016/j.jcmgh.2016.09.010
155. Flint HJ, Bayer EA, Rincon MT, et al. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol. 2008;6(2):121–131. doi:10.1038/nrmicro1817
156. Sonnenburg JL, Xu J, Leip DD, et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307(5717):1955–1959. doi:10.1126/science.1109051
157. Hoskins LC, Agustines M, McKee WB, et al. Mucin degradation in human colon ecosystems. Isolation and properties of fecal strains that degrade ABH blood group antigens and oligosaccharides from mucin glycoproteins. J Clin Invest. 1985;75(3):944–953. doi:10.1172/JCI111795
158. Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638. doi:10.1126/science.1110591
159. Clausen MR, Mortensen PB. Kinetic studies on colonocyte metabolism of short chain fatty acids and glucose in ulcerative colitis. Gut. 1995;37(5):684–689. doi:10.1136/gut.37.5.684
160. Segain JP, Raingeard de la Blétière D, Bourreille A, et al. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut. 2000;47(3):397–403. doi:10.1136/gut.47.3.397
161. Chang PV, Hao L, Offermanns S, et al. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014;111(6):2247–2252. doi:10.1073/pnas.1322269111
162. Morgan XC, Tickle TL, Sokol H, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13(9):R79. doi:10.1186/gb-2012-13-9-r79
163. Dalile B, Van Oudenhove L, Vervliet B, et al. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat Rev Gastroenterol Hepatol. 2019;16(8):461–478. doi:10.1038/s41575-019-0157-3
164. Lu Q, Yang MF, Liang YJ, et al. Immunology of Inflammatory Bowel Disease: molecular mechanisms and therapeutics. J Inflamm Res. 2022;15:1825–1844. doi:10.2147/JIR.S353038
165. Oncel S, Basson MD. Gut homeostasis, injury, and healing: new therapeutic targets. World J Gastroenterol. 2022;28(17):1725–1750. doi:10.3748/wjg.v28.i17.1725
166. Garrett WS, Gallini CA, Yatsunenko T, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8(3):292–300. doi:10.1016/j.chom.2010.08.004
167. Saez A, Gomez-Bris R, Herrero-Fernandez B, et al. Innate lymphoid cells in Intestinal Homeostasis and Inflammatory Bowel Disease. Int J Mol Sci. 2021;22(14):7618. doi:10.3390/ijms22147618
168. Liu CY, Cham CM, Chang EB. Epithelial wound healing in inflammatory bowel diseases: the next therapeutic frontier. Transl Res. 2021;236:35–51. doi:10.1016/j.trsl.2021.06.001
169. Sandborn WJ, Feagan BG, Rutgeerts P, et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2013;369(8):711–721. doi:10.1056/NEJMoa1215739
170. Feagan BG, Sandborn WJ, Gasink C, et al. Ustekinumab as induction and maintenance therapy for Crohn’s Disease. N Engl J Med. 2016;375(20):1946–1960. doi:10.1056/NEJMoa1602773
171. Chang X, Yang MF, Fan W, et al. Bioinformatic analysis suggests that three hub genes may be a vital prognostic biomarker in pancreatic ductal adenocarcinoma. J Comput Biol. 2020;27(11):1595–1609. doi:10.1089/cmb.2019.0367
172. Avansino JR, Chen DC, Woolman JD, et al. Engraftment of mucosal stem cells into murine jejunum is dependent on optimal dose of cells. J Surg Res. 2006;132(1):74–79. doi:10.1016/j.jss.2005.09.009
173. Yui S, Nakamura T, Sato T, et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5⁺ stem cell. Nat Med. 2012;18(4):618–623. doi:10.1038/nm.2695
174. Hou Q, Huang J, Ayansola H, et al. Intestinal stem cells and immune cell relationships: potential therapeutic targets for Inflammatory Bowel Diseases. Front Immunol. 2020;11:623691. doi:10.3389/fimmu.2020.623691
175. Che Z, Ye Z, Zhang X, et al. Mesenchymal stem/stromal cells in the pathogenesis and regenerative therapy of inflammatory bowel diseases. Front Immunol. 2022;13:952071. doi:10.3389/fimmu.2022.952071
176. Tian CM, Yang MF, Xu HM, et al. Stem cell-derived intestinal organoids: a novel modality for IBD. Cell Death Discov. 2023;9(1):255. doi:10.1038/s41420-023-01556-1
