")
Back to Journals » Journal of Inflammation Research » Volume 17
The AGEs/RAGE Signaling Pathway Regulates NLRP3-Mediated Neuronal Pyroptosis After MCAO Injury in Lepr−/− Obese Rats
Authors Zhao L, Li S, Wang X, Zhang L, Zhang J, Liu X, Hu Y , Xian X, Zhang F , Li W, Zhang M
Received 1 May 2024
Accepted for publication 23 September 2024
Published 1 October 2024 Volume 2024:17 Pages 6935—6954
DOI https://doi.org/10.2147/JIR.S476458
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Adam Bachstetter
Ling Zhao,1– 3,* Shichao Li,1,2,4,* Xiaoyu Wang,1,2 Lingyan Zhang,1,2 Jingge Zhang,1,2 Xiyun Liu,1,2 Yuyan Hu,1,2 Xiaohui Xian,1,2 Feng Zhang,5 Wenbin Li,1,2 Min Zhang1,2,6
1Department of Pathophysiology, Hebei Medical University, Shijiazhuang, Hebei Province, 050017, People’s Republic of China; 2Hebei Key Laboratory of Critical Disease Mechanism and Intervention, Shijiazhuang, Hebei Province, 050017, People’s Republic of China; 3Department of Obstetrics, The Fourth Hospital of Hebei Medical University, Shijiazhuang, Hebei Province, 050000, People’s Republic of China; 4Experimental Diagnostic Center for Infectious Diseases, The Second Hospital of Hebei Medical University, Shijiazhuang, Hebei Province, 050000, People’s Republic of China; 5Department of Rehabilitation Medicine, The Third Hospital of Hebei Medical University, Shijiazhuang, Hebei Province, 050051, People’s Republic of China; 6The Key Laboratory of Neural and Vascular Biology, Ministry of Education, Shijiazhuang, Hebei Province, 050017, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Min Zhang, Department of Pathophysiology, Key Laboratory of Critical Disease Mechanism and intervention of Hebei Province, The Key Laboratory of Neural and Vascular Biology, Ministry of Education, Hebei Medical University, No. 361 Zhongshan East Road, Shijiazhuang, Hebei, 050017, People’s Republic of China, Email [email protected]
Background: Obesity is recognized as a primary risk factor for cerebral ischemia, which has shown a significant increase in its incidence among obese patients. The exact mechanism by which obesity exacerbates cerebral ischemic injury is not fully understood though. The present study validated the hypothesis that obesity mediates pyroptosis by the AGEs/RAGE signaling pathway to exacerbate cerebral ischemic injury.
Methods: Leptin receptor knockout (Lepr−/−) rats were used in this study to construct an obesity model, and the middle cerebral artery occlusion (MCAO) models of ischemic stroke were established in Lepr−/− obese rats and their wild-type (WT) littermates respectively. Zea-Longa score, TTC and H&E staining were utilized to evaluate the neurological impairment. Western Blot, immunohistochemistry, and immunofluorescence were used to detect protein expressions. Transmission electron microscopy was used to observe the pores in the neuronal cell membrane in the ischemic penumbra cortex.
Results: Compared with WT littermates, Lepr−/− obese rats exhibited exacerbated neuronal injury after MCAO, with higher expressions of NLRP3 inflammasome and pyroptosis-related proteins in the cortical tissue of the penumbra. Moreover, more GSDMD pores were observed on the neuronal cell membranes of Lepr−/− obese rats according to the electron microscopy. Inhibition of NLRP3 inflammasome expression with MCC950 inhibited neuronal pyroptosis after cerebral ischemia in Lepr−/− obese rats, thus reducing neuronal injury. We also found that compared with WT littermates, the levels of AGEs and RAGE in the cortex of Lepr−/− obese rats are significantly higher, with further increase after cerebral ischemia. Inhibition of AGEs/RAGE signaling pathway with FPS-ZM1 reduced the NLRP3 inflammasome-mediated neuronal pyroptosis in Lepr−/− obese rats, thereby mitigating the neuronal damage after cerebral ischemia.
