")
Back to Journals » Clinical Ophthalmology » Volume 18
Second Primary Malignant Neoplasms in Survivors of Retinoblastoma in a Single Ocular Oncology Practice
Authors Wiseman Jr MT , Ebert JJ, Augsburger JJ, Di Nicola M, Correa ZM , Geller JI, Williams Jr BK
Received 14 August 2024
Accepted for publication 4 October 2024
Published 30 October 2024 Volume 2024:18 Pages 3103—3109
DOI https://doi.org/10.2147/OPTH.S484968
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Malcolm T Wiseman Jr,1 Jared J Ebert,1 James J Augsburger,1 Maura Di Nicola,1,2 Zelia M Correa,1,2 James I Geller,3 Basil K Williams Jr1,2
1Ocular Oncology Service, Department of Ophthalmology, University of Cincinnati College of Medicine, Cincinnati, OH, USA; 2Bascom Palmer Eye Institute, University of Miami Miller School of Medicine, Miami, FL, USA; 3Division of Oncology, Cincinnati Children’s Hospital Medical Center, University of Cincinnati, Cincinnati, OH, USA
Correspondence: Basil K Williams Jr, Email [email protected]
Introduction: A retrospective review of patients treated for retinoblastoma who developed a non-pineoblastoma second primary malignant neoplasm (SPMN) was performed.
Methods: The demographics, clinical features and treatments for retinoblastoma, pathologic types of non-pineoblastoma second primary malignant neoplasm (SPMN), intervals between the retinoblastoma diagnosis and treatment and diagnosis of non-pineoblastoma SPMN, treatment provided for the SPMN, and the survival outcomes of the patients were evaluated.
Results: Of 550 patients treated initially for retinoblastoma, this series used the 15 (2.7) that developed a non-pineoblastoma SPMN, 14 of which (93.3%) had been treated for bilateral retinoblastoma. All patients had carried a germline mutations in the RB1 gene. The median time from retinoblastoma diagnosis to SPMN diagnosis was 19.0 years (extremes 3.4 and 39.4 years). Six of the fifteen patients died during the follow-up of their SPMN. The median interval between initial retinoblastoma diagnosis and death in the 6 patients who died of their SPMN was 18.8 years (extremes 6.2 and 34.6 years) and between diagnosis of the SPMN and death was 1.2 years (extremes 0.25 and 4 years).
Discussion: Of the patients who had been treated with External Beam Radiotherapy (EBRT), 13 developed a SPMN within the previously irradiated field.
Keywords: external beam radiotherapy, pediatrics, osteosarcoma, retinoblastoma, family history, second primary malignant neoplasm
Introduction
Retinoblastoma (RB) is the most common primary intraocular malignancy in pediatric patients. The two genetic situations are: 1 germline + 1 somatic variant versus two somatic variants of the RB1 tumor suppressor gene located on chromosome 13q14.1–3 Hereditary retinoblastoma is associated with an increased lifetime risk of second primary malignant neoplasms (SPMNs), most of which are either pineoblastoma (ectopic intracranial retinoblastoma) or sarcomas. SPMNs are the leading cause of death in individuals with hereditary RB. While RB-associated pineoblastoma tends to occur during early pediatric patients, most RB-associated sarcomas occur years or even decades after initial retinoblastoma diagnosis and treatment.4,5 SPMNs are significantly higher likelihood to occur patients with hereditary retinoblastoma who have loss-of-function variant mutations in RB1.6 The reported incidence of SPMNs in the published literature varies due to differing definitions of SPMNs, differing lengths of follow-up, referral practice biases, and differences in retinoblastoma treatment.7 For this cohort, we decided to classify pineoblastoma as ectopic retinoblastoma and not a secondary tumor. Some reports mention it as an extension of the primary tumor while others consider it to be secondary tumor. This is due to Pineoblastoma being histopathologically very similar to retinoblastoma and some consider them to be ectopic intracranial retinoblastoma. SPMNs are now the leading cause of death in patients with hereditary retinoblastoma in high-income countries.8
Because SPMNs often occur decades following the initial diagnosis of retinoblastoma, long-term follow-up is necessary to accurately determine the frequency of and risk factors for the development of such outcomes in retinoblastoma survivors. In a case review covering 9 reports, a study by Mahoney et al found that out of 82 patients with hereditary RB, SPMN were found in the field of prior irradiation in 4 out of 14 (28.6%) patients. While other reports such as Marees et al showed that 89% of SPMNs were found in hereditary RB patients treated prior radiation therapy, while 40% were in-field of of prior irradiation (specifically soft tissue sarcomas, cancer of the bone, or melanoma).9 Overall this study concluded that although there is a clear increase of SMNs in irradiated patients, it did not find a significant association between exposure to ionizing radiation and the incidence of SMNs.