177. Fordham RP, Yui S, Hannan NR, et al. Transplantation of expanded fetal intestinal progenitors contributes to colon regeneration after injury. Cell Stem Cell. 2013;13(6):734–744. doi:10.1016/j.stem.2013.09.015
178. Yamaoka T, Yan F, Cao H, et al. Transactivation of EGF receptor and ErbB2 protects intestinal epithelial cells from TNF-induced apoptosis. Proc Natl Acad Sci U S A. 2008;105(33):11772–11777. doi:10.1073/pnas.0801463105
179. Zhao J, de Vera J, Narushima S, et al. R-spondin1, a novel intestinotrophic mitogen, ameliorates experimental colitis in mice. Gastroenterology. 2007;132(4):1331–1343. doi:10.1053/j.gastro.2007.02.001
180. Hong F, Haldeman BD, Jackson D, et al. Biochemistry of smooth muscle myosin light chain kinase. Arch Biochem Biophys. 2011;510(2):135–146. doi:10.1016/j.abb.2011.04.018
181. Yu L, Jin J, Xu Y, et al. Aberrant energy metabolism in Alzheimer’s Disease. J Transl Int Med. 2022;10(3):197–206. doi:10.2478/jtim-2022-0024
182. Parada Venegas D, De la Fuente MK, Landskron G, et al. Short Chain Fatty Acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for Inflammatory Bowel Diseases. Front Immunol. 2019;10:277. doi:10.3389/fimmu.2019.00277
183. Bilotta AJ, Ma C, Yang W, et al. Propionate enhances cell speed and persistence to promote intestinal epithelial turnover and repair. Cell Mol Gastroenterol Hepatol. 2021;11(4):1023–1044. doi:10.1016/j.jcmgh.2020.11.011
184. He Y, Li X, Yu H, et al. The functional role of fecal microbiota transplantation on dextran sulfate sodium-induced colitis in mice. Front Cell Infect Microbiol. 2019;9:393. doi:10.3389/fcimb.2019.00393
185. Liu M, Sun T, Li N, et al. BRG1 attenuates colonic inflammation and tumorigenesis through autophagy-dependent oxidative stress sequestration. Nat Commun. 2019;10(1):4614. doi:10.1038/s41467-019-12573-z
186. Ho GT, Theiss AL. Mitochondria and Inflammatory Bowel Diseases: toward a stratified therapeutic intervention. Annu Rev Physiol. 2022;84(1):435–459. doi:10.1146/annurev-physiol-060821-083306
187. Merkwirth C, Dargazanli S, Tatsuta T, et al. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008;22(4):476–488. doi:10.1101/gad.460708
188. Nijtmans LG, de Jong L, Artal Sanz M, et al. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO j. 2000;19(11):2444–2451. doi:10.1093/emboj/19.11.2444
189. Khaloian S, Rath E, Hammoudi N, et al. Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut. 2020;69(11):1939–1951. doi:10.1136/gutjnl-2019-319514
190. Vincent G, Novak EA, Siow VS, et al. Nix-mediated mitophagy modulates mitochondrial damage during intestinal inflammation. Antioxid Redox Signal. 2020;33(1):1–19. doi:10.1089/ars.2018.7702
191. James AM, Smith RA, Murphy MP. Antioxidant and prooxidant properties of mitochondrial Coenzyme Q. Arch Biochem Biophys. 2004;423(1):47–56. doi:10.1016/j.abb.2003.12.025
192. Dong L, Xie J, Wang Y, et al. Mannose ameliorates experimental colitis by protecting intestinal barrier integrity. Nat Commun. 2022;13(1):4804. doi:10.1038/s41467-022-32505-8
193. Duggan C, Gannon J, Walker WA. Protective nutrients and functional foods for the gastrointestinal tract. Am J Clin Nutr. 2002;75(5):789–808. doi:10.1093/ajcn/75.5.789
194. Kim MH, Kim H. The roles of glutamine in the intestine and its implication in intestinal diseases. Int J Mol Sci. 2017;18(5). doi:10.3390/ijms18051051
195. Shimshoni E, Yablecovitch D, Baram L, et al. ECM remodelling in IBD: innocent bystander or partner in crime? The emerging role of extracellular molecular events in sustaining intestinal inflammation. Gut. 2015;64(3):367–372. doi:10.1136/gutjnl-2014-308048
196. Charafeddine RA, Nosanchuk JD, Sharp DJ. Targeting microtubules for wound repair. Adv Wound Care. 2016;5(10):444–454. doi:10.1089/wound.2015.0658