Conclusion: The AGEs/RAGE signaling pathway is involved in the exacerbation of cerebral ischemic injury in Lepr−/− obese rats via regulating NLRP3-mediated neuronal pyroptosis.
Keywords: MCAO, neuronal pyroptosis, NLRP3, Lepr−/− obese rats, AGEs/RAGE signaling pathway
Graphical Abstract:

Introduction
The association between obesity and cerebral ischemia has been confirmed in numerous studies. For example, in 2015, Heuschmann et al reported that 35.5% of cerebral ischemia patients were obese among 881 patients with a median age of 66 years in a EUROASPIRE cross-sectional survey conducted across various European countries.1 In fact, the incidence of stroke in obese individuals is significantly higher than in normal-weight people. Some researchers suggest that obesity can increase the incidence of stroke by promoting inflammatory responses and oxidative stress, and the severity of brain tissue damage in obese patients after stroke is also greater than that in stroke patients with normal weight, ultimately resulting in poor prognosis for the patients.2,3 By contrast, other researchers propose the existence of an “obesity paradox”, whereby a higher body weight unexpectedly improves a patient’s prognosis and reduces the likelihood of mortality during stroke development.4–6 Therefore, it is of great significance to study the mechanism of obesity aggravating cerebral ischemia injury, so as to reduce neural damage and mortality in obese patients.
Pyroptosis is a form of pro-inflammatory programmed cell death characterized by cell swelling, membrane rupture, and the release of inflammatory mediators following activation of the inflammasome.7,8 The pathways of pyroptosis involve the classical pyroptosis pathway mediated by caspase-1 and the non-classical pathway mediated by caspase-4/5/11. More specifically, the classical pyroptosis pathway is triggered by the activation of caspase-1 through the action of the NLRP3 inflammasome. Cerebral ischemia-reperfusion leads to the activation of NLRP3, prompting the conversion of pro-caspase-1 into caspase-1, which then induces gasdermin D (GSDMD) to form pores in the cell membrane, releasing inflammatory factors and causing lytic cell death.9 In recent years, an increasing number of studies have suggested that inflammasomes and their mediated pyroptosis play a crucial role in the pathogenesis and neural functional impairment associated with ischemic stroke.10 Moreover, obesity can induce sustained low-grade inflammation, which triggers or promotes obesity-related diseases such as insulin resistance, enteritis, and cardiovascular disease.11,12 The expression of the NLRP3 inflammasome increases in obese patients and triggers the release of pro-inflammatory cytokines interleukin IL-1β and IL-18, thereby activating the inflammatory response. This mechanism plays a key role in obesity-related metabolic disorders and insulin resistance.13,14 However, it still remains unclear whether obesity aggravates cerebral ischemic injury by activating NLRP3-mediated pyroptosis.
Advanced glycation end products (AGEs) are products of non-enzymatic glycation and lipid oxidation of some reducing sugars such as glucose and fructose, which are widely distributed in tissues and cells within the body. When AGEs increase in the body, they primarily bind to their receptor RAGE, and then activate and promote inflammatory reactions and oxidative stress.15,16 Previous studies have found that AGEs are closely related to cerebral ischemia and neurological diseases. Ma et al found an increased expression of RAGE mRNA in the hippocampal CA1/2 region of rats with moderate ischemia and hypoxia for 72 hours by in situ hybridization, whereas there was no change in RAGE on the opposite side of ischemia.17 Reddy et al found that in an AD animal model, AGEs bind to RAGE and up-regulate the expression of RAGE mRNA and protein, leading to a significant increase in phosphorylated tau protein.18 Recent reports suggested that the expression levels of AGEs and RAGE proteins increased in the plasma and adipose tissues of obese patients. AGEs can induce the production of inflammatory mediators in adipocytes and macrophages through binding to RAGE, thereby mediating the occurrence of obesity-related complications.19 However, it has not yet been determined whether obesity exacerbates cerebral ischemic injury by upregulating AGEs/RAGE signaling pathway and overactivating NLRP3 to induce pyroptosis in neuronal cells.
