")
Back to Journals » Journal of Inflammation Research » Volume 17
Role of Lipopolysaccharides in the Inflammation and Pyroptosis of Alveolar Epithelial Cells in Acute Lung Injury and Acute Respiratory Distress Syndrome
Received 20 May 2024
Accepted for publication 22 August 2024
Published 30 August 2024 Volume 2024:17 Pages 5855—5869
DOI https://doi.org/10.2147/JIR.S479051
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Xiao Shen,1,* Linglin He,2,* Wanru Cai3
1The Second Clinical Medical College, Zhejiang Chinese Medical University, Hangzhou, 310053, People’s Republic of China; 2School of Basic Medical Sciences, Zhejiang Chinese Medical University, Hangzhou, 310053, People’s Republic of China; 3Department of Respiratory and Critical Care Medicine, The Second Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou, 310005, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wanru Cai, Department of Respiratory and Critical Care Medicine, The Second Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou, 310005, People’s Republic of China, Email [email protected]
Abstract: Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) represent a spectrum of common critical respiratory conditions characterized by damage and death of alveolar epithelial cells (AECs). Pyroptosis is a form of programmed cell death with inflammatory characteristics, and activation of pyroptosis markers has been observed in AECs of patients with ALI/ARDS. Lipopolysaccharides (LPS) possess strong pro-inflammatory effects and are a crucial pathological factor leading to ALI in patients and animals. In LPS-induced ALI models, AECs undergo pyroptosis. However, physiologically and pathologically relevant concentrations of LPS lead to minor effects on AEC cell viability and minimal induction of cytokine release in vitro and do not induce classical pyroptosis. Nevertheless, LPS can enter the cytoplasm directly and induce non-classical pyroptosis in AECs when assisted by extracellular vesicles from bacteria, HMGB1, and pathogens. In this review, we have explored the effects of LPS on AECs concerning inflammation, cell viability, and pyroptosis, analyzing key factors that influence LPS actions. Notably, we highlight the intricate response of AECs to LPS within the framework of ALI and ARDS, emphasizing the variable induction of pyroptosis. Despite the minimal effects of LPS on AEC viability and cytokine release in vitro, LPS can induce non-classical pyroptosis under specific conditions, presenting potential pathways for therapeutic intervention. Collectively, understanding these mechanisms is crucial for the development of targeted treatments that mitigate the inflammatory responses in ALI/ARDS, thereby enhancing patient outcomes in these severe respiratory conditions.
Keywords: alveolar cell death, inflammatory response, programmed cell death, pathological impact, cytokine activation
Introduction
Acute lung injury (ALI) is characterized by an uncontrolled inflammatory response that leads to acute noncardiogenic pulmonary edema and hypoxemia.1 With further deterioration, ALI can progress to acute respiratory distress syndrome (ARDS), which is clinically characterized by progressive respiratory distress and refractory hypoxemia.2 Currently, the primary drugs used in clinical ARDS treatment include steroids, monoclonal antibodies, and small-molecule inhibitors.2 However, these drugs have significant side effects, high treatment costs, and cannot reverse the condition in all patients with ARDS, with mortality rates persisting at 27.5–53%.3,4
Damage and death of alveolar epithelial cells (AECs) are typical features of ALI, and the severity of lung epithelial damage serves as a crucial prognostic factor in patients with ARDS.5–8 Various factors, including bacteria, viruses, acids, elevated oxygen levels, mechanical ventilation, and cytokines, can cause AEC injury.1 In a healthy alveolar–capillary barrier, the pulmonary epithelial structure comprises a tight layer of type I and scattered type II AECs. Disruption of this barrier through type I AEC damage permits extensive fluid entry into alveolar spaces, resulting in pulmonary edema. After injury, reduced surfactant protein secretion by type II AECs leads to alveolar collapse.9 AECs release pro-inflammatory cytokines (such as tumor necrosis factor alpha [TNF-α] and interleukin [IL]-1β) and damage-associated molecular patterns (such as nucleic acids and HMGB1), exacerbating pulmonary inflammation and leading to a detrimental cycle in the microenvironment.10,11
Lipopolysaccharides (LPS) are found in the cell walls of gram-negative bacteria and comprise three parts: O-specific side chains, core polysaccharides, and lipid A, with lipid A serving as the main toxic component. During gram-negative bacterial infections, the release of large amounts of LPS by these bacteria can lead to severe ALI and sepsis, positioning LPS as a pivotal pathological factor in sepsis and ALI/ARDS.12 The LPS model is widely utilized in animal studies of ALI/ARDS, reflecting pathological characteristics similar to those of human conditions.13 Both intraperitoneal and direct intratracheal LPS administrations induce ALI, albeit with distinct characteristics. Intratracheal administration substantially damages the alveolar epithelial structure, leading to significant death of type I and II AECs and subsequent hyaline membrane formation, whereas the vascular endothelial structure remains relatively intact.14 In contrast, intraperitoneal injection mimics sepsis-related ALI, where LPS stimulates an influx of inflammatory factors into the circulation, resulting in indirect lung injury characterized by vascular endothelium damage, pulmonary interstitial edema, and a relatively preserved pulmonary epithelial structure.14 Therefore, LPS is an important factor for inducing ALI in both clinical and experimental settings.
In this review, we explored the impact of LPS on AECs, focusing on inflammation, cellular activity, and pyroptosis, by reviewing prior studies. We have also summarized potential explanations for inconsistent findings regarding LPS effects on AECs. Collectively, our review highlights the necessity for further investigation into the immune role of AECs, which are among the first cells to encounter foreign stimuli.
Mechanism of LPS Signal Transduction
Overview of LPS Signal Transduction
Outside the cell membrane, the LPS-binding protein (LBP) binds to the lipid A portion of LPS, forming an LPS–LBP complex. LBP catalyzes the transfer of LPS via electrostatic interactions to cluster of differentiation 14 (CD14). Subsequently, CD14 transfers LPS to the toll-like receptor 4 (TLR4)/myeloid differentiation protein 2 (MD2) complex,15 leading to dimerization of the extracellular domain of the TLR4/MD-2 complex (Figure 1).
 |
Figure 1 Schematic Diagram Depicting LPS/TLR4 Signaling. LBP binds to LPS and transfers it to CD14, which then transfers LPS to the TLR4/MD-2 complex. TLR4 activates the MyD88-dependent pathway, promoting the expression of TNF-α, IL-1β, and IL-6. Subsequently, the TLR4/MD-2/LPS complex undergoes endocytosis and activates the TRIF-dependent pathway, promoting the expression of type I interferons and RANTES. (Created with BioRender.com). |
TLR4 belongs to a family of pattern recognition receptors responsible for identifying conserved pathogen-associated molecular patterns and initiating immune-inflammatory responses, with TLR4 serving as the primary receptor for LPS in mammals. TLR4 can bind to several adaptor proteins containing toll/interleukin-1 receptor domains (TIR domains), including myeloid differentiation primary response 88 (MyD88) and toll-like receptor adaptor molecule 1 (TRIF).16 Consequently, downstream signaling from TLR4 is mainly divided into MyD88-dependent and TRIF-dependent pathways. Upon binding with MyD88, TLR4 rapidly activates downstream mitogen-activated protein kinase families and nuclear factor kappa beta, leading to the production of numerous cytokines.16–18 Binding of TLR4 with TRIF activates the TRIF-dependent pathway, phosphorylating downstream interferon regulatory factor 3/7, thereby promoting the synthesis and release of type I interferons (IFN) (Figure 1).16–18
CD14 Plays a Key Role in LPS Signal Transduction
CD14 plays two major roles in LPS signaling. The first involves presenting LPS to the TLR4/MD-2 complex, enhancing the cellular response to low LPS concentrations. Upon stimulation with low concentrations of LPS (< 10 ng/mL), bone marrow-derived macrophages (BMDMs) from CD14−/− mice exhibit a significant reduction in TNF-α release, indicating that MyD88-mediated TNF-α release is CD14-dependent.18 However, when stimulated with high concentrations of LPS (> 100 ng/mL), TNF-α release from CD14−/− BMDMs is even greater than that from wild-type BMDMs.18 At low LPS concentrations, CD14 primarily facilitates the transfer of LPS to TLR4/MD2 complex. In contrast, at high LPS concentrations, the dependence of MyD88-dependent signaling on CD14 is overcome, potentially owing to the direct interaction between LPS and MD2.19,20 The second role involves controlling the endocytosis of the LPS receptor complex, thereby activating the TRIF-related adaptor molecule (TRAM)–TRIF pathway and producing type I IFN.17 TLR4 is the only receptor in the TLR family that activates both the MyD88-dependent and TRIF-dependent pathways.21 Although LPS binds to the TLR4–MD2 complex on the cell membrane surface, the LPS–TLR4 complex must be endocytosed into the cell to exert its full biological effect. Upon binding of LPS, facilitated by CD14, the TLR4–MD2 complex is recruited to the lipid rafts.22 Following this recruitment, TLR4 initiates the MyD88-dependent pathway, leading to an early and rapid inflammatory response.23 Subsequently, the entire receptor complex is internalized by CD14 to form endosomes, thereby activating the TRAM–TRIF pathway and producing type I IFN.17,18 The activation of the TRIF pathway and the production of type I IFN by LPS are highly dependent on CD14. In CD14−/− cells, even LPS concentrations of 1000 ng/mL fail to induce TRAM–TRIF-dependent regulated on activation, normal T cell expressed and secreted (RANTES) and type I IFN synthesis.18
Role of LPS in AECs
Pro-Inflammatory Effect of LPS on AECs
Owing to their roles in regeneration, immune regulation, and surfactant secretion, the role of alveolar type II (ATII) epithelial cells in inflammation has been well elucidated. Primary human ATII cells express functional TLR2 and TLR4 on their cell membranes. LPS stimulation induces the expression of TLR2/4, leading to increased expression on the cell membrane of ATII cells and secretion of cytokines such as IL-1β, TNF-α, and IL-6.24,25 Compared with macrophages, ATII cells produce lower levels of TNF-α and IL-1β but higher levels of chemokines such as monocyte chemoattractant protein-1 (MCP-1) and IL-8, suggesting that ATII cells may be an important source of chemokines.25,26 After LPS stimulation, ATI cells release more MCP-1 than IL-8, whereas ATII cells release significantly more IL-8 than MCP-1.25
Fluorescence-activated cell sorting (FACS) enables researchers to obtain high-purity primary cells. However, ATII cells sorted by FACS exhibit inflammatory characteristics that differ markedly from those reported in previous studies (Figure 2). Primary rat ATII cells treated with LPS produce only low levels of TNF-α, IL-6, and IL-1β. In contrast, ATI cells release much higher levels of these cytokines upon LPS stimulation.27 Non-FACS-sorted ATII cells release hundreds of times more TNF-α and IL-6 upon LPS stimulation compared with that from FACS-sorted ATII cells, suggesting contamination by macrophages.27 In addition, without 3D culture, the distinct lung epithelial phenotype of primary ATII cells disappears within 3–5 days of culture, which is evident from the loss of their cuboidal shape and decreased surfactant production.28 Notably, single-cell transcriptomic results from human tissues show that TLR4 expression levels are very low (< 1 nTPM) in both ATI and ATII cells.29 Considering that even minimal immune cell contamination can significantly affect the assessment of immune responses in ATII cells, researchers must exercise caution when interpreting results from studies involving primary human ATII cells.
