")
Back to Journals » Journal of Inflammation Research » Volume 18
Roles of Non-Coding RNA in Anthracycline Cardiotoxicity: A Narrative Review
Authors Xiao S , Li Y, Li B, Wang H, Wang K, Yang S
Received 19 March 2025
Accepted for publication 26 June 2025
Published 12 July 2025 Volume 2025:18 Pages 9129—9143
DOI https://doi.org/10.2147/JIR.S526611
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Shudan Xiao,* Yinghui Li,* Bo Li,* Haoxuan Wang, Kun Wang, SuMin Yang
Department of Cardiovascular Surgery, The Affiliated Hospital of Qingdao University, Institute of Chronic Diseases, College of Medicine, Qingdao University, Qingdao, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Kun Wang; SuMin Yang, Email [email protected]; [email protected]
Abstract: Anthracyclines, renowned for their efficacy and widespread application as antitumor agents, have substantially contributed to the enhancement of cancer patients’ survival rates. Nonetheless, anthracyclines predominantly account for the cardiotoxic effects, both acute and chronic, experienced by individuals undergoing cancer treatment. Anthracycline-induced cardiotoxicity (AIC) usually leads to progressive systolic dysfunction of the left ventricle, which may subsequently develop into heart failure and even death. Therefore, more stable, specific and sensitive auxiliary diagnostic biomarkers are needed. Non-coding RNAs, comprising long non-coding RNAs (lncRNAs), microRNAs (miRNAs), and circular RNAs (circRNAs), are pivotal in regulating cardiac metabolism, apoptosis, and oxidative stress, emerging as key targets for cardiovascular disease prevention and treatment. This review encapsulates the recent advancements in the field of non-coding RNA research in AIC, highlighting the prospect of non-coding RNAs as potential biomarkers and therapeutic targets for the condition. These molecules are known to regulate key cardiac processes including metabolism, apoptosis, and oxidative stress, which are central to the pathology of AIC. The potential of these molecules as diagnostic and prognostic indicators is significant, with miRNAs, for instance, being studied for their role in resistance to chemotherapeutic drugs and their application in therapeutic strategies. The review underscores the importance of understanding the intricate mechanisms by which non-coding RNAs influence gene expression and contribute to the development and progression of AIC, offering new avenues for targeted therapies and personalized treatment plans.
Keywords: anthracycline-induced cardiotoxicity, non-coding RNA, oxidative stress, apoptosis
Introduction
Anthracyclines, with doxorubicin as a notable example, have been approved by the US Food and Drug Administration for their exceptional anti-tumor efficacy and are widely utilized in cancer treatment, earning their place on the World Health Organization’s list of essential medicines, highlighting their indispensable role in addressing cancer globally.1–3 The anti-tumor mechanisms of anthracycline drugs include: 1) Anthracycline drugs can insert into the double strand of DNA, resulting in the distortion of the double helix structure of DNA, forming a stable anthracycline-DNA complex, blocking the replication of DNA and the transcription of RNA, and effectively curbs the multiplication of proliferation of cancer cells; 2) Anthracyclines disrupt DNA supercoiling by inhibiting topoisomerase II, thereby impeding DNA replication and transcription, and consequently, the proliferation of cancer cells; 3) Anthracyclines, by chelating with iron ions, create iron-anthracycline complexes that engage with oxygen, enhancing the generation of reactive oxygen species (ROS), which subsequently assail DNA, proteins, and cellular membranes, inflicting substantial cellular damage and further diminishing the viability of cancer cells.4,5 These mechanisms of action enable anthracycline drugs to effectively combat a variety of malignant tumors, such as breast cancer, multiple myeloma sarcoma, leukemia, lymphoma, gynecological cancer and other metastatic cancers. Among them, anthracyclines have significantly contributed to improved survival rates among cancer patients, particularly by raising the 5-year survival rate in childhood cancers to over 80%.6,7
Nonetheless, anthracyclines are a major contributor to both acute and chronic cardiotoxicity among oncology patients. AIC typically results in a progressive decline in the left ventricle’s systolic function, potentially escalating to heart failure, with chronic cardiotoxicity being responsible for mortality in approximately one-third of affected patients.8,9 The cardiotoxicity mechanism of anthracyclines involves oxidative stress (drug metabolism produces free radicals, reduces the level of antioxidant enzymes, and damages cardiomyocytes), abnormal iron metabolism (drugs activate iron regulatory proteins, increase intracellular iron deposition, catalyze oxygen formation, and damage myocardium), calcium overload (drugs activate Ca2+ channels, increase the concentration of Ca2+ in the cytoplasm, and affect myocardial function), DNA and chromatin damage (drugs embed in DNA, interfere with transcriptional replication, cause chromatin damage, and lead to apoptosis), and topoisomerase inhibition (drugs bind to enzymes to form covalent complexes, inhibit activity, and lead to DNA double-strand cleavage and cell dysfunction).9 Cardioprotective drugs are commonly used to treat patients with cardiotoxicity or at risk of cardiotoxicity, and these drugs can only provide symptomatic treatment.10 Despite their associated risks, anthracyclines continue to be a vital component in the therapeutic arsenal against a broad range of cancers.11 Therefore, more stable, specific and sensitive auxiliary diagnostic biomarkers are needed to regulate their expression and function to reduce or reverse the damage of drugs to cardiomyocytes.
At present, researchers are currently delving into novel diagnostic and therapeutic strategies to enhance the management of AIC, aiming to diversify and improve treatment options for this condition. Oncological cardiology, an emerging field, has investigated innovative therapeutic approaches to address AIC, offering fresh targets for both primary and secondary prevention of this adverse effect.
Within this domain, non-coding RNAs, such as lncRNAs, miRNAs, and circRNAs, are pivotal in modulating cardiac metabolism, ischemia, and inflammation, emerging as potent targets for the prophylaxis and management of cardiovascular diseases.12,13 This review encapsulates the recent advancements in the field of non-coding RNA research in AIC, highlighting the prospect of non-coding RNAs as potential biomarkers and therapeutic targets for the condition. These molecules are known to regulate key cardiac processes including metabolism, apoptosis, and oxidative stress, which are central to the pathology of AIC (Figure 1). The potential of these molecules as diagnostic and prognostic indicators is significant, with miRNAs, for instance, being studied for their role in resistance to chemotherapeutic drugs and their application in therapeutic strategies. The review underscores the importance of understanding the intricate mechanisms by which non-coding RNAs influence gene expression and contribute to the development and progression of AIC, offering new avenues for targeted therapies and personalized treatment plans.
 |
Figure 1 A summary of the relationship between non-coding RNA and AIC. Non-coding RNAs, comprising lncRNA, miRNA, and circRNA, are pivotal in regulating cardiac metabolism, apoptosis, and oxidative stress, emerging as key targets for AIC prevention and treatment. Abbreviations: AST, Astaxanthin; BMSC-Exos, Bone mesenchymal stem cell-derived exosomes; IPSC-MSCs, Human induced pluripotent stem cell-derived mesenchymal stem cells; IR, Irigenin; PEF, Paeoniflorin; RUT, Rutin; SMI, Shenmai Injection. |
miRNAs and Anthracycline Cardiotoxicity
miRNAs, conserved 20–22 nucleotide single-stranded ncRNAs, typically repress gene expression by binding to target mRNA, causing mRNA degradation or translation inhibition.14 The biogenesis of miRNAs begins with the transcription of pri-miRNAs in the cell nucleus by genes, which are then processed by the Drosha enzyme into pre-miRNAs.15 These pre-miRNAs are transported to the cytoplasm where they are further processed by Dicer into mature miRNAs.16 The mature miRNAs then bind to Argonaute proteins to form the RNA-Induced Silencing Complex (RISC), which specifically binds to the 3′UTR of target mRNAs to regulate gene expression.17,18 In the capacity of therapeutic targets for illnesses, miRNAs hold distinct benefits, especially in the realms of diagnosing and managing AIC. For instance, miRNAs, as prospective biomarkers, can function to gauge the susceptibility to and the effectiveness of treatments for AIC. When miRNA is central to AIC pathogenesis, pharmaceutical interventions can target its regulatory pathways, either by up-regulating or down-regulating miRNA expression to mitigate AIC. The significance of miRNA in the context of AIC is reflected in its multifaceted regulatory capabilities against cardiac toxicity. Firstly, miRNA can reduce oxidative stress levels, mitigate the accumulation of reactive oxygen species (ROS) caused by the drug, thereby protecting cardiomyocytes from damage. Additionally, by regulating apoptosis-related genes, miRNA inhibits unnecessary death of cardiomyocytes, helping to maintain cardiac function.19 miRNA also participates in modulating inflammatory responses, reducing inflammation-related cardiac toxicity.20 These advantages make miRNA a highly promising molecular target for the prevention and treatment of AIC, providing a scientific basis for the development of new therapies.