To elucidate the aforementioned problems, this study employed leptin receptor knockout (Lepr−/−) rats as an obesity model to investigate whether the Lepr−/− obese rats with MCAO exhibit aggravated cerebral ischemia-reperfusion injury via the upregulation of AGEs/RAGE and over-activation of the NLRP3 inflammasome, thus leading to an increased neuronal pyroptosis.
Materials and Methods
Experimental Animals
The parental Lepr+/- rats (Lepr knockout heterozygotes, Sprague-Dawley background) were donated by Professor Zhang Lianfeng from the Institute of Experimental Animals, Chinese Academy of Medical Sciences (SCXK 2019–0002). In patients with obesity and metabolic disorders, the leptin signaling pathway may be disabled due to leptin resistance, which is similar to LepR knockout rats. Therefore, leptin resistance in patients with obesity and metabolic disorders can be partially simulated in a LepR knockout rat model.20,21 The Lepr+/- rats, Lepr−/− rats, and wild-type SD rats (8 weeks old) were all housed under specific-pathogen-free (SPF) conditions at the Animal Center of Hebei Medical University (SYXK (Ji) 2019–005), with an ambient temperature of 20–25°C and a humidity of 40–70% for a 12 hour light/dark cycle, in transparent cages with standard laboratory food and water ad libitum. Due to the potential neuroprotective effects of estrogen in the context of cerebral ischemia-reperfusion injury,22 only male rats were used to minimize the variability in experimental data caused by gender differences. All the procedures were performed in accordance with the welfare and ethical requirements for experimental animals established by Hebei Medical University (ethical approval number LACUC-Hebmu-PD-201918).
Our decision on sample size was made with a combination of ethical considerations, statistical power, and the intricate challenges inherent in the model construction process. In the experimental design, the delicate balance between robust statistical analysis and ethical principles underlying animal welfare was meticulously considered. Statistical software GPower 3.1 was used to ensure the stability of the statistical method and meet the requirements of sample calculation power. Detailed information on sample size determination is listed in Supplemental Table 1.
Groups
The study primarily consisted of WT Sham group, WT MCAO group, Lepr−/− Sham group and Lepr−/− MCAO group to investigate whether Lepr−/− obesity can aggravate cerebral ischemic injury and pyroptosis in rats. The rats underwent neurological deficit testing 24 hours after MCAO, and then were sacrificed by decapitation under inhalation anesthesia with 4–5% isoflurane. The brain of the rats was obtained for the measurement of cerebral infarct volume by TTC staining, histological evaluation by H&E staining, GSDMD, N-terminal GSDMD, NLRP3, Pro-caspase-1, Cleaved caspase-1, Mature IL-18, Mature IL-1β, AGEs and RAGE protein expression by IF staining, IHC staining and Western blot, GSDMD, NLRP3 and RAGE mRNA expression by qRT-PCR and ultrastructural evaluation by transmission electron microscopy.
Furthermore, to investigate the effects of NLRP3 on neuronal pyroptosis and the relationship between AGEs/RAGE pathway and NLRP3-mediated neuronal pyroptosis in Lepr−/− obese rats during cerebral ischemia-reperfusion injury, MCC950 (the inhibitor of NLRP3) and FPS-ZM1 (the inhibitor of RAGE) were used to design MCC950 + Lepr−/− MCAO and FPS-ZM1 + Lepr−/− MCAO groups, respectively. In MCC950 + Lepr−/− MCAO group, MCC950 was administered by tail vein injection 1 hour before the MCAO at a dose of 5 mg/kg according to previous reports.23 A solvent control group was designed, in which DMSO was administered instead of MCC950 to the Lepr−/− MCAO rats. In FPS-ZM1 + Lepr−/− MCAO group, FPS-ZM1 (dissolved in a solvent consisting of 3% DMSO + 40% PEG300 + 1% Tween 80 + 54% saline) was administered via intraperitoneal injection at 5 minutes and 1 hour after MCAO with a dosage of 5 mg/kg each time according to previous reports.24 Simultaneously, a solvent control group was designed, in which the solvent of FPS-ZM1 was administered instead of FPS-ZM1 to the Lepr−/− MCAO rats. The rats were sacrificed after neurological deficit testing 24 hours after MCAO, and other treatments were the same with above mentioned.
