")
Back to Journals » Journal of Inflammation Research » Volume 17
Role of Cellular Senescence Genes and Immune Infiltration in Sepsis and Sepsis-Induced ARDS Based on Bioinformatics Analysis
Received 21 August 2024
Accepted for publication 14 November 2024
Published 19 November 2024 Volume 2024:17 Pages 9119—9133
DOI https://doi.org/10.2147/JIR.S488463
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Xiao-ling Wu, Ya-nan Guo
Intensive Care Unit, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China
Correspondence: Ya-nan Guo, Intensive Care Unit, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, 26 Shengli Street, Wuhan, 430014, People’s Republic of China, Tel +86-18186120762, Fax +86-27-82811446, Email [email protected]
Introduction: Sepsis is the leading cause of death in critically ill patients; it results in multiorgan dysfunction, including acute respiratory distress syndrome (ARDS). Our study was conducted to determine the role of cellular senescence genes and immune infiltration in sepsis and sepsis-induced ARDS via bioinformatic analyses.
Experimental Procedures: Datasets GSE66890 and GSE145227 were obtained from the Gene Expression Omnibus (GEO) database and utilized for bioinformatics analyses. Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of the differentially expressed genes (DEGs) were performed to identify the key functional modules. Two machine learning algorithms, least absolute shrinkage and selection operator (LASSO) and support vector machine recursive feature elimination (SVM-RFE), were used to screen for characteristic genes in sepsis and sepsis-induced ARDS. ROC curves were generated to evaluate the predictive ability of gene hubs. Differences in immune infiltration levels between the disease and control groups were compared via ssGSEA. The diagnostic value of the hub genes was verified via quantitative PCR (qPCR) in hospitalized patients.
Results: Four characteristic genes (ATM, CCNB1, CCNA1, and E2F2) were identified as biomarkers for the progression of sepsis-induced ARDS. E2F2 showed the highest predictive ability for the occurrence of ARDS in patients with sepsis. CD56bright and plasmacytoid dendritic cells exhibited high infiltration in the sepsis-induced ARDS group, whereas eosinophils, MDSCs, macrophages, and neutrophils exhibited low infiltration. In addition, ATM expression was lower in patients with sepsis than in those without sepsis (n = 6).
Conclusion: Sepsis-induced ARDS is correlated with circulating immune responses, and the expression of ATM, CCNB1, CCNA1, and E2F2 may serve as potential diagnostic biomarkers and therapeutic targets in sepsis-induced ARDS.
Keywords: sepsis, cellular senescence genes, immune infiltration, acute respiratory distress syndrome, bioinformatics analysis
Introduction
Sepsis is a life-threatening condition that results in organ dysfunction caused by a dysregulated host response to infection. Sepsis and septic shock are leading causes of death worldwide; they are fatal in approximately 25% of diagnosed patients.1 Patients with acute respiratory distress syndrome (ARDS) are at high risk of death in intensive care units (ICUs).2,3 Among the factors that influence the development of ARDS, sepsis is a major contributor. ARDS caused by sepsis is more severe than ARDS due to nonsepsis causes. Specifically, recovery from lung injury is worse, extubation success rates are lower, and mortality is greater.4 Sepsis-related ARDS has been demonstrated to be more severe than nonsepsis-related ARDS, as sepsis-induced ARDS causes more severe lung injuries, and its mortality is greater.5 Thus, key molecules involved in sepsis-related ARDS must be identified to treat sepsis more effectively.
Numerous studies have been conducted in recent years to determine whether genetic factors contribute to ARDS development.6,7 Clinical and biological markers are useful for predicting ARDS and its outcomes in patients with sepsis. Demand for biomarker-targeted therapies for ARDS is currently increasing,8,9 and previous studies have shown that specific markers may play important roles in detecting and preventing sepsis-induced ARDS. Specifically, cyclin B1 (CCNB1), cyclin B2 (CCNB2), DNA topoisomerase II alpha (TOP2A) and the transcription factor fork head box protein M1 (FOXM1) are potential targets for gene therapy.6 In this context, the analysis of gene expression profiles in relation to ARDS may yield breakthroughs to elucidate the pathological mechanisms underlying ARDS. Thus, cellular senescence may be involved in the development and onset of ARDS. Notably, inflammation, proliferation, and fibrosis may be causes or consequences of senescence. However, studies establishing a link between ARDS and cellular senescence are limited. Naturally occurring senescence in the early stages of acute injury in other tissues has been shown to be beneficial by limiting apoptosis and favouring tissue repair.10
In this study, we comprehensively evaluated the expression profiles of senescence genes in sepsis and sepsis-induced ARDS. Using the GEO database, the mRNA expression profiles of patients with sepsis and sepsis-related ARDS were analysed via differential expression, immune infiltration, gene expression, immune infiltration, and functional enrichment analyses. To effectively diagnose sepsis or sepsis-induced ARDS, we used two machine learning algorithms to predict outcomes. This study aimed to provide useful biological information for identifying the key cellular senescence genes involved in sepsis-induced ARDS.