Oxidative Stress
The mechanism underlying oxidative stress is the disruption of equilibrium between ROS/reactive nitrogen species (RNS) and the antioxidant system.21 Anthracyclines bind to a specific part of the enzyme endothelial cell-specific nitric oxide synthase (eNOS) in endothelial cells, which results in more oxygen-derived free radicals and superoxide being produced, and less nitric oxide (NO) being made. When there is too much doxorubicin in the mitochondria, it causes a big rise in ROS levels.22 Anthracyclines, being positively charged, target the negatively charged phospholipid cardiolipin within the mitochondrial inner membrane, forming a stable complex. This complex is susceptible to ROS-induced peroxidation, which can trigger the release of cytochrome c from mitochondria, ultimately causing cell damage.22,23 Various miRNAs such as miR-140-5p,24 miR-152,25 miR-200a,26 miR-24-3p,27 miR-375,28 miR-143,29 miR-128-3p30 have been associated with the modulation of oxidative stress in AIC (Figure 2).
 |
Figure 2 miRNA anthracyclines cardiotoxicity oxidative stress pathway. Anthracyclines regulate the generation of reactive oxygen species (ROS) by affecting multiple microRNAs (miRNAs) such as MiR-152, MiR-140-5p, MiR-200a, MiR-24-3P, MiR-375, MiR-143, and MiR-128-3P. These miRNAs, in turn, modulate the expression of key proteins like Keap1, Nrf2, GSH, PDK1, AKT, and PPAR-γ, thereby influencing ROS levels and impacting cellular oxidative stress responses. Abbreviations: AKT, Protein kinase B; GSH, Glutathione; Keap1, Kelch-like ECH-associated protein 1; Nrf2, nuclear erythroid factor 2-related factor 2; PDK1, phosphoinositide-dependent protein kinase 1; PPAR-γ, Peroxisome proliferator-activated receptorγ. |
For example, nuclear erythroid factor 2-related factor 2 (Nrf2), which belongs to the CNC transcription factor family, manages the cell’s antioxidant defense by adjusting its interaction with kelch-like ECH-associated protein 1 (Keap1).31,32 MiR-140-5p is significantly upregulated in response to DOX. Dual-luciferase reporter assays demonstrate that it directly targets Nrf2 and silent information regulator factor 2-related enzyme 2 (Sirt2), leading to myocardial oxidative damage. Nrf2 negatively regulates the polymerization or dissociation of Keap1, thereby influencing the expression of various antioxidant genes and enzymes that combat oxidative stress. Meanwhile, Sirt2 modulates oxidative stress by activating Forkhead box O3, which in turn upregulates superoxide dismutase (SOD) and reduces ROS levels.24 However, Zhang et al25 found that miR-152 is down-regulated in response to DOX treatment, which silences Keap1 and activates Nrf2 leading to decreased oxidative stress and apoptosis, providing a new therapeutic target for the treatment of DOX-related cardiac injury. Similarly, miR-200a26/MiR-24-3p27 was down-regulated in DOX and alleviated oxidative stress by up-regulating Nrf2. Protein kinase B (AKT), a principal modulator of apoptosis, has been discovered in new research to additionally manage oxidative stress triggered by DOX.33 MiR-375 levels was up-regulated after doxorubicin treatment, resulting in down-regulation of 3-phosphoinositide-dependent protein kinase 1 (PDK1) and inactivation of AKT, which promoted the development of doxorubicin-induced cardiotoxicity (DIC) oxidative damage in mice.28 Following doxorubicin treatment, miR-143 is up-regulated, leading to AKT inactivation, and this increase in miR-143 enhances oxidative stress and cardiomyocyte apoptosis through AKT inhibition.29 The transcriptional regulator peroxisome proliferator-activated receptor gamma (PPAR-γ) is significant for alleviating oxidative stress in the heart and for diminishing cardiac injury induced by DOX in mice; MiR-128-3p was found to be up-regulated following DOX administration, which reduced the expression of PPAR-γ.30
Cardiomyocyte Apoptosis
After anthracycline drugs enter cardiomyocytes, oxygen free radicals are produced through enzymatic and non-enzymatic pathways, resulting in lipid peroxidation of mitochondria and microsomes, and simultaneous inhibition of topoisomerase II and DNA binding to form a cleavage complex, resulting in DNA double-strand breaks, further affecting mitochondrial permeability, leading to mitochondrial function damage and cardiolipin peroxidation, resulting in cytochrome c release that activates the caspase-dependent apoptotic pathway and ultimately results in cardiomyocyte apoptosis.34,35 This process involves the interaction of multiple mechanisms, which together cause cardiotoxicity. Several miRNAs, including miR-495-3p,36 miR-25,37 miR-34a,38 miR-29b,39 miR-22,40 miR-200a-3p,41 miR-494-3p,42 miR-15b-5p,43 miR-34a-5p,44 miR-124,45 miR-21,46 miR-432-5p,47 miR-129-1-3p,48 miR-30,49 miR-23a,50 miR-133b,51 have been associated with cardiomyocyte apoptosis (Figure 3).
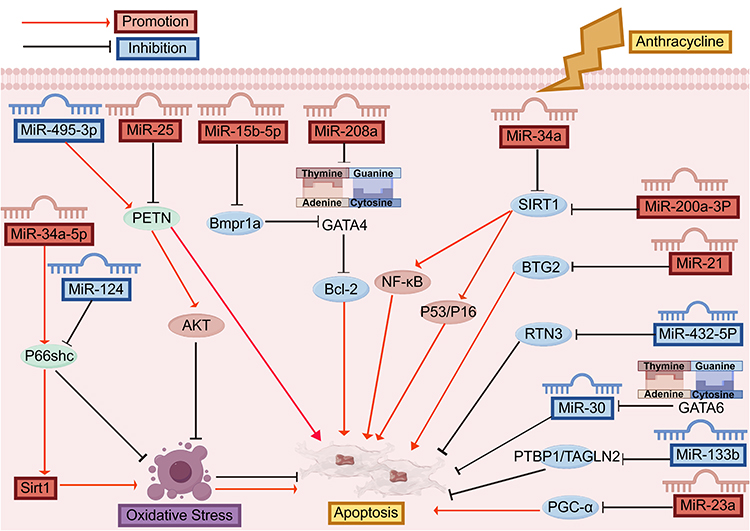 |
Figure 3 miRNA anthracyclines cardiotoxicity apoptosis pathway. Anthracycline-induced cardiotoxicity and apoptosis in cardiomyocytes involve multiple microRNAs (miRNAs) such as MiR-495-3p, MiR-25, MiR-15b-5p, MiR-208a, MiR-34a, MiR-200a-3P, MiR-21, MiR-432-5P, MiR-30, MiR-133b, and MiR-23a, which regulate key signaling molecules like PETN, Bmpr1a, GATA4, Bcl-2, NF-κB, P53/P16, SIRT1, BTG2, RTN3, PTBP1/TAGLN2, and PGC-α, further affecting oxidative stress and apoptosis processes. Abbreviations: AKT, Protein kinase B; Bcl-2, B-cell lymphoma-2; Bmpr1a, Bone Morphogenetic Protein Receptor Type 1A; BTG2, B cell translocation gene 2; GATA4, GATA Binding Protein 4; GATA-6, GATA Binding Protein 6; NF-κB, Nuclear Factor-kappa B; p53, proteins tumor protein p53; P66Shc, P66 Shc Protein; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator-1α; PTBP1, polypyrimidine tract binding protein 1; PETN, Phosphatidylethanolamine-binding protein; p16, p16 Inhibitor of Cyclin-Dependent Kinase 4a; RTN3, Reticulon3; SIRT1, silent mating type information regulation 2 homolog-1; TAGLN2, Transgelin-2. |
For instance, phosphate and tension homology deleted on chromosome ten (PTEN), a key suppressor of the phosphorylation and activation of protein kinase B (PKB/AKT), is upregulated in hearts treated with DOX, thereby diminishing AKT’s activation and intensifying oxidative stress-induced cardiomyocyte apoptosis;52 DOX downregulates miR-495-3p, which directly targets PTEN’s 3′-UTR, reducing PTEN expression, activating the AKT pathway, and protecting against DOX-induced oxidative stress and cardiac dysfunction.36 Li et al37 determined that miR-25 inhibition reduced the levels of phosphorylated phosphatidylinositol 3-kinase (p-PI3K) and phosphorylated AKT (p-AKT) in rats. B-cell lymphoma-2 (Bcl-2), an anti-apoptotic protein essential for cell survival in the heart, reduces DOX-induced cardiac cell apoptosis by activating silent mating type information regulation 2 homolog-1 (SIRT1), which decreases the acetylation levels of target proteins such as tumor protein p53 (p53), SMAD family member 2 and 3 (SMAD2/3), and Nuclear Factor-kappa B (NF-κB), thereby inactivating their downstream pathways and alleviating myocardial damage caused by DOX; The upregulation of miR-34a in DIC, and silencing miR-34a caused an increase