Focal Cerebral Ischemia
A model of transient focal cerebral ischemia was established by blocking and reopening the blood flow in the middle cerebral artery of rats.25 More specifically, the rats were subjected to a 12-hour fasting period, during which they had free access to water, prior to the surgery. After inducing anesthesia with isoflurane, the skin was incised along the midline of the neck, with layer by layer separation revealing the vagus nerve and the left common carotid artery (CCA). Moreover, the internal carotid artery (ICA) and external carotid artery (ECA) branches were also identified. The ECA was ligated using a silk thread, while the ICA was temporarily clamped using an artery clamp. The proximal end of the CCA was ligated with a silk thread, whereas the distal end was prepared using a movable knot. The proximal ligation line was gently elevated, and a small incision was made in the CCA between the two knots. A thread embolus was slowly inserted via this incision, passing through the bifurcation into the ICA. The artery clamp was opened when encountered.
The process was stopped when a slight resistance was felt at approximately 18–20 mm from the entrance, and then the ligature on the CCA was firmly tied. After 90 minutes of blocking the blood flow in the middle cerebral artery, while the rat remained under anesthesia, the embolus was gently withdrawn, initiating reperfusion. The Sham group underwent the same procedures, save for the insertion of the embolus. 1% procaine hydrochloride was locally administered to the wound before the wound suture to prevent postoperative pain, and the surgical site was thoroughly disinfected by local drops of gentamicin to reduce the risk of infection. Additionally, the rats were housed individually after surgery to prevent stress from contact with other animals, and a heating blanket was used to maintain the animals’ body temperature at 37±1 ° C to promote postoperative recovery. All surgical procedures were performed under deep anesthesia, and the recovery process strictly followed animal ethics guidelines to minimize pain and distress. Ad libitum access to water and food was provided.
Drug Administration
One hour before the MCAO surgery, the NLRP3 inhibitor MCC950 (5 mg/kg, Cat No. HY-12815A, Selleck Chemicals, USA) was administered via the tail vein. The control group received an equivalent volume of DMSO. Tail vein injection provides effective systemic delivery in rodent models, ensuring rapid and uniform distribution of the inhibitor throughout the body.26,27 MCC950 can cross the blood-brain barrier, making it appropriate in an acute ischemic stroke model. Li X23 showed that 5 mg/kg of MCC950 effectively inhibits NLRP3 inflammasome activation without significant off-target effects or toxicity, making it a suitable choice for our study. The RAGE inhibitor FPS-ZM1 (Cat No. HY-19370, Med Chem Express, USA) was dissolved in a solvent (3% DMSO + 40% PEG300 + 1% Tween 80 + 54% saline) and administered via intraperitoneal injection at 5 minutes and 1 hour after the surgery, with a dosage of 5 mg/kg each time according to previous report.24 Here, the control group received an equivalent volume of the solvent at the same two time points.
Neurological Deficit Testing
The neurological deficits of the rats were assessed at 24 hours after reperfusion according to the Longa method.25 To reduce the potential for bias, a double-blind design was followed (the experimental personnel did not know which group was being analyzed). The Zea-Longa assessment criteria were used to assess the neurologic deficits according to prior descriptions.25 Briefly, 0 points indicated no obvious neurological deficit, 1 point indicated the right forepaw failed to extend fully, 2 points indicated the rat rotated rightward, 3 points indicated the rat tilted to the right, and 4 points indicated the rat was unable to walk independently and lost consciousness.
Cerebral Infarct Volume Measurement
The rats were decapitated immediately after being anesthetized 24 hours after reperfusion. The whole-brain tissues were rapidly harvested and frozen at −20°C for 10 min, and they were then cut into 2 mm thick coronal sections and stained with 2% triphenyl tetrazolium chloride (TTC Cat No. A1010, Beijing, China) solution for 15 min at 37°C. The stained brain slices from the different groups were photographed using a microscope (Olympus BX63, Japan). Six serial coronal brain sections were obtained from each rat for the analysis of infarct volume. The sections were stained with 2% triphenyl tetrazolium chloride solution. The unstained areas were defined as infarct area, and the relative infarct volume was calculated using image analysis software (Image J software, National Institutes of Health, Bethesda, MD, USA)28 with the following formula: (the volume of contralateral hemisphere-noninfarct volume of ipsilateral hemisphere)/ (the volume contralateral hemisphere×2) × 100%.