197. Wang X, Chen JD. Therapeutic potential and mechanisms of sacral nerve stimulation for gastrointestinal diseases. J Transl Int Med. 2023;11(2):115–127. doi:10.2478/jtim-2023-0086
198. Zhang B, Li Q, Xu Q, et al. Polydopamine modified ceria nanorods alleviate inflammation in colitis by scavenging ROS and regulating macrophage M2 polarization. Int J Nanomed. 2023;18:4601–4616. doi:10.2147/IJN.S416049
199. Cao Y, Cheng K, Yang M, et al. Orally administration of cerium oxide nanozyme for computed tomography imaging and anti-inflammatory/anti-fibrotic therapy of inflammatory bowel disease. J Nanobiotechnol. 2023;21(1):21. doi:10.1186/s12951-023-01770-0
200. Liang X, Wen K, Chen Y, et al. Oral administration of therapeutic enzyme capsule for the management of Inflammatory Bowel Disease. Int J Nanomed. 2022;17:4843–4860. doi:10.2147/IJN.S378073
201. Bao M, Wang K, Li J, et al. ROS Scavenging and inflammation-directed polydopamine nanoparticles regulate gut immunity and flora therapy in inflammatory bowel disease. Acta Biomater. 2023;161:250–264. doi:10.1016/j.actbio.2023.02.026
202. Zhao Y, He R, Zang J, et al. Pathologically catalyzed physical coating restores the intestinal barrier for inflammatory bowel disease therapy. J Nanobiotechnology. 2023;21(1):444. doi:10.1186/s12951-023-02227-0
203. Jing S, Chen H, Liu E, et al. Oral pectin/oligochitosan microspheres for colon-specific controlled release of quercetin to treat inflammatory bowel disease. Carbohydr Polym. 2023;316:121025. doi:10.1016/j.carbpol.2023.121025
204. Zhuo Z, Guo K, Luo Y, et al. Targeted modulation of intestinal epithelial regeneration and immune response in ulcerative colitis using dual-targeting bilirubin nanoparticles. Theranostics. 2024;14(2):528–546. doi:10.7150/thno.87739
205. Hao W, Chen Z, Yuan Q, et al. Ginger polysaccharides relieve ulcerative colitis via maintaining intestinal barrier integrity and gut microbiota modulation. Int J Biol Macromol. 2022;219:730–739. doi:10.1016/j.ijbiomac.2022.08.032
206. Wang M, Cha R, Hao W, et al. Nanocrystalline cellulose modulates dysregulated intestinal barriers in ulcerative colitis. ACS Nano. 2023;17(19):18965–18978. doi:10.1021/acsnano.3c04569
207. Zhang X, Yuan Z, Wu J, et al. An orally-administered nanotherapeutics with carbon monoxide supplying for inflammatory bowel disease therapy by scavenging oxidative stress and restoring gut immune homeostasis. ACS Nano. 2023;17(21):21116–21133. doi:10.1021/acsnano.3c04819
208. Kotla NG, Isa ILM, Rasala S, et al. Modulation of gut barrier functions in ulcerative colitis by hyaluronic acid system. Adv Sci. 2022;9(4):e2103189. doi:10.1002/advs.202103189
209. Fitzpatrick JA, Melton SL, Yao CK, et al. Dietary management of adults with IBD - The emerging role of dietary therapy. Nat Rev Gastroenterol Hepatol. 2022;19(10):652–669. doi:10.1038/s41575-022-00619-5
210. Ashton JJ, Beattie RM. Treatment of active Crohn’s Disease with an ordinary food-based diet that replicates exclusive enteral nutrition. Gastroenterology. 2019;157(4):1160–1161. doi:10.1053/j.gastro.2019.01.277
211. Levine A, Wine E, Assa A, et al. Crohn’s Disease exclusion diet plus partial enteral nutrition induces sustained remission in a randomized controlled trial. Gastroenterology. 2019;157(2):440–450.e8. doi:10.1053/j.gastro.2019.04.021
212. Lewis JD, Sandler RS, Brotherton C, et al. A randomized trial comparing the specific carbohydrate diet to a Mediterranean diet in adults with Crohn’s Disease. Gastroenterology. 2021;161(3):837–852.e9. doi:10.1053/j.gastro.2021.05.047
213. Konijeti GG, Kim N, Lewis JD, et al. Efficacy of the autoimmune protocol diet for Inflammatory Bowel Disease. Inflamm Bowel Dis. 2017;23(11):2054–2060. doi:10.1097/MIB.0000000000001221