 |
Figure 2 FACS More Accurately Differentiates the Cytokine Secretion Abilities of ATI and ATII Cells. In traditional methods of extracting primary AECs, the lung tissue is digested using enzymes, and ATII cells are obtained through differential adhesion. However, the ATII cells produced using this method are contaminated with macrophages. Consequently, early studies found that ATII cells could secrete large amounts of IL-6, TNF-α, and IL-1β upon LPS stimulation. However, after purifying ATI and ATII cells using FACS, researchers discovered that ATII cells secrete only small amounts of IL-6 after LPS stimulation, while ATI cells secrete large amounts of IL-6, TNF-α, and IL-1β upon LPS stimulation. (Created with BioRender.com). |
A549 cells, derived from patients with lung alveolar epithelial carcinoma, exhibit characteristics similar to that of ATII epithelial cells. Owing to challenges in isolating and purifying primary human ATII cells, often contaminated by immune cells, A549 cells frequently serve as substitutes.30,31 Early studies in the 20th century demonstrated that A549 cells could secrete IL-6 and IL-8 in response to TNF-α, IL-1β, and other stimuli,30,32 indicating their potential for cytokine release. Nevertheless, data on whether A549 cells functionally express TLR4 and release inflammatory cytokines upon LPS stimulation remain contradictory. A549 cells reportedly express TLR4,33–38 MD-2,34,38 and CD14;38 the presence of TLR4 on the A549 cell membrane has been confirmed using flow cytometry and membrane protein isolation.38,39 The human bronchial epithelial cell line BEAS-2B and primary human bronchial epithelial cells also express TLR4.33,34 However, some studies suggest that although A549 and BEAS-2B cells express TLR4, this receptor may only reside in the cytoplasm, not on the cell membrane.34,35,40 Even after LPS stimulation, TLR2/4 is absent from the surface of A549 cells.40 Furthermore, multiple RNA-sequencing studies have indicated that A549 cells lack TLR4 mRNA under resting conditions.41–43 Respiratory syncytial virus induces the expression of TLR4 in various AECs, including A549 cells, and promotes the translocation of TLR4 to the cell membrane. After respiratory syncytial viral infection, LPS binds to the cell membrane of AECs,35 indicating that TLR4 activation in AECs may depend on pathological conditions, such as pathogen infection.
Although LPS stimulation induces the secretion of IL-8 and other cytokines in A549 and BEAS-2B cells,34,44–46 several studies have reported that LPS does not induce cytokine secretion in A549 cells.25,30,35,39,47 The low responsiveness of A549 cells to LPS may also be due to the lack of surface CD14,25 and they only secrete IL-8 under combined stimulation with LPS and soluble CD14.33,39 However, other studies have reported the presence of CD14 on the surface of A549 cell membranes and shown that biotinylated LPS can bind to CD14 on the cell membrane of A549 cells.40 Even in the absence of TLR4 or CD14 on the cell membrane, LPS can still enter cells through scavenger receptors with the help of LBP, and then bind to TLR4 in the cytoplasm, thereby exerting its pro-inflammatory effects.48,49
Despite being extensively studied in lung epithelial cell lines, A549 and BEAS-2B cells exhibit significant differences from primary cells in their physiological characteristics. Although A549 cells are often used as a substitute for ATII cells, they only exhibit ATII characteristics under specific culture conditions.50,51 Moreover, BEAS-2B cells cultured under serum-free and serum-containing conditions exhibit different phenotypes, with BEAS-2B cells cultured in serum lacking epithelial cell characteristics but exhibiting interstitial cells.52 These cells also exhibit an abnormal karyotype, which may render them unsuitable for use as “normal” cells.53
Physiological Concentration of LPS Does Not Inhibit AEC Cell Viability
Regulated cell death (RCD) is a crucial mechanism for eliminating damaged cells and maintaining internal homeostasis, including apoptosis, necroptosis, and pyroptosis.54 LPS induces various forms of RCD, including apoptosis,55–59 autophagy,55 pyroptosis,60 ferroptosis,61,62 and PANoptosis,63 in AECs. Cell viability indicates alterations in cell proliferation and death. Despite the ease of detection, studies report contradictory effects of LPS on AEC viability.
Under physiological conditions, the concentration of LPS in human serum ranges from 0–1.0 ng/mL; however, in pathological conditions, this concentration can increase to 10 ng/mL.64 In inflammation-related studies involving AECs, LPS is often used at concentrations exceeding 1 μg/mL, hundreds of times higher than those found clinically. Some studies have determined that stimulation with 1 μg/mL LPS for 24 h is sufficient to reduce A549 cell viability by 50%.65,66 However, other studies indicate that when stimulated for 24 h, the LPS concentration must exceed 15 μg/mL to achieve a 50% reduction in A549 cell viability.67,68 Researchers have also reported that at concentrations of 0–15 μg/mL, LPS had no effect on cell viability after 72 h of treatment.69 After stimulation with a high LPS concentration (500 µg/mL) for 24 h, A549 cell viability decreased only to 67% (Table 1).50 Interestingly, A549 cell viability increased with prolonged LPS stimulation, potentially owing to LPS tolerance and cell fusion.50
 |
Table 1 Effects of Different Concentrations of LPS on A549 Cell Viability |
In summary, pathologically significant LPS concentrations are unlikely to suppress AEC cell viability. Moreover, although high concentrations of LPS can reduce AEC cell viability and induce various forms of RCD,60–62,70–72 the pathological significance of RCD is unclear.