Experimental Procedures
Data Source
RNA-seq profiles and clinical features of the GSE66890 and GSE145227 datasets were accessed from the GEO website (https://www.ncbi.nlm.nih.gov/gds/?term=). GSE66890 includes 29 sepsis-induced ARDS samples and 28 sepsis samples collected on the platform GPL6244. GSE145227 includes 10 sepsis samples and 12 normal samples collected on the platform GPL23178. Senescence genes were obtained from the “cellular senescence” terms in the KEGG database (https://www.kegg.jp/entry/map04218).
Identification of DEGs
The R (version 4.1.1) package “preprocessCore” was used to homogenize the data prior to differential expression analysis. After homogenization, DEGs were screened with a threshold of p < 0.05 using the “limma” package. Genes that were significantly upregulated or downregulated in both GEO datasets were identified as DEGs.
Functional Enrichment Analysis
The Gene Ontology (GO) resource (http://geneontology.org) provides structured, computable knowledge regarding the functions of genes and gene products.11 Gene Ontology function enrichment includes three categories: molecular function (MF), biological process (BP) and cellular component (CC). The Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.ad.jp/kegg/)12 is a database of biological systems that collects genomic, chemical and systemic functional information. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed and visualized via the R package “clusterProfiler.”.13 Statistical significance was set at p < 0.05. Correlation analysis of the hub genes with all transcripts was conducted for gene set enrichment analysis (GSEA) of the hub genes. The R package “clusterProfiler” was used to perform GSEA via correlation analysis to explore the functions of the hub genes.
Machine Learning Analysis
To effectively diagnose sepsis or sepsis-induced ARDS, we used two machine learning algorithms to predict outcomes. The LASSO logistic regression14 and SVM-RFE15 algorithms are highly important for identifying key biomarkers. In recent years, these two algorithms have been widely used in research to identify diagnostic or prognostic factors. The LASSO regression algorithm was applied via the R package “glmnet” to determine genes significantly related to the screening ability of the disease group and the control group. A support vector machine (SVM) is a monitoring technology used for classification and regression analysis. To avoid overfitting, the RFE algorithm was used to screen hub genes from the metadata. Thus, we used SVM-RFE technology to select hub genes. The ROC curve was used to evaluate the predictive ability of the hub genes via the R package “pROC.”
Immune Infiltration Assessment
Single Sample Gene Set Enrichment Analysis (ssGSEA), an extension of the Gene Set Enrichment Analysis (GSEA) methodology, is extensively utilized in bioinformatics research, particularly in the context of immune infiltration studies.16 The ssGSEA algorithm within the R package “GSVA” was used to evaluate the infiltration levels of the 23 immune cells. The differences in immune infiltration levels between the disease and control groups were compared. The Spearman rank correlation coefficient was used to evaluate the correlation between the infiltration scores of immune cells and the expression levels of the hub genes.
qRT‒PCR
This study was approved by the Medical Ethics Committee of the Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology (WHZXKYL2022-007). Whole blood samples from six patients with sepsis and six patients without sepsis were collected from the intensive care unit of the Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, from October 20, 2023 to January 30, 2024. Sepsis was diagnosed on the basis of Sepsis version 3.0. Patients with neoplastic diseases or autoimmune disorders, as well as those taking oral immunosuppressants, were excluded from the study. Quantitative real-time polymerase chain reaction (qPCR) was performed to measure the expression levels of four characteristic genes (ATM, CCNB1, CCNA1, and E2F2).
Statistical Analysis
Student’s t test was used to compare the differences between two groups. Statistical analysis was performed via R software 4.1.1. Differences were considered statistically significant at *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results
Identification of DEGs
We first homogenized the mRNA expression data from the GSE66890 and GSE145227 datasets before performing differential expression analysis (Sup-Figure 1A-D). We identified 8655 upregulated DEGs and 11530 downregulated DEGs in GSE145227 (Figure 1A–B) and 1092 upregulated DEGs and 958 downregulated DEGs in GSE66890 (Figure 1C–D). DEGs that were simultaneously upregulated or downregulated in both GSE66890 and GSE145227 were selected as differentially expressed genes for subsequent analysis, which yielded 288 upregulated (Figure 1E) and 167 downregulated genes (Figure 1F).
 |
Figure 1 Identification of DEGs. A total of 8655 upregulated DEGs and 11530 downregulated DEGs were identified in GSE145227 (A-B), whereas 1092 upregulated DEGs and 958 downregulated DEGs were identified in GSE66890 (C-D), and 288 upregulated genes (E) and 167 downregulated genes (F) were identified in GSE66890 and GSE145227. |
Functional Enrichment Analysis
The functional enrichment of the DEGs was performed to identify possible biological functions. GO analysis revealed that the DEGs were related to neutrophil-mediated immunity in the biological process (BP), secretory granule lumen in the cellular component (CC), and kinase regulatory activity in the molecular function (MF) (Figure 2A). KEGG analysis revealed that genes related to Salmonella infection, the cell cycle, cellular senescence, and carbon metabolism were enriched (Figure 2B). These results suggest that cellular senescence-related functions and pathways play vital roles in sepsis-induced ARDS.
 |
Figure 2 Functional enrichment of DEGs. GO analysis (A); KEGG analysis (B). |
Identification of Cellular Senescence Genes Among the DEGs
Next, we intersected the DEGs with genes related to cellular senescence. Eleven cellular senescence genes were identified (Figure 3A). We further explored the functions and pathways of these genes. GO analysis revealed that these 11 genes were involved primarily in the regulation of protein serine/threonine kinase activity (BP and MF) and the cyclin-dependent protein kinase holoenzyme complex (CC) (Figure 3B). KEGG analysis revealed that the cellular senescence, cell cycle, and FoxO signalling pathways were enriched (Figure 3C–D).