214. Chiba M, Abe T, Tsuda H, et al. Lifestyle-related disease in Crohn’s disease: relapse prevention by a semi-vegetarian diet. World J Gastroenterol. 2010;16(20):2484–2495. doi:10.3748/wjg.v16.i20.2484
215. Sarbagili-Shabat C, Albenberg L, Van Limbergen J, et al. A novel UC exclusion diet and antibiotics for treatment of mild to moderate pediatric ulcerative colitis: a Prospective Open-Label Pilot Study. Nutrients. 2021;13(11):3736. doi:10.3390/nu13113736
216. Albenberg L, Brensinger CM, Wu Q, et al. A diet low in red and processed meat does not reduce rate of Crohn’s Disease Flares. Gastroenterology. 2019;157(1):128–136.e5. doi:10.1053/j.gastro.2019.03.015
217. Fritsch J, Garces L, Quintero MA, et al. Low-fat, high-fiber diet reduces markers of inflammation and dysbiosis and improves quality of life in patients with ulcerative colitis. Clin Gastroenterol Hepatol. 2021;19(6):1189–1199.e30. doi:10.1016/j.cgh.2020.05.026
218. Sandall AM, Cox SR, Lindsay JO, et al. Emulsifiers impact colonic length in mice and emulsifier restriction is feasible in people with Crohn’s Disease. Nutrients. 2020;12(9):2827. doi:10.3390/nu12092827
219. Yao CK, Muir JG, Gibson PR. Review article: insights into colonic protein fermentation, its modulation and potential health implications. Aliment Pharmacol Ther. 2016;43(2):181–196. doi:10.1111/apt.13456
220. Yamamoto-Furusho JK, Parra-Holguín NN. Emerging therapeutic options in inflammatory bowel disease. World J Gastroenterol. 2021;27(48):8242–8261. doi:10.3748/wjg.v27.i48.8242
221. Wang Q, More SK, Vomhof-DeKrey EE, et al. Small molecule FAK activator promotes human intestinal epithelial monolayer wound closure and mouse ulcer healing. Sci Rep. 2019;9(1):14669. doi:10.1038/s41598-019-51183-z
222. Oncel S, Gupta R, Wang Q, et al. ZINC40099027 promotes gastric mucosal repair in ongoing aspirin-associated gastric injury by activating focal adhesion kinase. Cells. 2021;10(4):908. doi:10.3390/cells10040908
223. Semiz A, Ozgun Acar O, Cetin H, et al. Suppression of inflammatory cytokines expression with bitter melon (Momordica charantia) in TNBS-instigated ulcerative colitis. J Transl Int Med. 2020;8(3):177–187. doi:10.2478/jtim-2020-0027
224. Lashner BA. Autologous hematopoietic stem cell transplantation for Crohn’s disease: high risk for a high reward. Inflamm Bowel Dis. 2005;11(8):778–779. doi:10.1097/01.MIB.0000171286.88690.ec
225. Xu J, Wang X, Chen J, et al. Embryonic stem cell-derived mesenchymal stem cells promote colon epithelial integrity and regeneration by elevating circulating IGF-1 in colitis mice. Theranostics. 2020;10(26):12204–12222. doi:10.7150/thno.47683
226. Clevers H. Modeling development and disease with organoids. Cell. 2016;165(7):1586–1597. doi:10.1016/j.cell.2016.05.082
227. Fukuda M, Mizutani T, Mochizuki W, et al. Small intestinal stem cell identity is maintained with functional Paneth cells in heterotopically grafted epithelium onto the colon. Genes Dev. 2014;28(16):1752–1757. doi:10.1101/gad.245233.114
228. Holmberg FE, Seidelin JB, Yin X, et al. Culturing human intestinal stem cells for regenerative applications in the treatment of inflammatory bowel disease. EMBO Mol Med. 2017;9(5):558–570. doi:10.15252/emmm.201607260
229. Lee M, Chang EB. Inflammatory Bowel Diseases (IBD) and the microbiome-searching the crime scene for clues. Gastroenterology. 2021;160(2):524–537. doi:10.1053/j.gastro.2020.09.056
230. Hadji H, Bouchemal K. Advances in the treatment of inflammatory bowel disease: focus on polysaccharide nanoparticulate drug delivery systems. Adv Drug Deliv Rev. 2022;181:114101. doi:10.1016/j.addr.2021.114101
231. Sahoo D, Swanson L, Sayed IM, et al. Artificial intelligence guided discovery of a barrier-protective therapy in inflammatory bowel disease. Nat Commun. 2021;12(1):4246. doi:10.1038/s41467-021-24470-5
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.