Effect of LPS on AEC Pyroptosis
Classical Pyroptosis and ALI
Pyroptosis is a form of programmed cell death characterized by cell swelling, membrane pore formation, and the release of inflammatory contents, presenting a “fried eggs” morphology upon rupture.73 In classical pyroptosis, various pattern recognition receptors (such as NLRP3 and AIM2) form inflammasomes by binding to scaffold proteins ASC and pro-caspase-1. Upon assembly of the inflammasome, pro-caspase-1 undergoes self-cleavage to form the active p33/p10 complex, which cleaves pro-IL-1β and pro-IL-18 to generate mature IL-1β and IL-18.74 Additionally, the p33/p10 complex can cleave gasdermin D (GSDMD) to form the GSDMD N-terminal domain, which binds to lipids on the cell membrane, creating pore structures and releasing intracellular contents (Figure 3).74–76
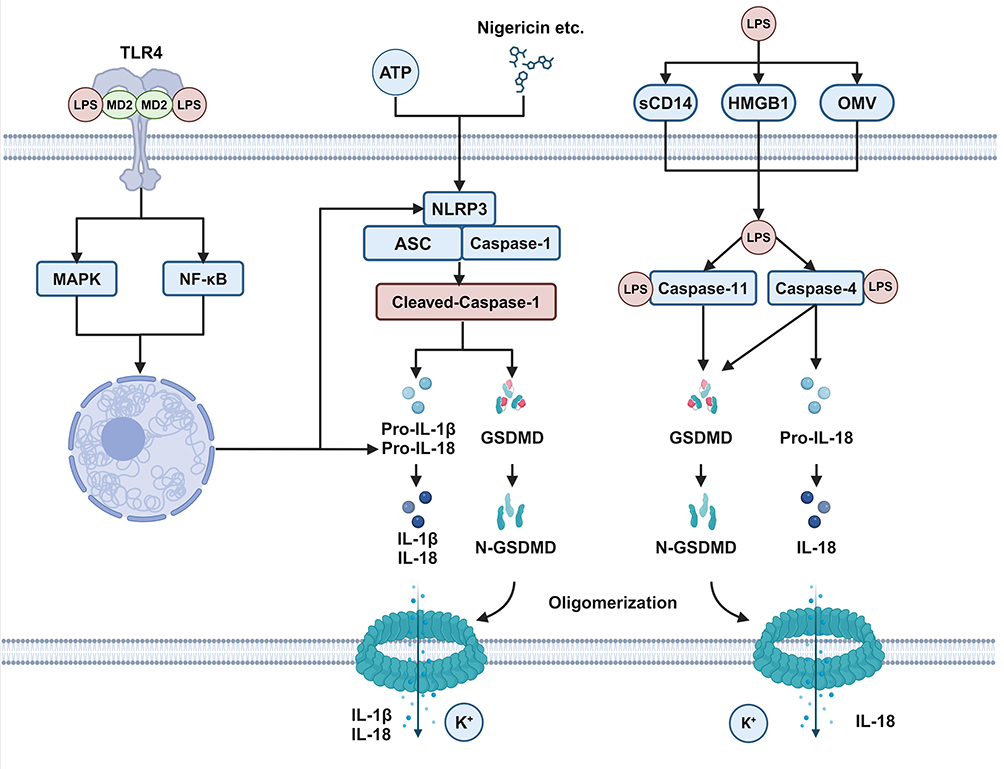 |
Figure 3 Schematic Diagram of LPS-induced Classical and Non-classical Pyroptosis in AECs. After LPS stimulation, the expression of NLRP3 and pro-IL-1β increases. When the cells are subsequently stimulated with ATP or Nigericin, the NLRP3-ASC-Caspase-1 complex forms in the cytoplasm. Within the complex, Caspase-1 undergoes autolysis and then cleaves pro-IL-1β, pro-IL-18, and GSDMD. Cleaved N-GSDMD oligomerizes and forms pores in the cell membrane, resulting in the release of active IL-1β, IL-18, and potassium ions. With the assistance of sCD14, HMGB1, OMV, and others, LPS can directly enter the cytoplasm and bind to cytoplasmic caspase-4/5 (human) or caspase-11 (mouse). Activated caspase-4/5/11 can cleave GSDMD, leading to cell membrane perforation. Additionally, caspase-4 can cleave IL-18, resulting in the release of active IL-18. (Created with BioRender.com). |
Pyroptosis is a crucial mechanism of the immune system for combating infections, wherein immune cells release a large number of cytokines, amplifying the inflammatory response against pathogenic infections.77 However, during pathogen infections, inflammation acts as a double-edged sword—excessive inflammation leads to tissue damage, whereas weak inflammation impedes pathogen clearance. For instance, in Yersinia infection, GSDMD−/− results in an increased bacterial load, exacerbating the risk of mortality in mice.78 In SARS-CoV-2 infection, GSDMD−/− does not alleviate infection-induced weight loss and lung damage.79 In Leishmania infection, NLRP3−/−, GSDMD−/−, and caspase-1/11−/− mice display significantly larger ear lesions.80 However, in H1N1 influenza infection, GSDMD−/− can reduce mortality rates and alleviate lung damage in mice.81,82 In sepsis caused by Candida albicans, GSDMD−/− mice exhibit reduced mortality rates, whereas caspase-1/11−/− mice experience increased mortality rates.83
In direct lung injury induced by LPS and sepsis-related lung injury, lacking interference from pathogens, the situation appears to be more “pure”. In LPS-induced sepsis models, GSDMD−/− improves mouse survival rates, whereas caspase-1−/− does not improve survival rates.84,85 Similarly, caspase-1−/− mice show more severe neutrophil infiltration in lung tissue following intratracheal LPS administration.86 However, caspase-1−/− can alleviate lung injury caused by high tidal volume mechanical ventilation.87 In cecal ligation and puncture-induced sepsis, GSDMD−/− improves mouse survival rates and reduces lung injury.88 In the LPS intratracheal administration model, neutrophil-specific GSDMD−/− mitigates lung injury by reducing neutrophil extracellular traps.89 Additionally, GSDMD−/− alleviates acute pancreatitis-related lung injury.90 Overall, GSDMD−/− can alleviate ALI caused by various etiologies, but caspase-1 may lack a strong role in mitigating this condition, possibly owing to the existence of the Caspase-8/GSDMD axis.78,91
NLRP3-mediated classical pyroptosis has been extensively studied in clinical and animal models of ALI. Targeted inhibition of NLRP3 using small molecule inhibitors showed good efficacy in ALI animal models.92–94 However, the activity of NLRP3 inflammasomes in AECs remains controversial. Clinically, elevated markers of pyroptosis have been detected in ALI/ARDS caused by various factors and are associated with poor prognosis in patients.95–97 Postmortem examinations of deceased patients with SARS-CoV-2 further confirm pyroptosis as a significant cause of pulmonary epithelial cell death.98 In animal models of ALI, pulmonary epithelial cells undergo NLRP3 inflammasome-mediated pyroptosis.99 Co-localization of caspase-1 p20 and an ATII marker (SPC) is observed in the lung tissues of ALI mice.99
A549 cells are commonly used as AECs in pyroptosis research. Numerous studies have shown that they express NLRP3, ASC, and Caspase-1, providing the molecular basis for classical pyroptosis, and can undergo classical pyroptosis when stimulated by agents such as LPS and SARS-CoV-2.100–104 SARS-CoV-2 activates NLRP3 inflammasome activity in A549 cells leading to pyroptosis.104–106 Notably, although A549 cells release IL-1β upon SARS-CoV-2 infection, the quantity is significantly less than that released by macrophages.106 Therefore, further elucidation of inflammasome activity in lung epithelial cells is required.
However, many studies have questioned the ability of AECs to undergo pyroptosis. Single-cell sequencing and transcriptomic studies suggest that primary human AEC and A549 cells either do not express NLRP3 and caspase-1 or express them at substantially low levels.29,107 Studies have found that primary human AECs and A549 cells do not show NLRP3 protein via Western blotting.108,109 Other studies have reported that caspase-1 is undetectable in lung epithelial cell lines (16-HBE, HBEC, and BEAS-2B cells) using Western blotting.110 Similarly, mouse lung epithelial cells (TC-1 cells) also lack the expression of NLRP3, ASC, and caspase-1.111 These studies provide contrasting evidence, suggesting that AECs may lack NLRP3 and caspase-1, and therefore cannot undergo classical pyroptosis.
Cytoplasmic LPS Induces Non-Classical Pyroptosis in AECs
After binding to TLR4, LPS enters the cells via lipid raft transport and is subsequently degraded in lysosomes without leaking into the cytoplasm. In contrast to the “gentle” extracellular action of LPS, entry into the cytoplasm can lead to non-classical pyroptosis.112 In human macrophages, epithelial cells, and endothelial cells, cytoplasmic LPS triggers caspase-4-dependent pyroptosis.85,113 Once inside the cytoplasm, LPS can directly bind to caspase-4/5 (human) or caspase-11 (mouse), leading to their self-cleavage and oligomerization.113,114 Activated caspase-4/5/11 cleaves GSDMD into NT-GSDMD, resulting in pyroptosis.115 Unlike the activation of caspase-1 in classical pyroptosis, caspase-4/5/11 lacks the activity to cleave pro-IL-1β.116,117 Notably, caspase-4 can cleave IL-18;114 moreover, activated caspase-11 can cleave pannexin-1, leading to cytoplasmic ATP release, which induces P2X purinoceptor 7-mediated classical pyroptosis.118 Furthermore, both GSDMD- and pannexin-1-dependent pores cause K+ efflux, which can activate NLRP3 inflammasomes, leading to the cleavage of pro-IL-1β and pro-IL-18 by caspase-1.85,119 However, the exact mechanism underlying the entry of LPS into the cytoplasm remains unclear. LPS may enter the cytoplasm by binding to HMGB1120–122 or CD14123 or through bacterially secreted outer membrane vesicles124,125 and host-derived extracellular vesicles,126 thereby inducing pyroptosis (Figure 3).