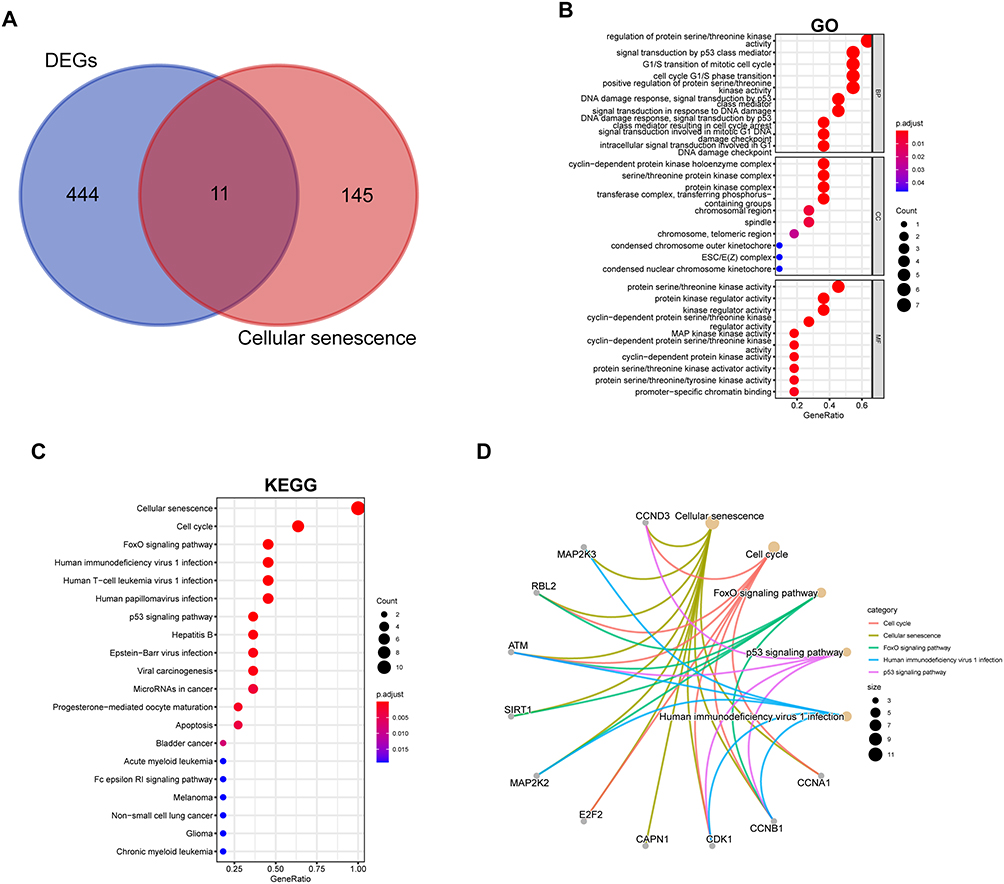 |
Figure 3 Identification of DEGs involved in cellular senescence. Eleven genes were identified (A). The enriched Gene Ontology (GO) terms in the molecular function (MF), biological process (BP) and cellular component (CC) categories (B). The results of the KEGG pathway enrichment analysis (C); the top 5 KEGG results and their relationships with the identified genes (D). |
Volcano plots and heatmaps were generated to depict the differences in the expression of cell senescence genes between the GSE66890 (Figure 4A–B) and GSE145227 datasets (Figure 4C–D). The differential expression of the 11 intersecting genes is displayed via data from the GSE66890 (Figure 5A) and GSE145227 datasets (Figure 5B). Eight genes (CCNA1, CCNB1, CDK1, CAPN1, E2F2, MAP2K2, MAP2K3, and CCND3) were upregulated in the sepsis-induced ARDS group, whereas three genes (SIRT1, ATM, and RBL2) were downregulated.