in its pro-survival targets Bcl-2 and SIRT1.38 Downregulation of miR-29b was accompanied by decreased Bcl-2 and increased Bax; miR-29b agomir treatment reversed this, enhancing Bcl-2 and diminishing Bax.39 In DOX-treated mouse hearts, miR-22 is markedly upregulated, and it binds directly to the 3′-untranslated region (3′-UTR) of Sirt1, leading to the downregulation of SIRT1.40 MiR-200a-3p exhibits high levels of expression in DIC, and miR-200a-3p inhibitors can negatively target paternally expressed gene 3 (PEG3) by up-regulating SIRT1 and down-regulating NF-κB to promote cardiomyocyte recovery.41 SIRT1 catalyzes the deacetylation of p53, a non-histone protein target; MDM4 messenger ribonucleic acid (MDM4) is an important upstream regulator of p53, and its high expression can inhibit apoptosis and promote cell proliferation, while THP can promote the expression of miR-494-3p, inhibit the expression of MDM4 mRNA, and promote the expression of p53.42 GATA Binding Protein 4 (GATA4), a transcriptional regulator predominantly found in the heart, is a key element in cardiac-specific gene expression, which is related to cardiomyocyte apoptosis; The up-regulation of miR-208a expression by DOX led to the down-regulation of GATA4, and the down-regulation of GATA4 led to the decrease of Bcl-2, followed by the increase of apoptosis.53 DOX caused a 2.46-fold up-regulation of miR-15b-5p and concurrently led to a significant reduction in the expression of bone morphogenetic protein receptor type 1a (Bmpr1a), a key receptor in the anaplastic lymphoma kinase (ALK) pathway, along with its downstream transcription factor GATA4 in H9c2 cardiomyocytes.43
P66 Shc Protein (P66Shc) is involved in the production of endogenous ROS and apoptosis.54 After anthracycline treatment, plasma miR-34a-5p increased, P66shc protein expression increased significantly, and Sirt1 expression decreased significantly.44 MiR-124 is down-regulated under DOX induction and can bind to the 3′UTR of Shc1 (gene encoding p66Shc) to reduce oxidative stress damage by inhibiting the p66Shc signaling pathway.45 The B-cell translocation gene 2 (BTG2), a member of the anti-proliferative (APRO) gene family, plays a role in the differentiation, growth, repair of DNA damage, and programmed cell death of cancer cells;55 Tong et al46 found that In the DOX group, miR-21’s expression notably rose to 1.8 times, while miR-21 markedly reduced BTG2 protein levels and encouraged cardiomyocyte apoptosis. Belonging to the RTN family, Reticulon3 (RTN3) predominantly resides in the endoplasmic reticulum and plays a role in apoptosis. When stimulated by Adriamycin, there was a notable reduction in miR-432-5p expression, leading to diminished stress in the endoplasmic reticulum and its autophagy by lowering RTN3 protein levels, impacting cardiomyocyte metabolism.47 The N-methyl-D-aspartate type subunit 2D (GRIN2D), a component of the NMDA (N-methyl-D-aspartate) receptor complex, creates a ligand-gated ion channel with significant calcium permeability. In a rat myocardial injury model induced by Pirarubicin (THP), THP reduced miR-129-1-3p levels, while miR129-1-3p directly controlled Glutamate Receptor, Ionotropic, GRIN2D, enhancing calcium overload and the apoptosis of cardiomyocytes triggered by THP attack.48 Cardiomyocytes increased sharply, and GATA-6 can inhibit the expression of miR-30 family, leading to DOX-induced down-regulation of miR-30, which is involved in the complex catecholamine/adrenergic pathway.
The heart’s GATA Binding Protein 6 (GATA-6), a transcription factor, shows high expression levels. Post-DOX treatment, there’s a significant rise in GATA-6 in cardiomyocytes, and GATA-6 can suppress miR-30 family expression, resulting in the DOX-triggered reduction of miR-30, a key player in the intricate catecholamine/adrenergic pathway.49 Furthermore, the expression of MiR-23a increased in cardiomyocytes treated with DOX, leading to doxorubicin-induced heart toxicity through the suppression of the peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α)/dynamin-related protein-1 (p-Drp1) route, which in turn triggered mitochondrial-dependent apoptosis.50 The Polypyrimidine tract binding protein 1 (PTBP1), or heterogeneous ribonucleoprotein 1, triggers apoptotic signals, leading to cardiomyocyte death; doxorubicin decreased miR-133b levels in HL-1 cardiomyocytes and cardiac tissues, while heightened PTBP1 levels negated miR-133b’s impact on apoptosis.51
lncRNAs and Anthracycline Cardiotoxicity
Non-coding RNA molecules known as lncRNAs, exceeding 200 nucleotides in length,56 play a crucial role in controlling gene expression and are significant in multiple biological functions, such as the emergence of AIC.57 In the context of cardiotoxicity of anthracycline drugs, lncRNA plays a role by regulating the expression of genes related to cardiac stress response, cell survival and cell death pathways.58 As a therapeutic target for cardiotoxicity of anthracycline drugs, lncRNA has some advantages in reducing drug-induced heart damage. As an instance, certain lncRNA may serve as a prospective biomarker for evaluating cardiotoxicity risks in patients treated with anthracyclines.59 Currently, research is underway on lncRNA as a potential target for the heart-damaging effects of anthracycline medications, concentrating on its involvement in anti-oxidative stress and cell death causing cardiac damage due to drugs. By targeting lncRNA, it is possible to develop new strategies to protect the heart from the toxic effects of anthracyclines, thereby improving the safety of cancer treatment60 (Figure 4).
 |
Figure 4 Involvement of lncRNA in the pathogenesis of AIC. Anthracyclines regulate the levels of reactive oxygen species (ROS) and apoptosis processes in cardiomyocytes by affecting the interactions between long non-coding RNAs (InRNA-P21, InRNA NORAD, InRNA PVT1, InRNA RMRP, InRNA 00339, InRNA Miat) and microRNAs (MiR-187-3P, MiR-484, and MiR-129-1-3p), and through various signaling pathways (Wnt/β-catenin, AGO1, P53, Apaf-1), collectively modulating the oxidative stress response and apoptosis in cardiomyocytes. Abbreviations: Apaf-1, apoptotic protease activating factor-1; AGO1, Argonaute 1; Nrf2, nuclear erythroid factor 2-related factor 2; p53, proteins tumor protein p53; PFN1, Profilin 1; Wnt/β-catenin, Wnt/β-catenin signaling pathway. |
Oxidative Stress
ROS plays an important role in the development of AIC. Activation of the Wnt/β-catenin signaling pathway in cardiomyocytes promotes cellular protective response and reduces oxidative stress-related damage;61 In the study of DIC, Xie et al62 found that the expression of lncRNA-p21 was increased, which could regulate oxidative stress through the Wnt/β-catenin signaling pathway, thereby protecting DOX-induced cardiac agingIn addition, the expression of lncRNA Mhrt in myocardial tissue decreased after DOX treatment, and overexpression of Mhrt promoted binding of H3 histone to the Nrf2 promoter region, enhanced Nrf2 transcription activity, and reduced oxidative stress damage in myocardial cells.63 These findings reveal the protective effect of lncRNA in DIC and provide new molecular targets for future cardiac protection strategies.