Herein we report the demographic and historical features of a series of patients with non-pineoblastoma SPMNs, the clinical features and treatment for the retinoblastoma in these cases, the types of SPMNs that occurred in these patients, and the treatment outcomes of these patients following SPMN diagnoses over a forty-three-year period in a single referral ocular oncology practice.
Methods
The authors performed a retrospective chart review of all patients in the Augsburger ocular oncology practice with a history of retinoblastoma who developed a SPMN between 1975 and 2022. A SPMN was defined as a histopathologically distinct solid malignant neoplasm that occurred after the onset of the primary retinoblastoma. The study was performed with the approval of the Institutional Review Board of the University of Cincinnati College of Medicine for retrospective analysis of deidentified clinical information contained in the charts of human patients evaluated in the practice and generated as part of standard patient care.
The authors abstracted the following information from the charts: demographic information, family history of retinoblastoma, features of the affected eye(s), therapeutic interventions for retinoblastoma, the interval between initial diagnosis of retinoblastoma and detection-diagnosis of the SPMN, age at diagnosis of the SPMN, pathologic type of SPMN, location of the SPMN, treatment provided for the SPMN, duration of follow-up after retinoblastoma diagnosis and after SPMN diagnosis and treatment, and life status of the patient through most recent follow-up. Because genetic testing was not available for all patients, hereditary disease was defined by bilateral disease, a positive family history, and/or a germline RB1 mutation detected on chromosomal/DNA analysis. Non-hereditary disease was defined by unifocal, unilateral disease, negative family history of RB, and/or chromosomal/DNA analysis that showed no evidence of a germline RB1 mutation, these are additive terms and not exclusive to define a nonhereditary disease. Since radiation field size data was not available for most patients in this series, SPMNs occurring in the head/neck region were defined as “in the field” whereas those occurring in the body or extremities were defined as “out of the field” of radiation.
The cases in this series fell into two discrete groups: Group 1 consisted of patients whose baseline diagnostic evaluation was performed and at least some of their initial retinoblastoma treatment was provided in the Augsburger ocular oncology practice and collaborating pediatric oncology practice, and Group 2 consisted of patients whose baseline diagnostic evaluation was performed and retinoblastoma treatment was provided at an outside center prior to referral to the Augsburger ocular oncology practice. For Group 1, we could determine both the baseline prognostic group (for ocular preservation) of the intraocular retinoblastoma of each affected eye (using both the Reese-Ellsworth7 and Murphree [ABCDE]8 classification systems) and the baseline stage of the retinoblastoma (using the AJCC tumor-node-metastasis [TNM] staging system, 2017 version9). For Group 2, this information was generally not available (ie, not provided by the outside center where the patient’s baseline diagnostic evaluation had been performed) and could not be determined from records of the baseline evaluation obtained from those centers.
Statistical analysis was performed using Microsoft Excel (Microsoft Corp, Redmond, WA). Continuous numeric variables were described using the median and extreme values. Categorical variables were described using numerical counts and percentages.