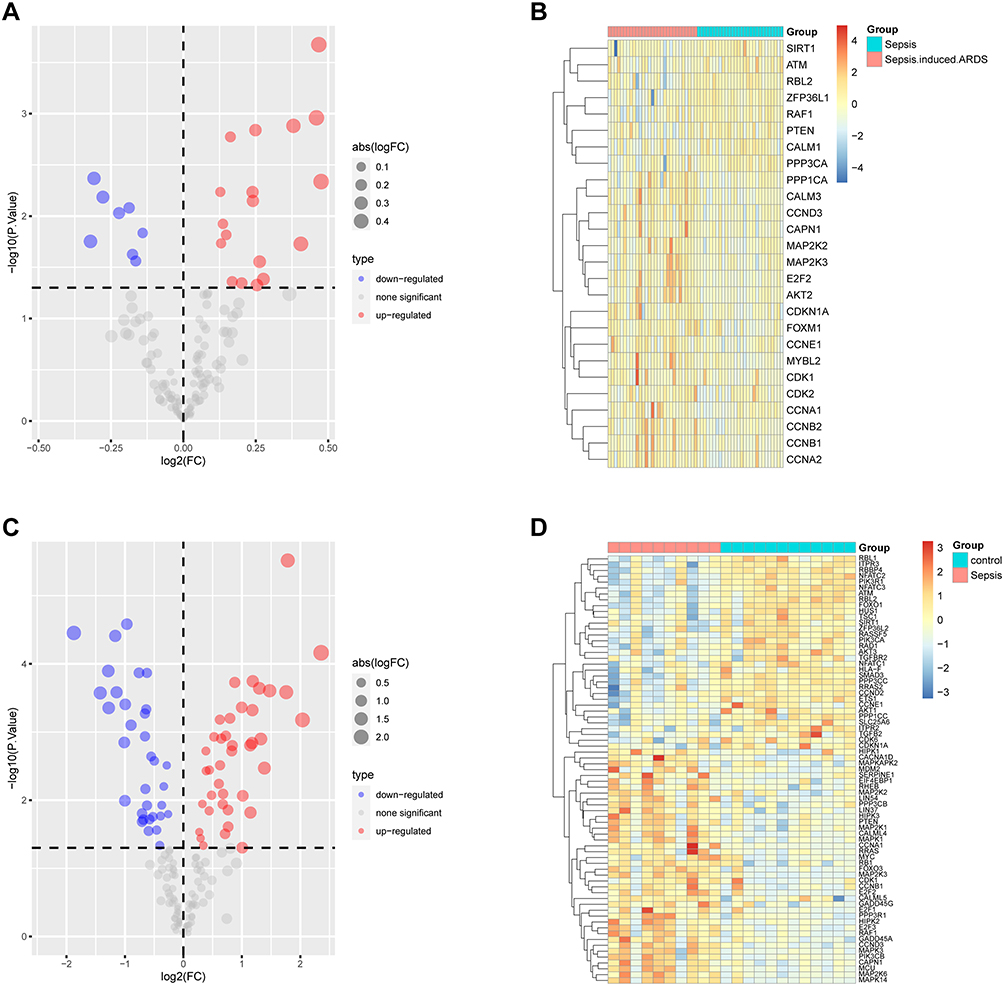 |
Figure 4 Volcano map and heatmap of cell senescence genes in the GSE66890 (A-B) and GSE145227 (C-D) datasets. |
 |
Figure 5 Differential expression of 11 cellular senescence genes in GSE66890 (A) and GSE145227 (B). *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. |
Identification of Hub Genes
Two machine learning algorithms were used to select biomarker genes. Using the LASSO regression algorithm, we screened for four hub genes as diagnostic markers (Figure 6A). Using the SVM-RFE algorithm, we identified 11 hub genes (Figure 6B). Finally, we selected four overlapping features (ATM, CCNB1, CCNA1, and E2F2) between the two algorithms (Figure 6C). These four genes may be involved in the progression of sepsis-induced ARDS.
 |
Figure 6 Machine learning algorithms. LASSO (A): SVM-RFE (B): overlapping features between these two algorithms (C). |
Predictive Ability of the Hub Genes
Figure 7A shows the log2-fold change values of the four hub genes in both datasets. The correlations between the hub genes based on the GSE66890 dataset are shown in Figure 7B. Notably, CCNA1 is positively correlated with CCNB1 and E2F2 and negatively correlated with ATM. The AUC value of the ROC curve indicates that E2F2 has the highest predictive ability (AUC = 0.749) for the occurrence of ARDS in patients with sepsis (Figure 7C).
 |
Figure 7 Predictive ability of the four hub genes. The log2(FC) values of 4 genes in GSE66890 and GSE145227 (A). Correlations between the hub genes in GSE66890 (B). The AUC value of the ROC curve for predicting the occurrence of ARDS in sepsis patients (C). |
Immune Infiltration Analysis
Because the immune system plays a crucial role in sepsis-induced ARDS, we assessed the infiltration levels of 23 immune cells in each patient. Figure 8A shows the relationship between each type of immune cell. We found that CD56bright structural killer cells and plasmacytoid dendritic cells were highly infiltrated in the sepsis-induced ARDS group, whereas eosinophils, MDSCs, macrophages, and neutrophils were poorly infiltrated (Figure 8B). Immune cell infiltration was correlated with the four hub genes (Figure 8C).