Currently, research on the relationship between non-classical pyroptosis and ALI is limited. Reportedly, caspase-11 knockout and endothelial-specific caspase-11 knockout can reduce mortality from endotoxic shock and alleviate lung damage secondary to endotoxic shock.84,85 Activation of the caspase-11/gasdermin D pathway induces mitochondrial DNA release, which hinders endothelial cell proliferation and impairs lung vascular repair.127 This implies that in ALI, caspase-11-mediated pyroptosis is a crucial mechanism leading to endothelial cell injury and inhibition of repair. Burkholderia pseudomallei, Shigella flexneri, and LPS electroporation can induce caspase-4-dependent pyroptosis in A549 cells;128,129 LPS transfection does not cause significant cell death in A549 cells.129 This is because A549 cells lack guanylate-binding proteins (GBPs), which directly bind and aggregate “free” LPS. IFN-γ can induce the expression of GBPs in A549 cells; therefore, IFN-γ-primed A549 cells can undergo pyroptosis after LPS transfection.129 Overall, the mechanism of non-classical pyroptosis in AECs needs to be further studied in primary AECs, and in vivo studies are needed to demonstrate the physiological significance of non-classical pyroptosis in AECs.
Factors Influencing the Effect of LPS on AECs
Effects of Extraction Methods and Bacterial Strain Sources on LPS Effects
The potency of LPS is influenced by the extraction method and bacterial strain. Common LPS is extracted using the phenol method and contains other bacterial components, such as lipoproteins, activating both TLR4 and TLR2. However, ultrapure LPS extracted through repeated enzymatic hydrolysis, followed by purification using the phenol–TEA–DOC method, only activates TLR4.130 Both TLR2 and TLR4 can activate the MyD88-dependent pathway, and TLR2 can also form a complex with RAC1, activating downstream nuclear factor kappa beta through the PI3K/AKT pathway.131 Common LPS additionally activates TLR2, making the signaling pathway exceptionally complex and, thus, unsuitable for studying downstream TLR signaling. However, LPS has been widely used in numerous studies, and many early studies did not effectively distinguish between LPS sources and purity.132
LPS from different bacterial strains also produces markedly different pro-inflammatory effects. LPS comprises lipid A, a core polysaccharide, and an O-polysaccharide. O-polysaccharides are polysaccharide chains composed of multiple oligosaccharide repeat units exhibiting high variability.133 Bacteria with long O-polysaccharide chains form smooth colonies and produce smooth LPS (sLPS), whereas those lacking long O-polysaccharide chains or with short O-polysaccharide chains form rough colonies and produce rough LPS (rLPS).133 At low LPS concentrations, neither sLPS nor rLPS activate TLR4 in the absence of CD14.134 However, at concentrations of ≥ 100 ng/mL, regardless of CD14 presence, rLPS can activate both the MyD88-dependent and TRIF-dependent pathways, whereas sLPS can only activate the MyD88-dependent pathway (Figure 4).134 It is worth mentioning that this study was conducted in RAW264 and J774A.1 cell lines, and the LPS concentration reached the μg/mL level, which may explain the difference from the results obtained by Zanoni et al.18 Thus, highlighting the extraction method and bacterial strain source of LPS is crucial to ensure the purity of LPS bioactivity and provide assurance of reproducibility by other researchers.
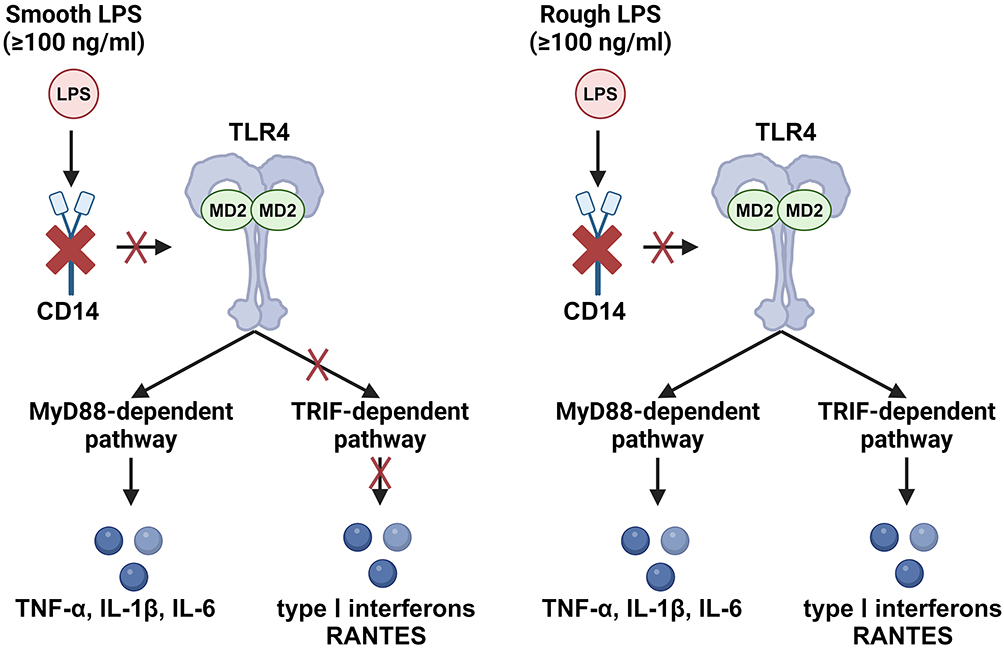 |
Figure 4 Smooth LPS and Rough LPS Exhibit Different Pro-inflammatory Activities in the Absence of CD14. In CD14-deficient cells or environments lacking CD14 (such as those with monoclonal antibodies that deplete CD14), smooth LPS at concentrations ≥100 ng/mL can activate the MyD88-dependent pathway but cannot activate the TRIF-dependent pathway. Under the same conditions, rough LPS can activate both the MyD88-dependent pathway and the TRIF-dependent pathway. (Created with BioRender.com). |
Effect of Serum Concentration on LPS Effects
The effect of serum on LPS cannot be overlooked because the concentration, manufacturer, and batch of serum can influence the biological activity of LPS. Fetal bovine serum (FBS) in the culture medium significantly increases LPS-induced release of TNF-α, MCP-1, and IL-8 in human primary lung macrophages and AECs.25 Moreover, at the same LPS concentration, higher concentrations of normal human serum (NHS) in the culture medium result in greater IL-8 and IL-6 release from AECs.33 This effect may be due to the presence of soluble CD14 in NHS. Under serum-free conditions, 200 ng/mL of CD14, combined with physiological concentrations of LPS (10 ng/mL), induces significant cytokine release from A549 and BEAS-2B cells.33 The amount of IL-8 produced by A549 cells stimulated with LPS+CD14 is no different from that produced by cells stimulated with 10% NHS + LPS; however, the amount of IL-8 produced by BEAS-2B cells stimulated with LPS+CD14 is only approximately one-tenth of that stimulated with 10% NHS + LPS,33 indicating that other substances in NHS may promote the LPS-induced cytokine secretion from BEAS-2B cells. Serum enhances not only the pro-inflammatory effects of LPS but also the inhibition of cell activity by LPS (Table 2).50,135,136 Tang et al136 observed that a FBS concentration of 5% was required for LPS to induce A549 cell death. Nova et al50 found that, compared with A549 cells cultured with 10% FBS, the inhibitory effect of LPS on cell activity was reduced by approximately 50% in cells cultured with 4% FBS.
 |
Table 2 Effects of Different Concentrations of FBS on A549 Cell Viability |
Serum from different manufacturers and different batches may lead to markedly different inflammatory responses.135,137 Serum from the same manufacturer, such as Gibco, but of different origins, can increase IL-8 release induced by LPS by thousands of times.135 Serum from different manufacturers or batches can induce different levels of IL-8 secretion in HCT-8 and HT-29 cells through extracellular signal-regulated kinase phosphorylation, which may be related to the presence of 1-palmitoyl-sn-glycero-3-phosphocholine in the serum.138
Owing to the potent effect of serum, which can mask multiple subtle changes, many studies have used serum starvation to eliminate serum interference.135,137 However, serum starvation lacks standardized criteria and can lead to the inhibition of proliferation, cell death, and phenotypic changes in AECs.51,139–141 The effect of serum starvation on cells is also time-dependent;139 thus, the duration of serum starvation may directly affect the responsiveness of AECs to LPS.