Cardiomyocyte Apoptosis
Researches have shown that AIC death involves a variety of pathways.64 In the pathogenesis of cardiovascular disease, lncRNA plays a role by regulating apoptosis. In particular, lncRNA NORAD, which promotes apoptosis through the miR-22-3p/PTEN axis and the miR-577/cordon-bleu WH2 repeat protein like 1 (COBLL1) axis, aggravates the progression of myocardial infarction, and provides a new direction for research.65,66 Recent studies have also shown that NORAD inhibits mitochondrial ROS levels, inhibits mitochondrial apoptotic protein apoptotic proteinase activator 1 (APAF-1), and inhibits classical apoptotic pathways following DOX intervention in cardiomyocytes.67 WNT1 induces signaling pathway protein 1 (WISP1), a multifunctional signaling protein, to participate in cell protection, cell proliferation, and extracellular matrix generation by interacting with multiple signaling pathways such as Wnt signaling, Notch1, and TGF-β;68–70 To explore the mechanism of action of DIC, specific lncRNA FOXC2-AS1 expression was reduced in mouse heart tissue, while overexpression of lncRNA FOXC2-AS1 enhanced survival rate of mouse cardiomyocytes, upregulated WISP1 expression, and provided a defense mechanism for Myocytes to reduce the cardiotoxic effect of DOX.71 Similarly, in cardiomyocytes, the increase of Argonaute 1 (AGO1) is related to the aggravation of apoptosis; Zhan et al found that the expression level of lncRNA PVT1 was significantly increased in the study of cardiotoxicity, and it formed a molecular sponge by binding to miR-187-3p, thereby reducing the activity of miR-187-3p, which in turn led to the up-regulation of AGO1 protein expression and increased cardiomyocyte apoptosis.58 Li et al found that in rat cardiomyocytes and H9c2 cell models, the expression level of lncRNA LINC00339 increased significantly under the stimulation of DOX, and it affected the regulation of miR-484 on downstream target genes by adsorbing miR-484, thereby inhibiting the apoptosis and proliferation of DOX-treated cardiac cells.72 In addition, lncRNA RMRP directly acts on the coding sequence (CDS) region of Profilin 1 (PFN1) mRNA, thereby inhibiting the expression of PFN1, reducing p53 protein and its phosphorylation level, and reducing apoptosis.73 Similarly, expression of lncRNA Miat was significantly increased in heart muscle cells after THP treatment, and miR-129-1-3p inhibition reduced THP-induced cardiomyocyte injury, thereby inhibiting apoptosis and reducing oxidative stress and calcium overload, providing a novel molecular target for future therapeutic strategies.74
Circular RNAs and Anthracycline Cardiotoxicity
circRNA is a class of non-coding, covalently closed single-stranded RNA with obvious tissue-specific and cell-specific expression patterns.75 As microRNA sponges, protein scaffolds, transcriptional regulators, protein synthesis templates, circRNAs have different biological functions in various tissues and organs including the cardiovascular system.76 For example, the CircSlc8a1-miR-133a interaction regulates cardiac hypertrophy and dilation.77 However, there are few studies on the involvement of circRNA in regulating the cardiotoxicity of anthracycline drugs (Figure 5).
 |
Figure 5 Involvement of circRNA in the pathogenesis of AIC. Anthracyclines affect the interaction between miRNAs (MiR-143, MiR-31-5p, MiR-1303, MiR-409-3p, MiR-107, and MiR-135a-5p) and their corresponding circRNAs (circ-0006332, circ-Pan3, circ-SKA3, circ-0001312, circ-LTBP1, and circ-Arhgap12), thereby influencing signaling pathways such as TLR2, TLR4, HMGB1, and QKI to promote or inhibit the generation of reactive oxygen species (ROS), which in turn affects the processes of apoptosis and pyroptosis in cells. Abbreviations: Brca1, breast cancer type 1 susceptibility protein; HMGB1, high-mobility group box 1; QKI, Quaking I; TLR2, Toll-like receptor 2; TLR4, toll-like receptor 4. |
Quaking I (QKI), an RNA-binding protein (RBP), regulates circRNA biogenesis, although its upstream regulatory mechanisms remain unclear and require further investigation;78 DOX treatment was found to increase miR-31-5p expression in cardiomyocytes and mouse heart tissues, directly targeting QKI. This resulted in the downregulation of circPan3, which was confirmed to be mediated by QKI silencing through miR-31-5p.79 In addition, Gupta et al80 demonstrated that AAV9-mediated cardiac overexpression of Qki5 prevented DOX-induced apoptosis and improved cardiac function. Mechanistically, lentivirus-mediated overexpression and CRISPR/Cas9 silencing of Qki5 revealed its regulation of specific circular RNAs derived from Ttn, Fhod3, and Strn. Inhibiting Ttn-derived circRNAs increased cardiomyocyte susceptibility to DOX. Lu et al81 identified circ-INSR as a highly conserved circRNA downregulated in DOX-induced cardiotoxicity (DIC) through circRNA sequencing. They showed that breast cancer type 1 susceptibility protein (Brca1) promoted circ-INSR formation, providing significant protective effects against DIC in rodents, human cardiomyocytes in vitro, and a chronic DIC mouse model. Conversely, circArhgap12 was upregulated in mouse heart tissue following DOX treatment, enhancing apoptosis rates. miR-135a-5p was shown to directly target circArhgap12, mitigating DOX-induced oxidative stress and apoptosis.82 In DOX-treated AC16 cells, circ-SKA3 was up-regulated and toll-like receptor 4 (TLR4) was up-regulated by targeting miR-1303, which promoted myocardial injury.83 DOX treatment also induced the expression of circ-0001312, which decreased miR-409-3p levels in cardiomyocytes but increased high-mobility group box 1 (HMGB1) expression. Functionally, miR-409-3p inhibition weakened the protective effect of circ-0001312 silencing, while HMGB1 overexpression counteracted these effects.84 Similarly, circ-LTBP1 expression was increased in DOX-treated AC16 cells, along with upregulated miR-107 and adenylate cyclase 1 (ADCY1). Silencing circ-LTBP1 reversed ADCY1 upregulation induced by DOX, and miR-107 inhibition neutralized the effects of circ-LTBP1 silencing.85 Moreover, circ-ZNF609 expression was significantly elevated in DIC, and its inhibition blocked m6A methylation increases. RNA m6A demethylase FTO was identified as a downstream factor of circ-ZNF609, and FTO inhibition abolished the protective effects of circ-ZNF609 knockdown, highlighting the role of circRNAs and m6A modifications in DIC.86 Finally, the study of Li et al87 showed that circ-0000098 inhibition in liver cancer cells reduced DOX resistance by lowering P-glycoprotein (P-gp, MDR1) expression and intracellular ATP levels. Differently, Zhang et al88 showed that circ-0006332 expression was significantly elevated in the myocardial tissues of DOX-treated rats, while miR-143 expression was decreased. Circ-0006332 stimulated cardiomyocyte pyroptosis by downregulating miR-143 and upregulating Toll-like receptor 2 (TLR2), thereby exacerbating DIC injury.
Treatment and Clinical Application
The translation of ncRNA research into clinical practice has opened new avenues for early diagnosis, prognosis, and therapeutic intervention in AIC. As mentioned above, ncRNA demonstrate tissue-specific and disease-specific expression patterns, making them ideal biomarkers. Not only that, it also plays a huge role in exosome therapy and drug therapy. As shown in Table 1.