 |
Figure 8 Immune infiltration analysis. Relationships between each immune cell type (A). Infiltrating immune cells in the sepsis group and sepsis-induced ARDS group (B). Correlations of the 4 hub genes with immune cell types (C). *p < 0.05, **p < 0.01 and ***p < 0.001. |
Exploration of the Hub Genes
Correlation analysis between the hub genes and all transcripts was performed based on data from GSE66890. The expression levels of the 50 genes most positively correlated with each hub gene are shown in Figure 9. GSEA was conducted on the basis of the correlation results. The 20 terms most enriched for the 50 genes correlated with each of the four hub genes are shown in Figure 10. For example, genes positively correlated with CCNA1 were enriched for functions involved in membrane trafficking, neutrophil degranulation, and vesicle-mediated transport terms and depleted for functions related to signal transduction and postmitotic nuclear pore complex (NPC) reformation. These results may facilitate in-depth investigations of the involvement of hub genes in sepsis-induced ARDS.
 |
Figure 9 The expression of the top 50 genes positively correlated with each hub gene. |
 |
Figure 10 The top 20 terms of the 4 hub genes identified via GSEA. |
Preliminary Validation Analysis of Hub Genes
We performed RT‒PCR analysis of peripheral blood mononuclear cells (PBMCs) from 6 sepsis patients and 6 nonsepsis patients to verify the expression of the four hub genes. The results revealed that only ATM expression significantly differed between the two groups (Figure 11), which was consistent with the results of the bioinformatics analysis. Further exploration of ATM genes in sepsis is therefore important.
 |
Figure 11 Expression of the ATM gene in sepsis patients and nonsepsis patients. *p < 0.05, P = 0.0327. |
Discussion
Owing to the lack of effective risk prediction, accurate genetic diagnosis, and molecular interventions, sepsis-induced acute respiratory distress syndrome adds to the global impact of sepsis, a life-threatening condition. The prognosis of patients with sepsis-induced ARDS is poor due to limited effective prevention and treatments.17 Because appropriate treatment for sepsis-induced ARDS relies heavily on early prediction, identification, and diagnosis, bioinformatic analysis can provide a useful tool for exploring the underlying mechanisms of sepsis-induced ARDS via large-scale gene expression data.18 To investigate the transcriptomic consequences of sepsis-induced ARDS, we conducted a bioinformatics analysis of human sepsis-related ARDS samples and sepsis samples. In total, 288 DEGs were upregulated, and 167 DEGs were downregulated. The differentially expressed transcripts were primarily involved in the cell cycle and cellular senescence, suggesting that cellular senescence-related functions and pathways play a vital role in sepsis-induced ARDS.
Pulmonary diseases may benefit from cellular senescence or may have detrimental effects. The senescence process can limit DNA damage during acute injury, preserve senescent cells, and prevent apoptosis. Following the removal of senescent cells, proliferation occurs to regenerate the injured tissue. Senescent cells may prevent recovery from lung injury if cell turnover slows because they decrease the proliferation rate and prolong proinflammatory states. Abundant evidence supports that senescence plays a role in chronic and age-related lung diseases, but less evidence is available for ALI.10 In recent years, animal and cell experiments have demonstrated that cellular senescence plays an important role in sepsis-induced lung injury. In a two-hit mouse model generated via caecal ligation and puncture (CLP) followed by sublethal Pseudomonas aeruginosa (PA) lung infection, a relatively high mortality rate was observed because of increased lung inflammation and cellular senescence.19 LPS can induce cellular senescence in alveolar epithelial cells, suggesting that LPS-induced cellular senescence plays a critical role in limiting tissue repair when sepsis occurs.20 Sepsis-induced ARDS has a multifactorial pathogenesis, among which cellular senescence is believed to play a crucial role.
In the present study, 11 cellular senescence genes were identified. The differential expression of these 11 genes was investigated via data from the GSE66890 and GSE145227 datasets. Eight genes, namely, CCNA1, CCNB1, CDK1, CAPN1, E2F2, MAP2K2, MAP2K3, and CCND3, were upregulated in the sepsis-induced ARDS group, whereas three genes, SIRT1, ATM, and RBL2, were downregulated. KEGG analysis revealed that the functions and pathways of these genes were enriched in the cellular senescence, cell cycle, and FoxO signalling pathways. These findings increase our understanding of sepsis-induced acute respiratory distress syndrome. To effectively diagnose sepsis or sepsis-induced ARDS, we used the LASSO regression algorithm and SVM-RFE algorithm to predict disease outcomes and identified four biomarkers (ATM, CCNB1, CCNA1, and E2F2) across both algorithms. These four genes may be involved in the progression of sepsis-induced ARDS. The AUC value of the ROC curve indicated that E2F2 had the highest predictive ability (AUC 0.749) for the occurrence of ARDS in patients with sepsis.
Targeted E2F2 knockdown causes disproportionate entry into the cell cycle upon T-cell stimulation in mice. This study suggests that E2F2 plays a critical role in regulating apoptosis and may help in the design of new approaches for treating immune-mediated diseases. Both in vitro and in vivo, E2F2 provides genomic stability to activated T lymphocytes and plays a specific antiapoptotic role.21 In LPS-induced acute lung injury, the processes of resolving lung injury and regulatory T (Treg) cells appear to be important mediators.22 Although E2F2 has not been studied previously, we speculate that E2F2 may be involved in the progression of ARDS by regulating T cells or antiapoptotic properties.