Conclusions and Future Perspectives
LPS is one of the most common triggers of ALI and ARDS in both humans and animals. AECs, as the most important cell group in the lungs, still hold many unknowns regarding the immune response to LPS. AECs can clearly produce a weak response to direct LPS stimulation. The intensity of this response largely depends on serum concentration, CD14, and LPS itself. Therefore, whether the weak response of AEC to LPS stimulation plays a significant role in disease requires further investigation. Clinical studies of ALI/ARDS have revealed that AECs undergo classical pyroptosis, characterized by caspase-1 and IL-1β cleavage. However, in vitro studies on primary AEC and lung epithelial cell lines have shown a lack of NLRP3 and caspase-1. Although SARS-CoV-2 can induce weak IL-1β release from AECs, the significance of this change in ALI/ARDS requires further investigation. Apart from its pro-inflammatory role, LPS also plays a vital role in inducing non-classical pyroptosis, which is gaining increasing attention in research aiming to improve our understanding of sepsis and ALI/ARDS. AECs can undergo non-classical pyroptosis under conditions such as bacterial infection or electroporation. However, the significance of AEC non-classical pyroptosis in ALI/ARDS requires confirmation through AEC-specific gene-knockout mice. Collectively, our review stresses the need for meticulous methodological consistency and further investigation into the immune functions of AECs, which play a critical role in the host defense against pathogenic threats. This enhanced understanding is crucial for developing targeted therapies that can effectively manage and mitigate conditions such as ALI and ARDS.
Acknowledgments
This work was supported by the Zhejiang Province Traditional Chinese Medicine Science and Technology Plan Project (grant no. 2017ZZ008) and the National Natural Science Foundation of China (grant no. 81603545). We thank Editage (www.editage.cn) for English language editing.
Disclosure
The authors declare that this study was conducted without any commercial or financial relationships that could be construed as conflicts of interest.
References
1. Matthay MA, Zemans RL, Zimmerman GA, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5(1):18. doi:10.1038/s41572-019-0069-0
2. Gorman EA, O’Kane CM, McAuley DF. Acute respiratory distress syndrome in adults: diagnosis, outcomes, long-term sequelae, and management. Lancet. 2022;400(10358):1157–1170. doi:10.1016/S0140-6736(22)01439-8
3. Gupta S, Wang W, Hayek SS, et al. Association between early treatment with tocilizumab and mortality among critically Ill patients with COVID-19. JAMA Intern Med. 2021;181(1):41–51. doi:10.1001/jamainternmed.2020.6252
4. Rein L, Calero K, Shah R, et al. Randomized phase 3 trial of ruxolitinib for COVID-19-associated acute respiratory distress syndrome. Crit Care Med. 2022;50(12):1701–1713. doi:10.1097/CCM.0000000000005682
5. Peukert K, Sauer A, Seeliger B, et al. Increased alveolar epithelial damage markers and inflammasome-regulated cytokines are associated with pulmonary superinfection in ARDS. J Clin Med. 2023;12(11):3649. doi:10.3390/jcm12113649
6. Spadaro S, Fogagnolo A, Campo G, et al. Markers of endothelial and epithelial pulmonary injury in mechanically ventilated COVID-19 ICU patients. Crit Care. 2021;25(1):74. doi:10.1186/s13054-021-03499-4
7. Atmowihardjo LN, Heijnen N, Smit MR, et al. Biomarkers of alveolar epithelial injury and endothelial dysfunction are associated with scores of pulmonary edema in invasively ventilated patients. Am J Physiol Lung Cell Mol Physiol. 2023;324(1):L38–L47. doi:10.1152/ajplung.00185.2022
8. de Souza Xavier Costa N, da Costa Sigrist G, Schalch AS, Belotti L, Dolhnikoff M, da Silva L. Lung tissue expression of epithelial injury markers is associated with acute lung injury severity but does not discriminate sepsis from ARDS. Respir Res. 2024;25(1):129. doi:10.1186/s12931-024-02761-x
9. Bos L, Ware LB. Acute respiratory distress syndrome: causes, pathophysiology, and phenotypes. Lancet. 2022;400(10358):1145–1156. doi:10.1016/S0140-6736(22)01485-4
10. Hsieh PC, Wu YK, Yang MC, Su WL, Kuo CY, Lan CC. Deciphering the role of damage-associated molecular patterns and inflammatory responses in acute lung injury. Life Sci. 2022;305:120782. doi:10.1016/j.lfs.2022.120782
11. Barnett KC, Xie Y, Asakura T, et al. An epithelial-immune circuit amplifies inflammasome and IL-6 responses to SARS-CoV-2. Cell Host Microbe. 2023;31(2):243–259.e6. doi:10.1016/j.chom.2022.12.005
12. Virzì GM, Mattiotti M, de Cal M, Ronco C, Zanella M, De Rosa S. Endotoxin in sepsis: methods for LPS detection and the use of omics techniques. Diagnostics. 2022;13(1):79. doi:10.3390/diagnostics13010079
13. Domscheit H, Hegeman MA, Carvalho N, Spieth PM. Molecular dynamics of lipopolysaccharide-induced lung injury in rodents. Front Physiol. 2020;11:36. doi:10.3389/fphys.2020.00036
14. Menezes SL, Bozza PT, Neto HC, et al. Pulmonary and extrapulmonary acute lung injury: inflammatory and ultrastructural analyses. J Appl Physiol. 2005;98(5):1777–1783. doi:10.1152/japplphysiol.01182.2004
15. Ryu JK, Kim SJ, Rah SH, et al. Reconstruction of LPS transfer cascade reveals structural determinants within LBP, CD14, and TLR4-MD2 for Efficient LPS recognition and transfer. Immunity. 2017;46(1):38–50. doi:10.1016/j.immuni.2016.11.007
16. Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. 2021;78(4):1233–1261.
17. Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9(4):361–368. doi:10.1038/ni1569
18. Zanoni I, Ostuni R, Marek LR, et al. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011;147(4):868–880. doi:10.1016/j.cell.2011.09.051
19. Rajaiah R, Perkins DJ, Ireland DD, Vogel SN. CD14 dependence of TLR4 endocytosis and TRIF signaling displays ligand specificity and is dissociable in endotoxin tolerance. Proc Natl Acad Sci U S A. 2015;112(27):8391–8396. doi:10.1073/pnas.1424980112
20. Akashi S, Saitoh S, Wakabayashi Y, et al. Lipopolysaccharide interaction with cell surface Toll-like receptor 4-MD-2: higher affinity than that with MD-2 or CD14. J Exp Med. 2003;198(7):1035–1042. doi:10.1084/jem.20031076
21. Akira S, Hoshino K. Myeloid differentiation factor 88-dependent and -independent pathways in toll-like receptor signaling. J Infect Dis. 2003;187(2):S356–363. doi:10.1086/374749
22. Płóciennikowska A, Hromada-Judycka A, Borzęcka K, Kwiatkowska K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. 2015;72(3):557–581. doi:10.1007/s00018-014-1762-5
23. Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006;125(5):943–955. doi:10.1016/j.cell.2006.03.047
24. Armstrong L, Medford AR, Uppington KM, et al. Expression of functional toll-like receptor-2 and −4 on alveolar epithelial cells. Am J Respir Cell Mol Biol. 2004;31(2):241–245. doi:10.1165/rcmb.2004-0078OC
25. Thorley AJ, Grandolfo D, Lim E, Goldstraw P, Young A, Tetley TD. Innate immune responses to bacterial ligands in the peripheral human lung--role of alveolar epithelial TLR expression and signalling. PLoS One. 2011;6(7):e21827. doi:10.1371/journal.pone.0021827
26. Thorley AJ, Ford PA, Giembycz MA, Goldstraw P, Young A, Tetley TD. Differential regulation of cytokine release and leukocyte migration by lipopolysaccharide-stimulated primary human lung alveolar type II epithelial cells and macrophages. J Immunol. 2007;178(1):463–473. doi:10.4049/jimmunol.178.1.463