 |
Table 1 Regulatory Effects of Treatment and Clinical Application |
Exosome Therapy
Exosomes Exo-and B-exo have a disc-shaped double-layer membrane structure, and after B-exo genetic engineering, miRNA-499a-5p delivery increased significantly and reached the heart. C-B-exo-miRNA-499a-5p can significantly reduce myocardial enzymes, serum, and myocardial cell factors, improve cardiac pathological changes, and inhibit Cluster of Differentiation 38 (CD38)/Mitogen-Activated Protein Kinase (MAPK) /NF-κB signaling pathway. In the DOX-induced cardiotoxicity model, miRNA-499a-5p was significantly down-regulated.90 By blocking the Vascular Peroxidase 1 (VPO1)/Extracellular Signal-Regulated Kinase (ERK) pathway, miR-9-5p extracted from human induced pluripotent stem cell-derived mesenchymal stem cells (iPSC-MSCs) and transferred from EXOs (iPSC-MSC-EXOs) reduces cardiomyocyte senescence and protects against DIC damage.91 Recent studies have revealed that lncRNA GHET1 expressed by Bone mesenchymal stem cell-derived exosomes (BMSC-Exos) can significantly inhibit pyroptosis, thus effectively preventing DOX-induced cardiotoxicity. Mechanistically, lncRNA GHET1 exerts its protective effect by reducing the expression of nod-like receptor protein 3 (NLRP3) inflammasome and binding to Insulin-like Growth Factor 2 mRNA-Binding Protein 1 (IGF2BP1).89 Sirt2, a crucial cellular deacetylase, plays a significant role in regulating antioxidant defenses and maintaining metabolic homeostasis within cells;92,93 Exosomal MIF mitigates DIC dysfunction by delivering lncRNA-NEAT1, while miR-221-3p targets the Sirt2 3′-untranslated region. Silencing lncRNA-NEAT1 in mesenchymal stem cells (MSCs), along with miR-221-3p overexpression and Sirt2 knockdown in cardiomyocytes, diminishes the anti-DOX aging effects mediated by exosomal MIF.94 Furthermore, the elevation of lncRNA-MALAT1 in exosomes facilitates its binding to miR-92a-3p, targeting the 3′UTR of Autophagy Related 4A Cysteine Peptidase (ATG4a) and activating ATG4a expression. This process enhances mitochondrial metabolism and suppresses DOX-induced cellular senescence.95
Drug Therapy
Rich in fruits and vegetables, rutin (RUT) is a significant edible flavonoid. In contrast to the THP model group, RUT significantly decreased ROS and apoptosis in HL-1 cells. Additionally, RUT significantly inhibited the expression level of miR-125b-1-3p, which in turn increased the expression of JunD proto-oncogene (JunD), altered the expression levels of Bax, Bcl-2, cleaved caspase-3, and cleaved caspase-9, and decreased THP-induced myocardial oxidative stress and apoptosis.96 Astaxanthin (AST) lowers THP-induced damage to H9c2 cells, inhibits p53, up-regulates MDM4, and down-regulates miR-494-3p to prevent THP-induced apoptosis of H9c2 cells.97 Shengmai Powder Injection produces Shenmai Injection (SMI), which is frequently used to treat cardiovascular conditions and malignancies; Cell survival, miR-30a expression, Beclin-1, microtubule-associated protein light chain 3 II (LC3-II), LC3-II/microtubule-associated protein light chain 3 I (LC3-I) expression, sequestosome 1 (p62) protein expression, apoptosis rate, and Bcl2 expression all increased in the SMI + miR-30a inhibitor group.98 The cardiovascular system benefits from the monoterpene glycoside known as paeoniflorin (PEF), which is isolated from the dry roots of Paeonia. DOX markedly increased the expression of miR-1, decreased the expression of Bcl-2, and was greatly suppressed by cells that had been pretreated with PEF.99 Our study showed that tanshinone IIA effectively improved cardiomyocyte apoptosis by inducing miR-133 and inhibiting caspase-9. Tanshinone IIA is the raw material of Salvia miltiorrhiza, whose roots have high medicinal value in traditional Chinese medicine and have been used to treat cardiovascular diseases.100 Irigenin (IR), which is extracted from the rhizome of Belamcanda chinensis, has the ability to stop the growth of cancer cells. It can also raise the expression of miR-425 in mouse heart and cardiomyocytes treated with DOX and decrease Receptor-interacting protein kinase 1 (RIPK1), which prevents inflammation, oxidative stress, and apoptosis.101
Conclusion and Prospect
The role of ncRNA in the pathogenesis of AIC in recent years has been thoroughly analyzed. Non-coding RNAs have emerged as critical regulators of apoptosis and oxidative stress, two fundamental mechanisms driving AIC. In addition, non-coding RNAs demonstrate high specificity and sensitivity in reflecting the molecular changes associated with AIC, making them promising candidates for early detection and disease monitoring. Despite the promising role of ncRNAs in AIC, several challenges remain. These include the need for large-scale clinical studies to validate ncRNA biomarkers, understanding the long-term effects of ncRNA-based therapies, and developing efficient delivery systems for RNA therapeutics. Additionally, the interplay between ncRNAs and other molecular pathways in AIC requires further exploration to uncover synergistic therapeutic targets. Overall, non-coding RNAs represent a transformative avenue for addressing AIC, providing a promising direction for the development of novel diagnostics and therapeutics. Their ability to bridge the gap between molecular pathogenesis and clinical application underscores their potential to revolutionize the management of cardiotoxicity in oncology patients. Continued research into ncRNAs will likely yield critical insights and groundbreaking treatments, paving the way for safer and more effective cancer therapies.
Abbreviation
AIC, Anthracycline-induced cardiotoxicity; AKT, Protein kinase B; APRO, anti-proliferative; Apaf-1, apoptotic protease activating factor-1; ADCY1, adenylate cyclase 1; AST, Astaxanthin; ALK, anaplastic lymphoma kinase; AGO1, Argonaute 1; ATG4a, Autophagy Related 4A Cysteine Peptidase; Bcl-2, B-cell lymphoma-2; Bmpr1a, Bone Morphogenetic Protein Receptor Type 1A; BTG2, B cell translocation gene 2; Brca1, breast cancer type 1 susceptibility protein; circRNA, circular RNA; CDS, coding sequence; COBLL1, cordon-bleu WH2 repeat protein like 1; CD38, Cluster of Differentiation 38; eNOS, endothelial cell-specific nitric oxide synthase; ERK, Extracellular Signal-Regulated Kinase; FTO, fat mass and obesity associated gene; JunD, JunD proto-oncogene; GRIN2D, glutamate ionotropic receptor N-methyl-D-aspartate type subunit 2D; GATA4, GATA Binding Protein 4; GATA-6, GATA Binding Protein 6; HMGB1, high-mobility group box 1; IPSC-MSCs, human induced pluripotent stem cell-derived mesenchymal stem cells; IR, Irigenin; IGF2BP1, Insulin-like Growth Factor 2 mRNA-Binding Protein 1; Keap1, Kelch-like ECH-associated protein 1; lncRNAs, long non-coding RNAs; LC3-II, microtubule-associated protein light chain 3 II; LC3-I, microtubule-associated protein light chain 3 I; MAPK, Mitogen-Activated Protein Kinase; miRNAs, microRNAs; MDM4, MDM4 messenger ribonucleic acid; Nrf2, nuclear erythroid factor 2-related factor 2; NF-κB, Nuclear Factor-kappa B; NMDA, N-methyl-D-aspartate; NLRP3, nod-like receptor protein 3; PDK1, phosphoinositide-dependent protein kinase 1; PPAR-γ, Peroxisome proliferator-activated receptor γ; PTEN, phosphate and tension homology deleted on chromosome ten; PKB, protein kinase B; p-PI3K, phosphotylinosital 3-kinase; p-AKT, phosphorylated AKT protein; p53, proteins tumor protein p53; P66Shc, P66 Shc Protein; PEG3, Paternally expressed gene 3; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator-1α; p-Drp1, dynamin-related protein-1; PTBP1, polypyrimidine tract binding protein 1; P-gp, P-glycoprotein; PFN1, Profilin 1; p62, sequestosome 1; ROS, reactive oxygen species; RIPK1, Receptor-interacting protein kinase 1; QKI, Quaking I; RNS, reactive nitrogen species; RTN3, Reticulon3; RUT, Rutin; RBP, RNA-binding protein; Sirt2, silent information regulator factor 2-related enzyme 2; SOD, superoxide dismutase; SIRT1, silent mating type information regulation 2 homolog-1; SMAD2/3, SMAD family member 2 and 3; SMI, Shenmai Injection; THP, Pirarubicin; TLR4, toll-like receptor 4; TLR2, Toll-like receptor; VPO, Vascular Peroxidase 1; WISP1, WNT1 inducible signaling pathway protein 1.
Data Sharing Statement
From PubMed Data base.
Acknowledgments
Fig Draw 2.0. drawing software was used for this research.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Open Project Program of State Key Laboratory of Frigid Zone Cardiovascular Diseases (SKLFZCD), Harbin Medical University (HDHY2024007); National Natural Science Foundation of China (82370291); Qingdao Science and Technology Benefiting the People Demonstration Project (24-1-8-smjk-7-nsh); Major Basic Research Projects in Shandong Province (ZR2024ZD46), Taishan Scholar Distinguished Expert.
Disclosure
The authors declare no competing interests.