Both CCNA1 and CCNB1 are cell cycle regulatory proteins that are involved in mitosis. A study reported that errors in mitosis can either kill the cell through apoptosis or cause potentially pathogenic mutations.23 GO functional analysis revealed that DEGs associated with the mitotic cell cycle and cellular senescence, such as CCNA1 and CCNB2, were the most significantly enriched biological processes among the upregulated genes. The abnormal cytoplasmic expression of cyclin B1 and T-cell cells is associated with immune responses specific to cyclin B1.24 A growing body of evidence suggests that the immune system plays a key role in recovery from lung diseases and acute lung injury.25 ARDS is driven by the activation of innate immunity via the binding of cell injury-associated endogenous molecules to pattern recognition receptors.26 However, neither CCNA1 nor CCNB2 are directly implicated in ARDS; therefore, we speculated that CCNA1 and CCNB1 might be closely related to ARDS development.
The ataxia-telangiectasia mutated (ATM) gene causes a rare autosomal recessive disorder that results in a defective repair of double-strand breaks in DNA.27 Ataxia-telangiectasia (A-T) is an autosomal recessive, progressive, multisystem disease caused by mutations in the ATM gene (11q22.3). This gene, which encodes ATM kinase, a kinase involved in cellular stress response signalling, is widely expressed. More than one hundred proteins related to the DNA damage response, cell cycle regulation, and other pathways are correlated with ATM expression. ATM plays several important roles in neuroprotection, innate immunity, inflammation, metabolism (eg, insulin signalling), longevity, and fertility.28 Several factors can contribute to the development of ataxia-telangiectasia (A-T), including recurrent sinus infections, immunodeficiency, and aspiration due to impaired swallowing caused by neurodegenerative progression.27,29 The immune deficiency in A-T is highly variable and affects both B and T cells. Therefore, we speculated that ATM downregulation might be related to sepsis-induced ARDS. In addition, ATM gene expression was significantly decreased in patients with sepsis (p = 0.0327).
Overall, our study revealed that the four identified cellular senescence genes exhibited marked differences and significant correlations with each other and with immune cell infiltration during the development of sepsis-induced ARDS in human transcriptomic studies. Recent studies have shown that LPS-induced ALI activates alveolar macrophages rather than causing systemic activation and that aged mice exhibit impaired adaptive immunity.30 Ageing and immune regulation are also inextricably linked. Immune regulation plays a crucial role in sepsis-induced ARDS.31,32 Multiple immune cells, including neutrophils and monocytes, are considered closely involved.33 Therefore, to explore the underlying immune mechanisms, we performed GSEA and immune infiltration assays. According to the GSEA results, the high-risk groups presented dysregulated expression of genes that were significantly enriched in neutrophil degranulation and innate immune pathways. We found that CD56bright structural killer cells and plasmacytoid dendritic cells were highly infiltrated in the sepsis-induced ARDS group, whereas eosinophils, MDSCs, macrophages, and neutrophils were poorly infiltrated.
To further explore the pathogenesis of ARDS, correlation analysis between the four hub genes and all transcripts was performed on the GSE66890 dataset. These analyses revealed that CCNA1 was positively correlated with membrane trafficking, neutrophil degranulation, and vesicle-mediated transport terms and negatively associated with signal transduction and postmitotic nuclear pore complex (NPC) reformation terms. These results may facilitate in-depth investigations of the hub genes involved in sepsis-induced ARDS.
Although this study produced some positive results, it also has several limitations. First, our findings were not verified in cells or tissues; we did not combine expression profiles to confirm our findings, and the sample size was limited. Furthermore, mRNA expression profiles were obtained from blood samples in this study and not from other important cells in ARDS, such as the epithelial and endothelial cells of the lungs. Hence, further studies involving other ARDS cell types with experimental verification and diverse samples are necessary.
Conclusions
In conclusion, the results of the present study revealed that ATM, CCNB1, CCNA1, and E2F2 can predict the progression of ARDS in sepsis patients. This study provides a comprehensive understanding of the genes involved in sepsis-induced ARDS pathogenesis.
Data Sharing Statement
The data used to support the findings of this study are available from the GEO website (https://www.ncbi.nlm.nih.gov/gds/?term=) and KEGG database (https://www.kegg.jp/entry/map04218), and the code is supplied by the corresponding author upon request. This paper has been uploaded to ResearchGate as a preprint: https://www.researchsquare.com/article/rs-4476919/v1.
Ethics Approval and Consent to Participate
Our study adhered to the principles outlined in the Declaration of Helsinki concerning ethical considerations in medical research involving human subjects. All procedures performed in this study involving human participants were conducted in accordance with the ethical standards of the institutional and/or national research committee and the 1964 helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all participants included in this study. The participants were informed of the nature of the study and its objectives, procedures, potential risks, and benefits. They were assured that their participation was voluntary and that they could withdraw from the study at any time without any consequences. The participants were also informed of the confidentiality of their data and how the data would be used and shared. Written consent was obtained from each participant before inclusion in the study. This study was approved by the Ethics Committee of the Central Hospital of Wuhan (Ethics approval NO: WHZXKYL2022-007).