Results
Of 550 pediatric patients with retinoblastoma encountered in this practice during the study period, our series used the 15 patients who developed a SPMN (2.7%), 2 of whom (patient 4 and 7) developed 2 distinct SPMNs. Patient 4 diagnosed with both a left temporal fossa rhabdomyosarcoma and a left orbital osteosarcoma, both inside the field of radiation. Patient 7 developed osteosarcoma of both the left proximal tibia and the right hip outside the field of radiation. The median age at retinoblastoma diagnosis was 9.7 months (extremes 1.6 and 33.6 months). All 15 patients had a positive family history of retinoblastoma. Twelve patients were male (80%) and three were female (20%). Bilateral disease was present in 14 of the 15 (93.0%) patients, and the genetic nature of the one unilateral case (case 7, Table 1) was established by germline RB1 mutation on genetic analysis.
 |
Table 1 Demographics and Baseline Characteristics of Retinoblastoma |
Demographics and characteristics of each patient’s retinoblastoma are presented in Table 1. Eleven patients received treatment prior to referral, including enucleation (11/30 eyes; 36.6%), and external beam radiation therapy (EBRT) (12/30 eyes; 40.0%). Following completion of their retinoblastoma treatment course, a total of 15/30 (50.0%) eyes were enucleated, and 14/15 (93.3%) patients underwent EBRT. In 13 patients, retinoblastoma was diagnosed, and treatment was initiated prior to the adoption of carboplatin-etoposide-vincristine (CEV) and intravenous chemotherapy at our center (in 1995). All 15 patients were diagnosed and underwent treatment for their retinoblastoma prior to the availability of selective ophthalmic intra-arterial chemotherapy at our institution (in 2008). The 2 patients diagnosed after the introduction of primary systemic chemotherapy both received intravenous chemotherapy during their treatment course. None of the patients developed metastasis from retinoblastoma and no patient died of their retinoblastoma.
The characteristics of the SPMNs are reported in Table 2. Thirteen of the 14 patients (92.9%) who underwent EBRT developed their SPMN within the field of prior radiation. The histopathologic type of SPMN was osteosarcoma in 7, rhabdomyosarcoma in 3, malignant fibrous histiocytoma in 3, and a malignant astrocytoma, liposarcoma, esthesioneuroblastoma and a leiomyosarcoma in 1 case each. Treatment data for SPMN management was available for 11 of the 15 patients and included a combination of surgical resection in 11, intravenous chemotherapy in 6, and radiation therapy in 4. The median time from initial retinoblastoma diagnosis to development of SPMN was 19.0 years (extremes 3.4 and 39.4 years). The median time from retinoblastoma diagnosis to death in the 6 patients who died of their SPMN was 18.8 years (extremes 6.2 and 34.6 years), and the median interval between SPMN diagnosis and death from the neoplasm in these 6 patients was 1.2 years (extremes 0.25 and 4 years). In contrast, the median duration of follow-up after retinoblastoma in the 9 surviving patients was 32.0 years (extremes 12.5 and 39.3 years) and the median follow-up interval after diagnosis of the SPMN in these patients was 8.9 years (extremes 1.6 and 27.6 years).