References
1. Mattioli R, Ilari A, Colotti B, Mosca L, Fazi F, Colotti G. Doxorubicin and other anthracyclines in cancers: activity, chemoresistance and its overcoming. Mol Aspects Med. 2023;93:101205. doi:10.1016/j.mam.2023.101205
2. Abrahams C, Woudberg NJ, Lecour S. Anthracycline-induced cardiotoxicity: targeting high-density lipoproteins to limit the damage? Lipids Health Dis. 2022;21(1):85. doi:10.1186/s12944-022-01694-y
3. Curry HL, Parkes SE, Powell JE, Mann JR. Caring for survivors of childhood cancers: the size of the problem. Eur J Cancer. 2006;42(4):501–508. doi:10.1016/j.ejca.2005.11.003
4. Huang J, Wu R, Chen L, Yang Z, Yan D, Li M. Understanding Anthracycline Cardiotoxicity From Mitochondrial Aspect. Front Pharmacol. 2022;13:811406. doi:10.3389/fphar.2022.811406
5. Saleh Y, Abdelkarim O, Herzallah K, Abela GS. Anthracycline-induced cardiotoxicity: mechanisms of action, incidence, risk factors, prevention, and treatment. Heart Fail Rev. 2021;26(5):1159–1173. doi:10.1007/s10741-020-09968-2
6. Narezkina A, Narayan HK, Zemljic-Harpf AE. Molecular mechanisms of anthracycline cardiovascular toxicity. Clin Sci. 2021;135(10):1311–1332. doi:10.1042/CS20200301
7. Bernstein D. Anthracycline Cardiotoxicity: worrisome Enough to Have You Quaking? Circ Res. 2018;122(2):188–190. doi:10.1161/CIRCRESAHA.117.312395
8. Cardinale D, Iacopo F, Cipolla CM. Cardiotoxicity of anthracyclines. Front Cardiovascular Med. 2020;7:26. doi:10.3389/fcvm.2020.00026
9. Dempke WCM, Zielinski R, Winkler C, Silberman S, Reuther S, Priebe W. Anthracycline-induced cardiotoxicity - are we about to clear this hurdle? Eur J Cancer. 2023;185:94–104. doi:10.1016/j.ejca.2023.02.019
10. Rawat PS, Jaiswal A, Khurana A, Bhatti JS, Navik U. Doxorubicin-induced cardiotoxicity: an update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed Pharmacother. 2021;139:111708. doi:10.1016/j.biopha.2021.111708
11. Aix SP, Ciuleanu TE, Navarro A. Combination lurbinectedin and doxorubicin versus physician’s choice of chemotherapy in patients with relapsed small-cell lung cancer (ATLANTIS): a multicentre, randomised, open-label, Phase 3 trial. Lancet Respir Med. 2023;11(1):74–86. doi:10.1016/S2213-2600(22)00309-5
12. Täubel J, Hauke W, Rump S. Novel antisense therapy targeting microRNA-132 in patients with heart failure: results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur Heart J. 2021;42(2):178–188. doi:10.1093/eurheartj/ehaa898
13. Batkai S, Genschel C, Viereck J. CDR132L improves systolic and diastolic function in a large animal model of chronic heart failure. Eur Heart J. 2021;42(2):192–201. doi:10.1093/eurheartj/ehaa791
14. Wang L, Lv Y, Li G, Xiao J. MicroRNAs in heart and circulation during physical exercise. J Sport Health Sci. 2018;7(4):433–441. doi:10.1016/j.jshs.2018.09.008
15. Bartel DP. Metazoan MicroRNAs. Cell. 2018;173(1):20–51. doi:10.1016/j.cell.2018.03.006
16. Nguyen T, Park J, Dang T, Choi Y-G, Kim V. Microprocessor depends on hemin to recognize the apical loop of primary microRNA. Nucleic Acids Res. 2018;46(11):5726–5736. doi:10.1093/nar/gky248
17. Alisi L, Giovannetti F, Armentano M, Lucchino L, Lambiase A, Bruscolini A. Challenging corneal diseases and microRNA expression: focus on rare diseases and new therapeutic frontiers. Surv Ophthalmol. 2025;70(1):121–131. doi:10.1016/j.survophthal.2024.09.002
18. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15(8):509–524. doi:10.1038/nrm3838
19. Kuang Z, Wu J, Tan Y, Zhu G, Li J, Wu M. MicroRNA in the Diagnosis and Treatment of Doxorubicin-Induced Cardiotoxicity. Biomolecules. 2023;13(3):568. doi:10.3390/biom13030568
20. Chen L, Xu Y. MicroRNAs as Biomarkers and Therapeutic Targets in Doxorubicin-Induced Cardiomyopathy: a Review. Front Cardiovasc Med. 2021;8:740515. doi:10.3389/fcvm.2021.740515
21. Cheung EC, Vousden KH. The role of ROS in tumour development and progression. Nat Rev Cancer. 2022;22(5):280–297. doi:10.1038/s41568-021-00435-0
22. Kong CY, Guo Z, Song P. Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: oxidative Stress and Cell Death. Int J Biol Sci. 2022;18(2):760–770. doi:10.7150/ijbs.65258
23. Vitale R, Marzocco S, Popolo A. Role of Oxidative Stress and Inflammation in Doxorubicin-Induced Cardiotoxicity: a Brief Account. Int J Mol Sci. 2024;25(13):7477. doi:10.3390/ijms25137477
24. Zhao L, Qi Y, Xu L. MicroRNA-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. 2018;15:284–296. doi:10.1016/j.redox.2017.12.013
25. Zhang WB, Lai X, Guo XF. Activation of Nrf2 by miR-152 Inhibits Doxorubicin-Induced Cardiotoxicity via Attenuation of Oxidative Stress, Inflammation, and Apoptosis. Oxid Med Cell Longev. 2021;2021(1):8860883. doi:10.1155/2021/8860883
26. Hu X, Liu H, Wang Z, Hu Z, Li L. miR-200a Attenuated Doxorubicin-Induced Cardiotoxicity through Upregulation of Nrf2 in Mice. Oxid Med Cell Longev. 2019;2019:1512326. doi:10.1155/2019/1512326
27. Fan D, Chen H-B, Leng Y, Yang S-J. MiR-24-3p Attenuates Doxorubicin-induced Cardiotoxicity via the Nrf2 Pathway in Mice. Curr Med Sci. 2022;42(1):48–55. doi:10.1007/s11596-022-2536-1
28. Zhang H, Tian Y, Liang D. The Effects of Inhibition of MicroRNA-375 in a Mouse Model of Doxorubicin-Induced Cardiac Toxicity. Med Sci Monit. 2020;26:e920557. doi:10.12659/MSM.920557
29. Li XQ, Liu Y-K, Yi J. MicroRNA-143 Increases Oxidative Stress and Myocardial Cell Apoptosis in a Mouse Model of Doxorubicin-Induced Cardiac Toxicity. Med Sci Monit. 2020;26:e920394. doi:10.12659/MSM.920394
30. Zhang WB, Zheng YF, Wu YG. Inhibition of miR-128-3p Attenuated Doxorubicin-Triggered Acute Cardiac Injury in Mice by the Regulation of PPAR-γ. PPAR Res. 2021;2021:7595374. doi:10.1155/2021/7595374
31. Luo C, Urgard E, Vooder T, Metspalu A. The role of COX-2 and Nrf2/ARE in anti-inflammation and antioxidative stress: aging and anti-aging. Med Hypoteses. 2011;77(2):174–178. doi:10.1016/j.mehy.2011.04.002
32. Wang F, Pu C, Zhou P. Cinnamaldehyde prevents endothelial dysfunction induced by high glucose by activating Nrf2. Cell Physiol Biochem. 2015;36(1):315–324. doi:10.1159/000374074
33. Zhang X, Hu C, Kong C-Y. FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2020;27(2):540–555. doi:10.1038/s41418-019-0372-z
34. Dai S. Emodin attenuates cardiomyocyte pyroptosis in doxorubicin-induced cardiotoxicity by directly binding to GSDMD. Phytomedicine. 2023;121:155105. doi:10.1016/j.phymed.2023.155105
35. Kitakata H. Therapeutic Targets for DOX-Induced Cardiomyopathy: role of Apoptosis vs. Ferroptosis Int J Mol Sci. 2022;23(3):1.