27. Wong MH, Johnson MD. Differential response of primary alveolar type I and type II cells to LPS stimulation. PLoS One. 2013;8(1):e55545. doi:10.1371/journal.pone.0055545
28. Sucre J, Jetter CS, Loomans H, et al. Successful establishment of primary type II alveolar epithelium with 3D organotypic coculture. Am J Respir Cell Mol Biol. 2018;59(2):158–166. doi:10.1165/rcmb.2017-0442MA
29. Karlsson M, Zhang C, Méar L, et al. A single-cell type transcriptomics map of human tissues. Sci Adv. 2021;7(31):eabh2169. doi:10.1126/sciadv.abh2169
30. Standiford TJ, Kunkel SL, Basha MA, et al. Interleukin-8 gene expression by a pulmonary epithelial cell line. A model for cytokine networks in the lung. J Clin Invest. 1990;86(6):1945–1953. doi:10.1172/JCI114928
31. Lieber M, Smith B, Szakal A, Nelson-Rees W, Todaro G. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int, J, Cancer. 1976;17(1):62–70. doi:10.1002/ijc.2910170110
32. Crestani B, Cornillet P, Dehoux M, Rolland C, Guenounou M, Aubier M. Alveolar type II epithelial cells produce interleukin-6 in vitro and in vivo. Regulation by alveolar macrophage secretory products. J Clin Invest. 1994;94(2):731–740. doi:10.1172/JCI117392
33. Schulz C, Farkas L, Wolf K, Kratzel K, Eissner G, Pfeifer M. Differences in LPS-induced activation of bronchial epithelial cells (BEAS-2B) and type II-like pneumocytes (A-549). Scand J Immunol. 2002;56(3):294–302. doi:10.1046/j.1365-3083.2002.01137.x
34. Guillot L, Medjane S, Le-Barillec K, et al. Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J Biol Chem. 2004;279(4):2712–2718. doi:10.1074/jbc.M305790200
35. Monick MM, Yarovinsky TO, Powers LS, et al. Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J Biol Chem. 2003;278(52):53035–53044. doi:10.1074/jbc.M308093200
36. Li D, Jin Y, Sun Y, Lei J, Liu C. Knockdown of toll-like receptor 4 inhibits human NSCLC cancer cell growth and inflammatory cytokine secretion in vitro and in vivo. Int J Oncol. 2014;45(2):813–821. doi:10.3892/ijo.2014.2479
37. De S, Zhou H, DeSantis D, Croniger CM, Li X, Stark GR. Erlotinib protects against LPS-induced endotoxicity because TLR4 needs EGFR to signal. Proc Natl Acad Sci U S A. 2015;112(31):9680–9685. doi:10.1073/pnas.1511794112
38. MacRedmond R, Greene C, Taggart CC, McElvaney N, O’Neill S. Respiratory epithelial cells require Toll-like receptor 4 for induction of human beta-defensin 2 by lipopolysaccharide. Respir Res. 2005;6(1):116. doi:10.1186/1465-9921-6-116
39. Abate W, Alghaithy AA, Parton J, Jones KP, Jackson SK. Surfactant lipids regulate LPS-induced interleukin-8 production in A549 lung epithelial cells by inhibiting translocation of TLR4 into lipid raft domains. J Lipid Res. 2010;51(2):334–344. doi:10.1194/jlr.M000513
40. Tsutsumi-Ishii Y, Nagaoka I. Modulation of human beta-defensin-2 transcription in pulmonary epithelial cells by lipopolysaccharide-stimulated mononuclear phagocytes via proinflammatory cytokine production. J Immunol. 2003;170(8):4226–4236. doi:10.4049/jimmunol.170.8.4226
41. Klijn C, Durinck S, Stawiski EW, et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol. 2015;33(3):306–312. doi:10.1038/nbt.3080
42. Ghandi M, Huang FW, Jané-Valbuena J, et al. Next-generation characterization of the cancer cell line encyclopedia. Nature. 2019;569(7757):503–508. doi:10.1038/s41586-019-1186-3
43. Pontén F, Jirström K, Uhlen M. The human protein atlas--A tool for pathology. J Pathol. 2008;216(4):387–393. doi:10.1002/path.2440
44. Manoury B, Nenan S, Leclerc O, et al. The absence of reactive oxygen species production protects mice against bleomycin-induced pulmonary fibrosis. Respir Res. 2005;6(1):11. doi:10.1186/1465-9921-6-11
45. Wang F, Li W, Liu Z, Yu R, Wang D. LPS-induced inflammatory response and apoptosis are mediated by Fra-1 upregulation and binding to YKL-40 in A549 cells. Exp Ther Med. 2021;22(6):1474. doi:10.3892/etm.2021.10909
46. Hattar K, Savai R, Subtil FS, et al. Endotoxin induces proliferation of NSCLC in vitro and in vivo: role of COX-2 and EGFR activation. Cancer Immunol Immunother. 2013;62(2):309–320. doi:10.1007/s00262-012-1341-2
47. Li J, Qin Y, Chen Y, et al. Mechanisms of the lipopolysaccharide-induced inflammatory response in alveolar epithelial cell/macrophage co-culture. Exp Ther Med. 2020;20(5):76. doi:10.3892/etm.2020.9204
48. Dunzendorfer S, Lee HK, Soldau K, Tobias PS. TLR4 is the signaling but not the lipopolysaccharide uptake receptor. J Immunol. 2004;173(2):1166–1170. doi:10.4049/jimmunol.173.2.1166
49. Dunzendorfer S, Lee HK, Soldau K, Tobias PS. Toll-like receptor 4 functions intracellularly in human coronary artery endothelial cells: roles of LBP and sCD14 in mediating LPS responses. FASEB J. 2004;18(10):1117–1119. doi:10.1096/fj.03-1263fje
50. Nova Z, Skovierova H, Strnadel J, Halasova E, Calkovska A. Short-term versus long-term culture of A549 cells for evaluating the effects of lipopolysaccharide on oxidative stress, surfactant proteins and cathelicidin LL-37. Int J Mol Sci. 2020;21(3):1148. doi:10.3390/ijms21031148
51. Chary A, Groff K, Stucki AO, et al. Maximizing the relevance and reproducibility of A549 cell culture using FBS-free media. Toxicol In Vitro. 2022;83:105423. doi:10.1016/j.tiv.2022.105423
52. Han X, Na T, Wu T, Yuan BZ. Human lung epithelial BEAS-2B cells exhibit characteristics of mesenchymal stem cells. PLoS One. 2020;15(1):e0227174. doi:10.1371/journal.pone.0227174
53. Lee Y, Ryu YJ. Morphologically and karyotypically atypical cells of ‘normal’ human bronchial epithelial cell line (Beas-2B). Ultrastruct Pathol. 2023;47(6):470–477. doi:10.1080/01913123.2023.2262561
54. Bedoui S, Herold MJ, Strasser A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol. 2020;21(11):678–695. doi:10.1038/s41580-020-0270-8
55. Liu H, Wang S, Gong L, et al. SIRT6 ameliorates LPS-induced apoptosis and tight junction injury in ARDS through the ERK1/2 pathway and autophagy. Int J Med Sci. 2023;20(5):581–594. doi:10.7150/ijms.80920
56. Shi Q, Li Z, Dong Y, Yang G, LncRNA THRIL LM. transcriptionally activated by AP-1 and stabilized by METTL14-mediated m6A modification, accelerates LPS-evoked acute injury in alveolar epithelial cells. Int Immunopharmacol. 2023;123:110740. doi:10.1016/j.intimp.2023.110740
57. Wen XP, Li M, Zhang RQ, Wan QQ. Insulin reverses impaired alveolar fluid clearance in ARDS by inhibiting LPS-induced autophagy and inflammatory. Front Immunol. 2023;14:1162159. doi:10.3389/fimmu.2023.1162159
58. Hou L, Zhang J, Liu Y, et al. MitoQ alleviates LPS-mediated acute lung injury through regulating Nrf2/Drp1 pathway. Free Radic Biol Med. 2021;165:219–228. doi:10.1016/j.freeradbiomed.2021.01.045
59. Xi X, Yao Y, Liu N, Li P. MiR-297 alleviates LPS-induced A549 cell and mice lung injury via targeting cyclin dependent kinase 8. Int Immunopharmacol. 2020;80:106197. doi:10.1016/j.intimp.2020.106197
60. Kang JY, Xu MM, Sun Y, et al. Melatonin attenuates LPS-induced pyroptosis in acute lung injury by inhibiting NLRP3-GSDMD pathway via activating Nrf2/HO-1 signaling axis. Int Immunopharmacol. 2022;109:108782. doi:10.1016/j.intimp.2022.108782
61. Xu B, Wang H, Chen Z. Puerarin inhibits ferroptosis and inflammation of lung injury caused by sepsis in LPS induced lung epithelial cells. Front Pediatr. 2021;9:706327. doi:10.3389/fped.2021.706327