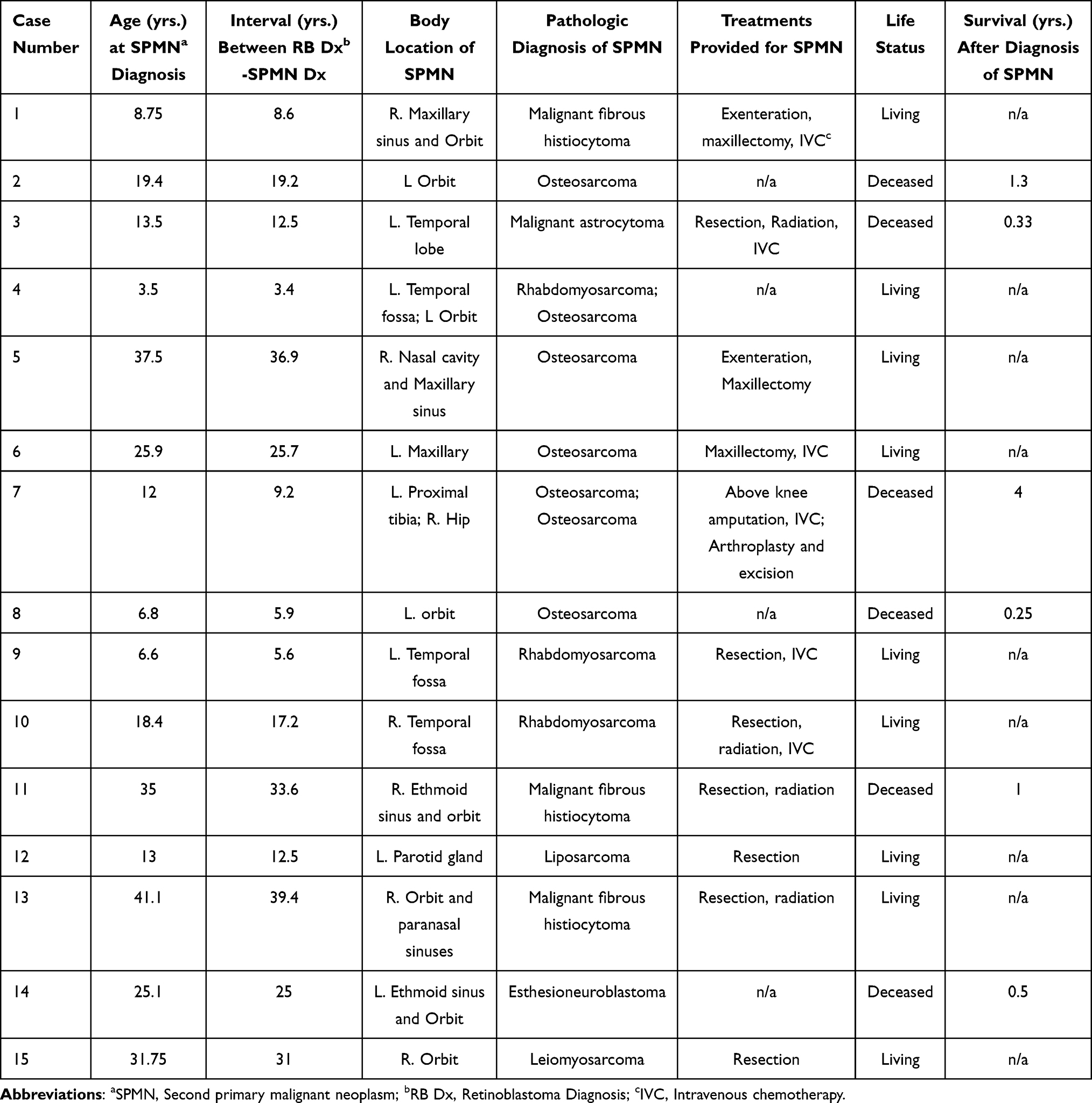 |
Table 2 Second Primary Malignancy Characteristics |
Discussion
Following the advancement of therapeutic options for retinoblastoma, the survival rate for retinoblastoma is >95% in high-income countries.7,10 As the rate of mortality from retinoblastoma has decreased, SPMNs have become the leading cause of death for patients with hereditary retinoblastoma.8 The cumulative actuarial incidence of SPMNs in hereditary disease has been reported to be 15.7% at 20 years and around 30% at 40 years.7,11–13At 60 years, a Danish cohort had a cumulative incidence of SPMNs of 51% for hereditary disease versus 13% for nonhereditary disease.8 The variance in published rates is due to a multitude of factors, including different lengths of follow-up, different definitions of SPMNs (some include pineoblastomas and non-melanoma skin cancers), different treatments provided for retinoblastoma, non-equivalent hereditary versus nonhereditary disease ratios, and use of national population-based studies versus tertiary referral center studies.7
Prior to the introduction of primary intravenous chemotherapy, EBRT was the predominant strategy for globe-salvaging therapy. EBRT for retinoblastoma has been associated with high rates of soft tissue and bony sarcomas in the field of radiation. In our study, 13 of 15 patients were diagnosed and treated prior to an effective and low toxicity CEV regimen of systemic chemotherapy, and all 15 patients were diagnosed and treated prior to the availability of selective ophthalmic intra-arterial chemotherapy at our center. This is reflected in the high rate of soft tissue and bony sarcomas in the field of radiation in our study (11 of 15 patients; 13 of 17 tumors). Additionally, 14 patients developed a SPMN in the head/neck region, 13 of whom had a history of prior EBRT.