36. Meng J, Xu C. MicroRNA-495-3p diminishes doxorubicin-induced cardiotoxicity through activating AKT. J Cell Mol Med. 2022;26(7):2076–2088. doi:10.1111/jcmm.17230
37. Li Z, Li H, Liu B. Inhibition of miR-25 attenuates doxorubicin-induced apoptosis, reactive oxygen species production and DNA damage by targeting PTEN. Int J Med Sci. 2020;17(10):1415–1427. doi:10.7150/ijms.41980
38. Piegari E, Cozzolino A, Ciuffreda LP. Cardioprotective effects of miR-34a silencing in a rat model of doxorubicin toxicity. Sci Rep. 2020;10(1):12250. doi:10.1038/s41598-020-69038-3
39. Jing X, Yang J, Jiang L, Chen J, Wang H. MicroRNA-29b Regulates the Mitochondria-Dependent Apoptotic Pathway by Targeting Bax in Doxorubicin Cardiotoxicity. Cell Physiol Biochem. 2018;48(2):692–704. doi:10.1159/000491896
40. Xu C, Liu CH, Zhang DL. MicroRNA-22 inhibition prevents doxorubicin-induced cardiotoxicity via upregulating SIRT1. Biochem Biophys Res Commun. 2020;521(2):485–491. doi:10.1016/j.bbrc.2019.10.140
41. Fu Q. MiR-200a-3p Aggravates DOX-Induced Cardiotoxicity by Targeting PEG3 Through SIRT1/NF-κB Signal Pathway. Cardiovasc Toxicol. 2021;21(4):302–313. doi:10.1007/s12012-020-09620-3
42. Huang P, Zhang Y, Wang F. MiR −494-3p aggravates pirarubicin-induced cardiomyocyte injury by regulating MDM4 /p53 signaling pathway. Environ Toxicol: Int J. 2023;38(10):2499–2508. doi:10.1002/tox.23888
43. Wan GX, Cheng L, Qin H-L, Zhang Y-Z, Wang L-Y, Zhang Y-G. MiR-15b-5p is Involved in Doxorubicin-Induced Cardiotoxicity via Inhibiting Bmpr1a Signal in H9c2 Cardiomyocyte. Cardiovasc Toxicol. 2019;19(3):264–275. doi:10.1007/s12012-018-9495-6
44. Zhu JN, Fu Y-H, Hu Z-Q. Activation of miR-34a-5p/Sirt1/p66shc pathway contributes to doxorubicin-induced cardiotoxicity. Sci Rep. 2017;7(1):11879. doi:10.1038/s41598-017-12192-y
45. Liu Y, Li Y, Ni J, Shu Y, Wang H, Hu T. MiR-124 attenuates doxorubicin-induced cardiac injury via inhibiting p66Shc-mediated oxidative stress. Biochem Biophys Res Commun. 2020;521(2):420–426. doi:10.1016/j.bbrc.2019.10.157
46. Tong Z, Jiang B, Wu Y. MiR-21 Protected Cardiomyocytes against Doxorubicin-Induced Apoptosis by Targeting BTG2. Int J Mol Sci. 2015;16(7):14511–14525. doi:10.3390/ijms160714511
47. Geng W, Yan S, Sang D. Downregulating miR-432-5p exacerbates Adriamycin-induced cardiotoxicity via activating the RTN3 signaling pathway. Aging. 2024;16(16):11904–11916. doi:10.18632/aging.206062
48. Li Q, Qin M, Tan Q. MicroRNA-129-1-3p protects cardiomyocytes from pirarubicin-induced apoptosis by down-regulating the GRIN2D-mediated Ca 2+ signalling pathway. J Cell Mol Med. 2020;24(3):2260–2271. doi:10.1111/jcmm.14908
49. Roca-Alonso L, Castellano L, Mills A. Myocardial MiR-30 downregulation triggered by doxorubicin drives alterations in β-adrenergic signaling and enhances apoptosis. Cell Death Dis. 2015;6(5):e1754. doi:10.1038/cddis.2015.89
50. Du J, Hang P, Pan Y. Inhibition of miR-23a attenuates doxorubicin-induced mitochondria-dependent cardiomyocyte apoptosis by targeting the PGC-1α/Drp1 pathway. Toxicol Appl Pharmacol. 2019;369:73–81. doi:10.1016/j.taap.2019.02.016
51. Li Z, Ye Z, Ma J, Gu Q, Teng J, Gong X. MicroRNA‑133b alleviates doxorubicin‑induced cardiomyocyte apoptosis and cardiac fibrosis by targeting PTBP1 and TAGN2. Int J Mol Med. 2021;48(1). doi:10.3892/ijmm.2021.4958
52. Yun X, Fang Y, Lv C. Inhibition of the activation of γδT17 cells through PPARγ-PTEN/Akt/GSK3β/NFAT pathway contributes to the anti-colitis effect of madecassic acid. Cell Death Dis. 2020;11(9):752. doi:10.1038/s41419-020-02969-x
53. Tony H, Yu K, Qiutang Z. MicroRNA-208a Silencing Attenuates Doxorubicin Induced Myocyte Apoptosis and Cardiac Dysfunction. Oxid Med Cell Longev. 2015;2015:597032. doi:10.1155/2015/597032
54. Giorgio M, Migliaccio E, Orsini F. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122(2):221–233. doi:10.1016/j.cell.2005.05.011
55. Mao B, Zhang Z, Wang G. BTG2: a rising star of tumor suppressors (review). Int J Oncol. 2015;46(2):459–464. doi:10.3892/ijo.2014.2765
56. Bridges MC, Daulagala AC, Kourtidis A. LNCcation: lncRNA localization and function. J Cell Biol. 2021;220(2). doi:10.1083/jcb.202009045
57. Herman AB, Tsitsipatis D, Gorospe M. Integrated lncRNA function upon genomic and epigenomic regulation. Mol Cell. 2022;82(12):2252–2266. doi:10.1016/j.molcel.2022.05.027
58. Zhan J, Hu P, Wang Y. lncRNA PVT1 aggravates doxorubicin-induced cardiomyocyte apoptosis by targeting the miR-187-3p/AGO1 axis. Mol Cell Probes. 2020;49:101490. doi:10.1016/j.mcp.2019.101490
59. Fa HG, Chang W-G, Zhang X-J, Xiao -D-D, Wang J-X. Noncoding RNAs in doxorubicin-induced cardiotoxicity and their potential as biomarkers and therapeutic targets. Acta Pharmacol Sin. 2021;42(4):499–507. doi:10.1038/s41401-020-0471-x
60. Tian C, Yang Y, Bai B. Potential of exosomes as diagnostic biomarkers and therapeutic carriers for doxorubicin-induced cardiotoxicity. Int J Biol Sci. 2021;17(5):1328–1338. doi:10.7150/ijbs.58786
61. Chen KH, Chen C-H, Wallace CG. Combined therapy with melatonin and exendin-4 effectively attenuated the deterioration of renal function in rat cardiorenal syndrome. Am J Transl Res. 2017;9(2):214–229.