62. Jiao Y, Yong C, Zhang R, Qi D, Wang D. Hepcidin alleviates LPS-induced ARDS by regulating the ferritin-mediated suppression of ferroptosis. Shock. 2022;57(6):274–281. doi:10.1097/SHK.0000000000001941
63. Guo J, Luo Y, Zuo J, Teng J, Shen B, Liu X. Echinacea polyphenols inhibit NLRP3-dependent pyroptosis, apoptosis, and necroptosis via suppressing NO production during lipopolysaccharide-induced acute lung injury. J Agric Food Chem. 2023;71(19):7289–7298. doi:10.1021/acs.jafc.2c08382
64. Nighot M, Al-Sadi R, Guo S, et al. Lipopolysaccharide-induced increase in intestinal epithelial tight permeability is mediated by toll-like receptor 4/myeloid differentiation primary response 88 (MyD88) activation of myosin light chain kinase expression. Am J Pathol. 2017;187(12):2698–2710. doi:10.1016/j.ajpath.2017.08.005
65. Chuang CY, Chen TL, Cherng YG, Tai YT, Chen TG, Chen RM. Lipopolysaccharide induces apoptotic insults to human alveolar epithelial A549 cells through reactive oxygen species-mediated activation of an intrinsic mitochondrion-dependent pathway. Arch Toxicol. 2011;85(3):209–218. doi:10.1007/s00204-010-0585-x
66. Zhao J, Li X, Zou M, et al. miR-135a inhibition protects A549 cells from LPS-induced apoptosis by targeting Bcl-2. Biochem Biophys Res Commun. 2014;452(4):951–957. doi:10.1016/j.bbrc.2014.09.025
67. Huang C, Zheng H, He W, et al. Ghrelin ameliorates the human alveolar epithelial A549 cell apoptosis induced by lipopolysaccharide. Biochem Biophys Res Commun. 2016;474(1):83–90. doi:10.1016/j.bbrc.2016.04.074
68. Li S, Guo L, Qian P, et al. Lipopolysaccharide induces autophagic cell death through the PERK-dependent branch of the unfolded protein response in human alveolar epithelial A549 cells. Cell Physiol Biochem. 2015;36(6):2403–2417. doi:10.1159/000430202
69. Ding Z, Wu X, Wang Y, et al. Melatonin prevents LPS-induced epithelial-mesenchymal transition in human alveolar epithelial cells via the GSK-3β/Nrf2 pathway. Biomed Pharmacother. 2020;132:110827. doi:10.1016/j.biopha.2020.110827
70. Kim CO, Huh AJ, Han SH, Kim JM. Analysis of cellular senescence induced by lipopolysaccharide in pulmonary alveolar epithelial cells. Arch Gerontol Geriatr. 2012;54(2):e35–41. doi:10.1016/j.archger.2011.07.016
71. Li S, Zhao L, Li X, et al. Mir-204 Regulates LPS-Induced A549 cell damage by targeting FOXK2. J Healthc Eng. 2021;2021:7404671. doi:10.1155/2021/7404671
72. Zhang Z, Chen Z, Liu R, et al. Bcl-2 proteins regulate mitophagy in lipopolysaccharide-induced acute lung injury via PINK1/parkin signaling pathway. Oxid Med Cell Longev. 2020;2020:6579696. doi:10.1155/2020/6579696
73. Chen X, He WT, Hu L, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016;26(9):1007–1020. doi:10.1038/cr.2016.100
74. Boucher D, Monteleone M, Coll RC, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med. 2018;215(3):827–840. doi:10.1084/jem.20172222
75. Xu J, Núñez G. The NLRP3 inflammasome: activation and regulation. Trends Biochem Sci. 2023;48(4):331–344. doi:10.1016/j.tibs.2022.10.002
76. Sharif H, Wang L, Wang WL, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature. 2019;570(7761):338–343. doi:10.1038/s41586-019-1295-z
77. Sundaram B, Tweedell RE, Prasanth Kumar S, Kanneganti TD. The NLR family of innate immune and cell death sensors. Immunity. 2024;57(4):674–699. doi:10.1016/j.immuni.2024.03.012
78. Demarco B, Grayczyk JP, Bjanes E, et al. Caspase-8-dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci Adv. 2020;6(47):eabc3465. doi:10.1126/sciadv.abc3465
79. Eltobgy MM, Zani A, Kenney AD, et al. Caspase-4/11 exacerbates disease severity in SARS-CoV-2 infection by promoting inflammation and immunothrombosis. Proc Natl Acad Sci U S A. 2022;119(21):e2202012119. doi:10.1073/pnas.2202012119
80. de Sá K, Amaral LA, Rodrigues TS, et al. Gasdermin-D activation promotes NLRP3 activation and host resistance to Leishmania infection. Nat Commun. 2023;14(1):1049. doi:10.1038/s41467-023-36626-6
81. Speaks S, McFadden MI, Zani A, et al. Gasdermin D promotes influenza virus-induced mortality through neutrophil amplification of inflammation. Nat Commun. 2024;15(1):2751. doi:10.1038/s41467-024-47067-0
82. Rosli S, Harpur CM, Lam M, et al. Gasdermin D promotes hyperinflammation and immunopathology during severe influenza A virus infection. Cell Death Dis. 2023;14(11):727. doi:10.1038/s41419-023-06258-1
83. Ding X, Kambara H, Guo R, et al. Inflammasome-mediated GSDMD activation facilitates escape of Candida albicans from macrophages. Nat Commun. 2021;12(1):6699. doi:10.1038/s41467-021-27034-9
84. Hu JJ, Liu X, Xia S, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21(7):736–745. doi:10.1038/s41590-020-0669-6
85. Cheng KT, Xiong S, Ye Z, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. 2017;127(11):4124–4135. doi:10.1172/JCI94495
86. Rowe SJ, Allen L, Ridger VC, Hellewell PG, Whyte MK. Caspase-1-deficient mice have delayed neutrophil apoptosis and a prolonged inflammatory response to lipopolysaccharide-induced acute lung injury. J Immunol. 2002;169(11):6401–6407. doi:10.4049/jimmunol.169.11.6401
87. Dai M, Li Q, Pan P. The modulation of interferon regulatory factor-1 via caspase-1-mediated alveolar macrophage pyroptosis in ventilator-induced lung injury. Mediators Inflamm. 2022;2022:1002582. doi:10.1155/2022/1002582
88. Chen H, Li Y, Wu J, et al. RIPK3 collaborates with GSDMD to drive tissue injury in lethal polymicrobial sepsis. Cell Death Differ. 2020;27(9):2568–2585. doi:10.1038/s41418-020-0524-1
89. Xie J, Zhu CL, Wan XJ, et al. GSDMD-mediated NETosis promotes the development of acute respiratory distress syndrome. Eur J Immunol. 2023;53(1):e2250011. doi:10.1002/eji.202250011
90. Wu J, Zhang J, Zhao J, Chen S, Zhou T, Xu J. Treatment of severe acute pancreatitis and related lung injury by targeting gasdermin D-mediated pyroptosis. Front Cell Dev Biol. 2021;9:780142. doi:10.3389/fcell.2021.780142
91. Hughes SA, Lin M, Weir A, et al. Caspase-8-driven apoptotic and pyroptotic crosstalk causes cell death and IL-1β release in X-linked inhibitor of apoptosis (XIAP) deficiency. EMBO J. 2023;42(5):e110468. doi:10.15252/embj.2021110468
92. Xu Y, Biby S, Guo C, et al. Characterization of a small molecule inhibitor of the NLRP3 inflammasome and its potential use for acute lung injury. Bioorg Chem. 2024;150:107562. doi:10.1016/j.bioorg.2024.107562
93. Cao F, Chen G, Xu Y, et al. METTL14 contributes to acute lung injury by stabilizing NLRP3 expression in an IGF2BP2-dependent manner. Cell Death Dis. 2024;15(1):43. doi:10.1038/s41419-023-06407-6
94. Wang L, Lei W, Zhang S, Yao L. MCC950, a NLRP3 inhibitor, ameliorates lipopolysaccharide-induced lung inflammation in mice. Bioorg Med Chem. 2021;30:115954. doi:10.1016/j.bmc.2020.115954
95. Peukert K, Fox M, Schulz S, et al. Inhibition of caspase-1 with tetracycline ameliorates acute lung injury. Am J Respir Crit Care Med. 2021;204(1):53–63. doi:10.1164/rccm.202005-1916OC
96. Xiong S, Hong Z, Huang LS, et al. IL-1β suppression of VE-cadherin transcription underlies sepsis-induced inflammatory lung injury. J Clin Invest. 2020;130(7):3684–3698. doi:10.1172/JCI136908
97. Dolinay T, Kim YS, Howrylak J, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med. 2012;185(11):1225–1234. doi:10.1164/rccm.201201-0003OC
98. Schifanella L, Anderson J, Wieking G, et al. The defenders of the alveolus succumb in COVID-19 Pneumonia to SARS-CoV-2, necroptosis, pyroptosis and panoptosis. bioRxiv. 2022;2022:503050.