Following the advent of an effective CEV intravenous chemotherapy as a treatment for retinoblastoma, there has been a change in patterns of SPMNs encountered. While hematologic SPMNs were rare prior to the introduction of chemotherapy, there has been an increased risk of hematologic SPMNs (most commonly acute myelogenous leukemia), especially in patients who received higher doses of chemotherapy.14–16 When used in combination with EBRT, chemotherapy may lead to higher rates of bone cancers and leiomyosarcomas in patients with hereditary disease compared to either treatment alone.17 Two patients in our study received intravenous chemotherapy during their treatment course. Patient 7 received both EBRT and a cyclophosphamide-based intravenous chemotherapy regimen, while patient 12 (the only patient that did not undergo EBRT) was treated with vincristine, etoposide, and carboplatin. Neither patient treated with systemic chemotherapy developed a hematologic SPMN.
Limitations of this study include its retrospective nature, the lack of follow-up information on the patients in the total group of 550 patients who did not develop a SPMN during available follow-up, the lack of baseline classification data on patients diagnosed and treated elsewhere prior to referral to the practice, the lack of information regarding the precise field of radiation, radiation dose, and fractionation schedule in most of the cases, and the referral bias of a single practice ocular oncology tertiary referral practice. An association of SPMNs with mutational status was not available in most cases in this series because genetic testing was not performed routinely during retinoblastoma management during the era of treatment for most patients in this cohort.
In conclusion, we describe our experience with non-pineoblastoma SPMNs in a tertiary referral practice. The vast majority of SPMNs in this series occurred in patients who had been treated by EBRT, and most occurred in the field of prior radiation. SPMNs occurred on average nearly two decades following the original diagnosis of retinoblastoma.
Human Studies and Informed Consent
The Institutional Review Board of the University of Cincinnati College of Medicine granted a waiver for informed consent in this retrospective chart review. All appropriate steps were taken.
Data Sharing Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Ethics Statement
This study complies with the Declaration of Helsinki.
Acknowledgments
No financial or material support was used. There are no funding sources.
This paper has been uploaded to Authorea as a preprint: https://www.authorea.com/users/713562/articles/697866-second-primary-malignant-neoplasms-in-survivors-of-retinoblastoma-in-a-single-ocular-oncology-practice‘.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Maura Di Nicola consults for EyePoint Pharmaceuticals and reports personal fees from SpringWorks Therapeutics, outside the submitted work. Basil K Williams Jr consults for Alcon, Allergan/Abbvie, Alimera, Astellas, Castle Biosciences, EyePoint Pharmaceuticals, Genentech/Roche, Immunocore, and Regeneron; owns stock options of Lumata Health. Malcolm T Wiseman Jr, Jared J Ebert, James J Augsburger, Zelia M Correa, and James I Geller declare that they have no conflicts of interest in this work.