Acknowledgments
We would like to express our deepest gratitude to all those who provided support and assistance for the completion of this project.
Author Contributions
All authors made a significant contribution to the work reported, including the conception, study design, execution, acquisition of data, analysis and interpretation; participated in drafting, revising or critically reviewing the article; gave final approval of the version to be published; agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work.
Funding
We thank everyone who helped us in this study. This study was supported by the Wuhan Central Hospital Discipline Fund (2021XK005) and the Wuhan Municipal Health Commission (WZ22Q45).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could influence the work reported in this study.
References
1. Evans L, Rhodes A, Alhazzani W, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 2021;47(11):1181–1247. doi:10.1007/s00134-021-06506-y
2. Fowler AA 3rd, Truwit JD, Hite RD, et al. Effect of vitamin C infusion on organ failure and biomarkers of inflammation and vascular injury in patients with sepsis and severe acute respiratory failure: the CITRIS-ALI randomized clinical trial. JAMA. 2019;322(13):1261–1270. doi:10.1001/jama.2019.11825
3. De Freitas Caires N, Gaudet A, Portier L, Tsicopoulos A, Mathieu D, Lassalle P. Endocan, sepsis, pneumonia, and acute respiratory distress syndrome. Crit Care. 2018;22(1):280. doi:10.1186/s13054-018-2222-7
4. Abe T, Ogura H, Shiraishi A, et al. Characteristics, management, and in-hospital mortality among patients with severe sepsis in intensive care units in Japan: the FORECAST study. Critical Care. 2018;22(1):322. doi:10.1186/s13054-018-2186-7
5. Sheu CC, Gong MN, Zhai R, et al. Clinical characteristics and outcomes of sepsis-related vs non-sepsis-related ARDS. Chest. 2010;138(3):559–567. doi:10.1378/chest.09-2933
6. Wang M, Yan J, He X, Zhong Q, Zhan C, Li S. Candidate genes and pathogenesis investigation for sepsis-related acute respiratory distress syndrome based on gene expression profile. Biol Res. 2016;49(1):25. doi:10.1186/s40659-016-0085-4
7. Chen Y, Qiu C, Cai W. Identification of key immune genes for sepsis-induced ARDS based on bioinformatics analysis. Bioengineered. 2022;13(1):697–708. doi:10.1080/21655979.2021.2012621
8. Mahida RY, Price J, Lugg ST, et al. CD14-positive extracellular vesicles in bronchoalveolar lavage fluid as a new biomarker of acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2022;322(4):L617–L624. doi:10.1152/ajplung.00052.2022
9. Fang Q, Wang Q, Zhou Z, Xie A. Consensus analysis via weighted gene co-expression network analysis (WGCNA) reveals genes participating in early phase of acute respiratory distress syndrome (ARDS) induced by sepsis. Bioengineered. 2021;12(1):1161–1172. doi:10.1080/21655979.2021.1909961
10. Huidobro C, Martin-Vicente P, Lopez-Martinez C, et al. Cellular and molecular features of senescence in acute lung injury. Mech Ageing Dev. 2021;193:111410. doi:10.1016/j.mad.2020.111410
11. Gene Ontology Consortium. The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 2019;47(D1):D330–D338. doi:10.1093/nar/gky1055
12. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi:10.1093/nar/28.1.27
13. Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
14. Tibshirani R. Regression shrinkage and selection via the lasso. J Royal Stat Soc Series B. 1996;58(1):267–288. doi:10.1111/j.2517-6161.1996.tb02080.x
15. Suykens JAK, Lukas L, Van P, De DB, Vandewalle MJ Least squares support vector machine classifiers: a large scale algorithm.
16. Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–112. doi:10.1038/nature08460
17. Hu Q, Hao C, Tang S. From sepsis to acute respiratory distress syndrome (ARDS): emerging preventive strategies based on molecular and genetic researches. Biosci Rep. 2020;40(5). doi:10.1042/BSR20200830
18. Liu AC, Patel K, Vunikili RD, et al. Sepsis in the era of data-driven medicine: personalizing risks, diagnoses, treatments and prognoses. Brief Bioinform. 2020;21(4):1182–1195. doi:10.1093/bib/bbz059
19. Li H, Luo YF, Wang YS, Xiao YL, Cai HR, Xie CM. Pseudomonas aeruginosa induces cellular senescence in lung tissue at the early stage of two-hit septic mice. Pathog Dis. 2018;76(9). doi:10.1093/femspd/ftz001
20. Kim CO, Huh AJ, Han SH, Kim JM. Analysis of cellular senescence induced by lipopolysaccharide in pulmonary alveolar epithelial cells. Arch Gerontol Geriatr. 2012;54(2):e35–41. doi:10.1016/j.archger.2011.07.016
21. Mustafa N, Mitxelena J, Infante A, et al. E2f2 attenuates apoptosis of activated T lymphocytes and protects from immune-mediated injury through repression of Fas and FasL. Int J Mol Sci. 2021;23(1):311. doi:10.3390/ijms23010311
22. Zhou M, Fang H, Du M, et al. The modulation of regulatory T cells via HMGB1/PTEN/β-catenin axis in LPS induced acute lung injury. Front Immunol. 2019;10:1612. doi:10.3389/fimmu.2019.01612
23. Diederichs S, Baumer N, Schultz N, et al. Expression patterns of mitotic and meiotic cell cycle regulators in testicular cancer and development. Int J Cancer. 2005;116(2):207–217. doi:10.1002/ijc.21034
24. Ersvaer E, Zhang J-Y, McCormack E, et al. Cyclin B1 is commonly expressed in the cytoplasm of primary human acute myelogenous leukemia cells and serves as a leukemia-associated antigen associated with autoantibody response in a subset of patients. Eur J Haematol. 2007;79(3):210–225. doi:10.1111/j.1600-0609.2007.00899.x
25. Opitz B, van Laak V, Eitel J, Suttorp N. Innate immune recognition in infectious and noninfectious diseases of the lung. Am J Respir Crit Care Med. 2010;181(12):1294–1309. doi:10.1164/rccm.200909-1427SO
26. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. New Engl J Med. 2017;377(6):562–572. doi:10.1056/NEJMra1608077
27. Duecker R, Baer P, Eickmeier O, et al. Oxidative stress-driven pulmonary inflammation and fibrosis in a mouse model of human ataxia-telangiectasia. Redox Biol. 2018;14:645–655. doi:10.1016/j.redox.2017.11.006
28. Bhatt JM, Bush A, van Gerven M, et al. Ataxia telangiectasia: why should the ERS care? Europ resp J. 2015;46(6):1557–1560. doi:10.1183/13993003.01456-2015
29. Bhatt JM, Bush A, van Gerven M, et al. ERS statement on the multidisciplinary respiratory management of ataxia telangiectasia. Eur Respir Rev. 2015;24(138):565–581. doi:10.1183/16000617.0066-2015
30. Brandenberger C, Kling KM, Vital M, Christian M. The role of pulmonary and systemic immunosenescence in acute lung injury. Aging Dis. 2018;9(4):553–565. doi:10.14336/AD.2017.0902
31. Root-Bernstein R. Innate receptor activation patterns involving TLR and NLR synergisms in COVID-19, ALI/ARDS and sepsis cytokine storms: a review and model making novel predictions and therapeutic suggestions. Int J Mol Sci. 2021;22(4):2108. doi:10.3390/ijms22042108
32. Zhang J, Wang C, Wang H, Li X, Xu J, Yu K. Loganin alleviates sepsis-induced acute lung injury by regulating macrophage polarization and inhibiting NLRP3 inflammasome activation. Int Immunopharmacol. 2021;95:107529. doi:10.1016/j.intimp.2021.107529
33. Ming T, Dong M, Song X, et al. Integrated analysis of gene co-expression network and prediction model indicates immune-related roles of the identified biomarkers in sepsis and sepsis-induced acute respiratory distress syndrome. Front Immunol. 2022;13:897390. doi:10.3389/fimmu.2022.897390
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Identification of an Immune-Related Gene Diagnostic Model and Potential Drugs in Sepsis Using Bioinformatics and Pharmacogenomics Approaches
Chen P, Chen J, Ye J, Yang L
Infection and Drug Resistance 2023, 16:5665-5680
Published Date: 28 August 2023
ITGA11, a Prognostic Factor Associated with Immunity in Gastric Adenocarcinoma
Yang X, Wei M, Huang Y, Yang X, Yuan Z, Huang J, Wei J, Tian L
International Journal of General Medicine 2024, 17:471-483
Published Date: 5 February 2024
Bioinformatics Analysis and Experimental Validation of Mitochondrial Autophagy Genes in Knee Osteoarthritis
Tang K, Sun L, Chen L, Feng X, Wu J, Guo H, Zheng Y
International Journal of General Medicine 2024, 17:639-650
Published Date: 23 February 2024
Cuproptosis-Related Biomarkers and Characterization of Immune Infiltration in Sepsis
Wang Y, Qiu X, Liu J, Liu X, Pan J, Cai J, Liu X, Qu S
Journal of Inflammation Research 2024, 17:2459-2478
Published Date: 22 April 2024
Effect of Neutrophil Elastase Inhibitor (Sivelestat Sodium) on Oxygenation in Patients with Sepsis-Induced Acute Respiratory Distress Syndrome
Wu T, Wang T, Jiang J, Tang Y, Zhang L, Jiang Z, Liu F, Kong G, Zhou T, Liu R, Guo H, Xiao J, Sun W, Li Y, Zhu Y, Liu Q, Xie W, Qu Y, Wang X
Journal of Inflammation Research 2025, 18:4449-4458
Published Date: 27 March 2025