62. Xie Z, Xia W, Hou M. Long intergenic non‑coding RNA‑p21 mediates cardiac senescence via the Wnt/β‑catenin signaling pathway in doxorubicin-induced cardiotoxicity. Mol Med Rep. 2018;17(2):2695–2704. doi:10.3892/mmr.2017.8169
63. Li HQ, Wu Y-B, Yin C-S, Chen L, Zhang Q, Hu L-Q. Obestatin attenuated doxorubicin-induced cardiomyopathy via enhancing long noncoding Mhrt RNA expression. Biomed Pharmacother. 2016;81:474–481. doi:10.1016/j.biopha.2016.04.017
64. Christidi E, Brunham LR. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021;12(4):339. doi:10.1038/s41419-021-03614-x
65. Li C, Zhang L, Bu X, Chu G, Zhao X, Liu Y. LncRNA NORAD promotes the progression of myocardial infarction by targeting the miR-22-3p/PTEN axis. Acta Biochim Biophys Sin. 2022;54(4):463–473. doi:10.3724/abbs.2022037
66. Xiong X, Liu J, He Q. Long non-coding RNA NORAD aggravates acute myocardial infarction by promoting fibrosis and apoptosis via miR −577/ COBLL1 axis. Environ Toxicol: Int J. 2021;36(11):2256–2265. doi:10.1002/tox.23339
67. Guan X, Wang Y, Li W. The effects and mechanism of LncRNA NORAD on doxorubicin-induced cardiotoxicity. Toxicology. 2023;494:153587. doi:10.1016/j.tox.2023.153587
68. Berschneider B, Königshoff M. WNT1 inducible signaling pathway protein 1 (WISP1): a novel mediator linking development and disease. Int J Biochem Cell Biol. 2011;43(3):306–309. doi:10.1016/j.biocel.2010.11.013
69. Shao H, Cai L, Moller M. Notch1-WISP-1 axis determines the regulatory role of mesenchymal stem cell-derived stromal fibroblasts in melanoma metastasis. Oncotarget. 2016;7(48):79262–79273. doi:10.18632/oncotarget.13021
70. Berschneider B, Ellwanger DC, Baarsma HA. miR-92a regulates TGF-β1-induced WISP1 expression in pulmonary fibrosis. Int J Biochem Cell Biol. 2014;53:432–441. doi:10.1016/j.biocel.2014.06.011
71. Zhang S, Yuan Y, Zhang Z. LncRNA FOXC2-AS1 protects cardiomyocytes from doxorubicin-induced cardiotoxicity through activation of WNT1-inducible signaling pathway protein-1. Biosci Biotechnol Biochem. 2019;83(4):653–658. doi:10.1080/09168451.2018.1553606
72. Li J, Li L, Li X, Wu S. Long noncoding RNA LINC00339 aggravates doxorubicin-induced cardiomyocyte apoptosis by targeting MiR-484. Biochem Biophys Res Commun. 2018;503(4):3038–3043. doi:10.1016/j.bbrc.2018.08.090
73. Li J, Zhou L, Jiang Y, Gao H, Maierhaba T, Gong H. Long noncoding RNA RMRP ameliorates doxorubicin-induced apoptosis by interacting with PFN1 in a P53-Dependent manner. Mol Cell Probes. 2023;72:101937. doi:10.1016/j.mcp.2023.101937
74. Huang P, Zhang W, Ji J. LncRNA M iat knockdown protects against pirarubicin-induced cardiotoxicity by targeting miRNA −129-1-3p. Environ Toxicol: Int J. 2023;38(11):2751–2760. doi:10.1002/tox.23910
75. Patop IL, Wüst S, Kadener S. Past, present, and future of circ RNA s. THE EMBO Journal. 2019;38(16):e100836. doi:10.15252/embj.2018100836
76. Aufiero S, Reckman YJ, Pinto YM, Creemers EE. Circular RNAs open a new chapter in cardiovascular biology. Nat Rev Cardiol. 2019;16(8):503–514. doi:10.1038/s41569-019-0185-2
77. Lim TB, Aliwarga E, Luu TDA. Targeting the highly abundant circular RNA circSlc8a1 in cardiomyocytes attenuates pressure overload induced hypertrophy. Cardiovasc Res. 2019;115(14):1998–2007. doi:10.1093/cvr/cvz130
78. Conn SJ, Pillman K, Toubia J. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015;160(6):1125–1134. doi:10.1016/j.cell.2015.02.014
79. Ji X, Ding W, Xu T. MicroRNA-31-5p attenuates doxorubicin-induced cardiotoxicity via quaking and circular RNA Pan3. J Mol Cell Cardiol. 2020;140:56–67. doi:10.1016/j.yjmcc.2020.02.009
80. Gupta SK, Garg A, Bär C. Quaking Inhibits Doxorubicin-Mediated Cardiotoxicity Through Regulation of Cardiac Circular RNA Expression. Circ Res. 2018;122(2):246–254. doi:10.1161/CIRCRESAHA.117.311335
81. Lu D, Chatterjee S, Xiao K. A circular RNA derived from the insulin receptor locus protects against doxorubicin-induced cardiotoxicity. Eur Heart J. 2022;43(42):4496–4511. doi:10.1093/eurheartj/ehac337
82. Wang X, Cheng Z, Xu J. Circular RNA Arhgap12 modulates doxorubicin-induced cardiotoxicity by sponging miR-135a-5p. Life Sci. 2021;265:118788. doi:10.1016/j.lfs.2020.118788
83. Li B, Cai X, Wang Y. Circ-SKA3 Enhances Doxorubicin Toxicity in AC16 Cells Through miR-1303/TLR4 Axis. Int Heart J. 2021;62(5):1112–1123. doi:10.1536/ihj.20-809
84. Hu X, Liao W, Teng L, Ma R, Li H. Circ_0001312 Silencing Suppresses Doxorubicin-Induced Cardiotoxicity via MiR-409-3p/HMGB1 Axis. Int Heart J. 2023;64(1):71–80. doi:10.1536/ihj.22-379
85. Li C, Zhang L, Bu X, Wang J, Li L, Yang Z. Circ-LTBP1 is involved in doxorubicin-induced intracellular toxicity in cardiomyocytes via miR-107/ADCY1 signal. Mol Cell Biochem. 2022;477(4):1127–1138. doi:10.1007/s11010-022-04360-0
86. Yu P, Wang J, Xu G-E. RNA m(6)A-Regulated circ-ZNF609 Suppression Ameliorates Doxorubicin-Induced Cardiotoxicity by Upregulating FTO. JACC Basic Transl Sci. 2023;8(6):677–698. doi:10.1016/j.jacbts.2022.12.005
87. Li Y, Wu A, Chen L. Hsa_circ_0000098 is a novel therapeutic target that promotes hepatocellular carcinoma development and resistance to doxorubicin. J Exp Clin Cancer Res. 2022;41(1):267. doi:10.1186/s13046-022-02482-3
88. Zhang P, Liu Y, Zhan Y. Circ-0006332 stimulates cardiomyocyte pyroptosis via the miR-143/TLR2 axis to promote doxorubicin-induced cardiac damage. Epigenetics. 2024;19(1):2380145. doi:10.1080/15592294.2024.2380145
89. Zhai X, Zhou J, Huang X. LncRNA GHET1 from bone mesenchymal stem cell-derived exosomes improves doxorubicin-induced pyroptosis of cardiomyocytes by mediating NLRP3. Sci Rep. 2024;14(1):19078. doi:10.1038/s41598-024-70151-w
90. Ma C, Yang Z, Wang J. Exosomes miRNA-499a-5p targeted CD38 to alleviate anthraquinone induced cardiotoxicity: experimental research. Int J Surg. 2024;110(4):1992–2006. doi:10.1097/JS9.0000000000001118
91. Zheng H, Liang X, Liu B. Exosomal miR-9-5p derived from iPSC-MSCs ameliorates doxorubicin-induced cardiomyopathy by inhibiting cardiomyocyte senescence. J Nanobiotechnol. 2024;22(1):195. doi:10.1186/s12951-024-02421-8
92. Shimizu K, Quillinan N, Orfila JE, Herson PS. Sirtuin-2 mediates male specific neuronal injury following experimental cardiac arrest through activation of TRPM2 ion channels. Exp Neurol. 2016;275(Pt 1):78–83. doi:10.1016/j.expneurol.2015.10.014
93. Sarikhani M, Maity S, Mishra S. SIRT2 deacetylase represses NFAT transcription factor to maintain cardiac homeostasis. J Biol Chem. 2018;293(14):5281–5294. doi:10.1074/jbc.RA117.000915
94. Zhuang L, Xia W, Chen D. Exosomal LncRNA-NEAT1 derived from MIF-treated mesenchymal stem cells protected against doxorubicin-induced cardiac senescence through sponging miR-221-3p. J Nanobiotechnol. 2020;18(1):157. doi:10.1186/s12951-020-00716-0
95. Xia W, Chen H, Xie C, Hou M. Long-noncoding RNA MALAT1 sponges microRNA-92a-3p to inhibit doxorubicin-induced cardiac senescence by targeting ATG4a. Aging. 2020;12(9):8241–8260. doi:10.18632/aging.103136
96. Li Q, Qin M, Li T. Rutin protects against pirarubicin-induced cardiotoxicity by adjusting microRNA-125b-1-3p-mediated JunD signaling pathway. Mol Cell Biochem. 2020;466(1–2):139–148. doi:10.1007/s11010-020-03696-9
97. Huang P, Zhang Y, Wang F. Astaxanthin protects against pirarubicin-induced H9c2 cardiomyocytes by adjusting microRNA-494-3p-mediated MDM4/p53 signalling pathway. J Pharm Pharmacol. 2023;75(12):1521–1529. doi:10.1093/jpp/rgad084
98. Li Y, Fan L, Wang X, Lv S. Shenmai injection ameliorates doxorubicin-induced myocardial injury by suppressing autophagy-apoptosis via miR-30a. Aging. 2023;15(21):12400–12412. doi:10.18632/aging.205188
99. Li J-Z, Tang X-N, Li TT. Paeoniflorin inhibits doxorubicin-induced cardiomyocyte apoptosis by downregulating microRNA-1 expression. Exp Ther Med. 2016;11(6):2407–2412. doi:10.3892/etm.2016.3182
100. Song T, Yao Y, Wang T, Huang H, Xia H. Tanshinone IIA ameliorates apoptosis of myocardiocytes by up-regulation of miR-133 and suppression of Caspase-9. Eur J Pharmacol. 2017;815:343–350. doi:10.1016/j.ejphar.2017.08.041
101. Guo L, Zheng X, Wang E, Jia X, Wang G, Wen J. Irigenin treatment alleviates doxorubicin (DOX)-induced cardiotoxicity by suppressing apoptosis, inflammation and oxidative stress via the increase of miR-425. Biomed Pharmacother. 2020;125:109784. doi:10.1016/j.biopha.2019.109784
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.