References
1. Grossniklaus HE. Retinoblastoma. fifty years of progress. The LXXI Edward Jackson memorial lecture. Am J Ophthalmol. 2014;158(5):875–891. doi:10.1016/j.ajo.2014.07.025
2. Hong FD, Huang HJ, To H, et al. Structure of the human retinoblastoma gene. Proc Natl Acad Sci U S A. 1989;86(14):5502–5506. doi:10.1073/pnas.86.14.5502
3. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820–823. doi:10.1073/pnas.68.4.820
4. Yamanaka R, Hayano A, Takashima Y. Trilateral retinoblastoma: a systematic review of 211 cases. Neurosurg Rev. 2019;42(1):39–48. doi:10.1007/s10143-017-0890-4
5. Woo KI, Harbour JW. Review of 676 second primary tumors in patients with retinoblastoma: association between age at onset and tumor type. Arch Ophthalmol. 2010;128(7):865–870. doi:10.1001/archophthalmol.2010.126
6. Dommering CJ, Marees T, van der Hout AH, et al. RB1 mutations and second primary malignancies after hereditary retinoblastoma. Fam Cancer. 2012;11(2):225–233. doi:10.1007/s10689-011-9505-3
7. Fabius AWM, van Hoefen Wijsard M, van Leeuwen FE, Moll AC. Subsequent malignant neoplasms in retinoblastoma survivors. Cancers. 2021;13(6):1200. doi:10.3390/cancers13061200
8. Gregersen PA, Olsen MH, Urbak SF, et al. Incidence and mortality of second primary cancers in Danish patients with retinoblastoma, 1943-2013. JAMA Network Open. 2020;3(10):e2022126–e2022126. doi:10.1001/jamanetworkopen.2020.22126
9. Figueiredo D, Marques IA, Pires AS, et al. Risk of second tumors in retinoblastoma survivors after ionizing radiation: a review. Cancers. 2023;15(22):5336. doi:10.3390/cancers15225336
10. Moll AC, Kuik DJ, Bouter LM, et al. Incidence and survival of retinoblastoma in The Netherlands: a register based study 1862-1995. Br J Ophthalmol. 1997;81(7):559–562. doi:10.1136/bjo.81.7.559
11. Marees T, Moll AC, Imhof SM, de Boer MR, Ringens PJ, van Leeuwen FE. Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. J Natl Cancer Inst. 2008;100(24):1771–1779. doi:10.1093/jnci/djn394
12. Kleinerman RA, Tucker MA, Tarone RE, et al. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. J Clin Oncol. 2005;23(10):2272–2279. doi:10.1200/jco.2005.05.054
13. Uemura Y, Matsuda H, Uyama M, Matsuo N, Choshi K, UENO H Survival rate and risk factors for patients with retinoblastoma in Japan. The committee for the national registry of retinoblastoma. Jpn J Ophthalmol. 1992;36(2):121–131.
14. Turaka K, Shields CL, Meadows AT, Leahey A. Second malignant neoplasms following chemoreduction with carboplatin, etoposide, and vincristine in 245 patients with intraocular retinoblastoma. Pediatr Blood Cancer. 2012;59(1):121–125. doi:10.1002/pbc.23278
15. Gombos DS, Hungerford J, Abramson DH, et al. Secondary acute myelogenous leukemia in patients with retinoblastoma: is chemotherapy a factor? Ophthalmology. 2007;114(7):1378–1383. doi:10.1016/j.ophtha.2007.03.074
16. Smith MA, Rubinstein L, Anderson JR, et al. Secondary leukemia or myelodysplastic syndrome after treatment with epipodophyllotoxins. J Clin Oncol. 1999;17(2):569–577. doi:10.1200/jco.1999.17.2.569
17. Wong JR, Morton LM, Tucker MA, et al. Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. J Clin Oncol. 2014;32(29):3284–3290. doi:10.1200/jco.2013.54.7844
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.