99. Zhang T, Li M, Zhao S, et al. CaMK4 Promotes acute lung injury through NLRP3 Inflammasome activation in type II alveolar epithelial cell. Front Immunol. 2022;13:890710. doi:10.3389/fimmu.2022.890710
100. Wang Q, Wen W, Zhou L, et al. LL-37 improves sepsis-induced acute lung injury by suppressing pyroptosis in alveolar epithelial cells. Int Immunopharmacol. 2024;129:111580. doi:10.1016/j.intimp.2024.111580
101. Chittasupho C, Umsumarng S, Srisawad K, et al. Inhibition of SARS-CoV-2-Induced NLRP3 inflammasome-mediated lung cell inflammation by triphala-loaded nanoparticle targeting spike glycoprotein S1. Pharmaceutics. 2024;16(6):751. doi:10.3390/pharmaceutics16060751
102. Xie WM, Su W, Liu XY, et al. FTO deficiency alleviates LPS-induced acute lung injury by TXNIP/NLRP3-mediated alveolar epithelial cell pyroptosis. Am J Respir Cell Mol Biol. 2024;70(5):351–363. doi:10.1165/rcmb.2023-0251OC
103. Xie C, Zhou X, Chen W, et al. Diallyl trisulfide induces pyroptosis and impairs lung CSC-like properties by activating the ROS/Caspase 1 signaling pathway. Chem Biol Interact. 2024;397:111083. doi:10.1016/j.cbi.2024.111083
104. Xu H, Akinyemi IA, Chitre SA, et al. SARS-CoV-2 viroporin encoded by ORF3a triggers the NLRP3 inflammatory pathway. Virology. 2022;568:13–22. doi:10.1016/j.virol.2022.01.003
105. Sun X, Liu Y, Huang Z, et al. SARS-CoV-2 non-structural protein 6 triggers NLRP3-dependent pyroptosis by targeting ATP6AP1. Cell Death Differ. 2022;29(6):1240–1254. doi:10.1038/s41418-021-00916-7
106. Lécuyer D, Nardacci R, Tannous D, et al. The purinergic receptor P2X7 and the NLRP3 inflammasome are druggable host factors required for SARS-CoV-2 infection. Front Immunol. 2023;14:1270081. doi:10.3389/fimmu.2023.1270081
107. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. doi:10.1038/nature11003
108. Planès R, Pinilla M, Santoni K, et al. Human NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Mol Cell. 2022;82(13):2385–2400.e9. doi:10.1016/j.molcel.2022.04.033
109. Bodnar-Wachtel M, Huber AL, Gorry J, et al. Inflammasome-independent NLRP3 function enforces ATM activity in response to genotoxic stress. Life Sci Alliance. 2023;6(4):e202201494. doi:10.26508/lsa.202201494
110. Gillette DD, Shah PA, Cremer T, et al. Analysis of human bronchial epithelial cell proinflammatory response to Burkholderia cenocepacia infection: inability to secrete il-1β. J Biol Chem. 2013;288(6):3691–3695. doi:10.1074/jbc.C112.430298
111. Wang J, Sahoo M, Lantier L, et al. Caspase-11-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLoS Pathog. 2018;14(5):e1007105. doi:10.1371/journal.ppat.1007105
112. Moretti J, Jia B, Hutchins Z, et al. Caspase-11 interaction with NLRP3 potentiates the noncanonical activation of the NLRP3 inflammasome. Nat Immunol. 2022;23(5):705–717. doi:10.1038/s41590-022-01192-4
113. Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–192.
114. Wang K, Sun Q, Zhong X, et al. Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell. 2020;180(5):941–955.e20. doi:10.1016/j.cell.2020.02.002
115. Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi:10.1038/nature18629
116. Bibo-Verdugo B, Snipas SJ, Kolt S, Poreba M, Salvesen GS. Extended subsite profiling of the pyroptosis effector protein gasdermin D reveals a region recognized by inflammatory caspase-11. J Biol Chem. 2020;295(32):11292–11302. doi:10.1074/jbc.RA120.014259
117. Devant P, Dong Y, Mintseris J, et al. Structural insights into cytokine cleavage by inflammatory caspase-4. Nature. 2023;624(7991):451–459.
118. Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity. 2015;43(5):923–932. doi:10.1016/j.immuni.2015.10.009
119. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–1153. doi:10.1016/j.immuni.2013.05.016
120. Deng M, Tang Y, Li W, et al. The endotoxin delivery protein HMGB1 mediates caspase-11-dependent lethality in sepsis. Immunity. 2018;49(4):740–753.e7. doi:10.1016/j.immuni.2018.08.016
121. Wang X, Li Z, Bai Y, et al. A small molecule binding HMGB1 inhibits caspase-11-mediated lethality in sepsis. Cell Death Dis. 2021;12(4):402. doi:10.1038/s41419-021-03652-5
122. Tang Y, Wang X, Li Z, et al. Heparin prevents caspase-11-dependent septic lethality independent of anticoagulant properties. Immunity. 2021;54(3):454–467.e6. doi:10.1016/j.immuni.2021.01.007
123. Vasudevan SO, Russo AJ, Kumari P, Vanaja SK, Rathinam VA. A TLR4-independent critical role for CD14 in intracellular LPS sensing. Cell Rep. 2022;39(5):110755. doi:10.1016/j.celrep.2022.110755
124. Vanaja SK, Russo AJ, Behl B, et al. Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell. 2016;165(5):1106–1119. doi:10.1016/j.cell.2016.04.015
125. Santos JC, Dick MS, Lagrange B, et al. LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. EMBO J. 2018;37(6):e98089. doi:10.15252/embj.201798089
126. Kumari P, Vasudevan SO, Russo AJ, et al. Host extracellular vesicles confer cytosolic access to systemic LPS licensing non-canonical inflammasome sensing and pyroptosis. Nat Cell Biol. 2023;25(12):1860–1872. doi:10.1038/s41556-023-01269-8
127. Huang LS, Hong Z, Wu W, et al. mtDNA Activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity. 2020;52(3):475–486.e5. doi:10.1016/j.immuni.2020.02.002
128. Srisaowakarn C, Pudla M, Ponpuak M, Utaisincharoen P. Caspase-4 mediates restriction of Burkholderia pseudomallei in human alveolar epithelial cells. Infect Immun. 2020;88(3):e00868–00819. doi:10.1128/IAI.00868-19
129. Dickinson MS, Kutsch M, Sistemich L, et al. LPS-aggregating proteins GBP1 and GBP2 are each sufficient to enhance caspase-4 activation both in cellulo and in vitro. Proc Natl Acad Sci U S A. 2023;120(15):e2216028120. doi:10.1073/pnas.2216028120
130. Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000;165(2):618–622. doi:10.4049/jimmunol.165.2.618
131. Arbibe L, Mira JP, Teusch N, et al. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat Immunol. 2000;1(6):533–540. doi:10.1038/82797
132. Parusel R, Steimle A, Lange A, et al. An important question: which LPS do you use. Virulence. 2017;8(8):1890–1893. doi:10.1080/21505594.2017.1361100
133. Gorman A, Golovanov AP. Lipopolysaccharide structure and the phenomenon of low endotoxin recovery. Eur J Pharm Biopharm. 2022;180:289–307. doi:10.1016/j.ejpb.2022.10.006
134. Borzęcka K, Płóciennikowska A, Björkelund H, Sobota A, Kwiatkowska K. CD14 mediates binding of high doses of LPS but is dispensable for TNF-α production. Mediators Inflamm. 2013;2013:824919. doi:10.1155/2013/824919
135. Yang N, Sin DD, Dorscheid DR. Various factors affect lipopolysaccharide sensitization in cell cultures. Biotechniques. 2020;69(2):126–132. doi:10.2144/btn-2020-0043
136. Tang PS, Tsang ME, Lodyga M, et al. Lipopolysaccharide accelerates caspase-independent but cathepsin B-dependent death of human lung epithelial cells. J Cell Physiol. 2006;209(2):457–467. doi:10.1002/jcp.20751
137. Wu S, Duan S, Zhao S, Cai Y, Chen P, Fang X. Atorvastatin reduces lipopolysaccharide-induced expression of cyclooxygenase-2 in human pulmonary epithelial cells. Respir Res. 2005;6(1):27. doi:10.1186/1465-9921-6-27
138. Liu S, Yang W, Li Y, Sun C. Fetal bovine serum, an important factor affecting the reproducibility of cell experiments. Sci Rep. 2023;13(1):1942. doi:10.1038/s41598-023-29060-7
139. Dong S, Khoo A, Wei J, et al. Serum starvation regulates E-cadherin upregulation via activation of c-Src in non-small-cell lung cancer A549 cells. Am J Physiol Cell Physiol. 2014;307(9):C893–899. doi:10.1152/ajpcell.00132.2014
140. Rashid MU, Coombs KM. Serum-reduced media impacts on cell viability and protein expression in human lung epithelial cells. J Cell Physiol. 2019;234(6):7718–7724. doi:10.1002/jcp.27890
141. Nakhjavani M, Nikounezhad N, Ashtarinezhad A, Shirazi FH. Human lung carcinoma reaction against metabolic serum deficiency stress. Iran J Pharm Res. 2016;15(4):817–823.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.