Hematoxylin and Eosin (H&E) Staining
The rats were transcardially perfused with 4% paraformaldehyde under deep anesthesia at 24 hours after reperfusion. Their brains were then rapidly removed, fixed in 4% paraformaldehyde for 48 hours, and cut into 5-μm-thick continuous coronal brain slices, which were dehydrated using xylene and ethanol and then stained with H&E (Cat No. G1120, Beijing, China). The stained brain slices from the different groups were photographed using a microscope (Olympus BX63, Japan). Five images were taken from each rat. The observations were conducted across five independent batches.
Western Blot Analysis
Western blot analysis was performed using electrophoresis apparatus, as reported previously.29 The total protein was harvested from the cerebral ischemic penumbra and extracted using RIPA lysis buffer (Cell Signaling Technology, USA) on ice from the supernatants following centrifugation at 12,000 rpm for 15 min. The protein concentrations were determined via the BCA protein assay. Next, the protein samples were separated with 8%, 12%, or 15% SDS-PAGE gel and then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, USA). These membranes were probed overnight at 4°C using primary antibodies. The primary antibodies were as follows: rabbit anti-NLRP3 polyclonal antibody (1:1000, Cat No. 19771-1-AP, Proteintech, China, RRID: AB_10646484), rabbit anti-GSDMD (1:1000, Cat No: AB219800, Abcam, UK), rabbit anti-N-GSDMD polyclonal antibody (1:1000, Cat No. AF4012, Afnity Biosciences, China), rabbit anti-pro-caspase-1 polyclonal antibody (1:1000, Cat No. ET1608-69, HUABIO, China), rabbit anti-cleaved caspase-1 polyclonal antibody (1:1000, Cat No. E2G2I, Cell Signaling, USA), mouse anti-IL-1β (1:200, Cat No: sc-12742, Santa, USA), rabbit anti-IL-18 (1:1000, Cat No: 10663-1-AP, Proteintech, USA, RRID: AB_2123636), rabbit anti-AGEs (1:1000, Cat No: AB23722, Abcam, UK), rabbit anti-RAGE (1:1000, Cat No: AB37647, Abcam, UK), mouse anti-β-actin (1:3000, Cat No: 66009-1-lg, Proteintech, USA, RRID: AB_2687938), and rabbit anti-GAPDH polyclonal antibody (1:5000, Cat No. 10494-1-AP, Proteintech, China, RRID: AB_2263076). The membranes were washed and then treated with the secondary antibodies for 60 minutes at 37°C. The secondary antibodies were as follows: goat anti-rabbit IgG (1:3000, Cat No. SA00001-2, Proteintech, China, RRID: AB_2722564) for NLRP3, GSDMD, N-GSDMD, Pro-caspase1, Cleaved-caspase1, IL-18, AGEs, RAGE, and GAPDH; goat anti-mouse IgG (1:2000, Cat No. SA00001-1, Proteintech, China, RRID: AB_2722564) for IL-1β and β-actin. The primary antibodies of GSDMD, IL-18, AGEs, RAGE have been confirmed its specific by knockout test. The specificity of other primary antibodies were selected from high-quality articles of similar studies.
The bands were visualized by means of enzyme-linked chemiluminescence in a detection system (Azure C400, USA). Quantitative analysis of the protein bands was conducted using ImageJ software. The relative expression levels of the target proteins or genes were normalized to housekeeping proteins (GAPDH or β-actin) and expressed as a fold change relative to the control group.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Each rat’s cerebral ischemic penumbra tissues were separated and homogenized using TRIzol reagent (Invitrogen, USA). The PrimeScript RT Reagent Kit (RR047A, Takara, Japan) was used for the reverse transcription of the RNA. We performed qPCR to detect the mRNA levels using the SYBR Premix Ex Taq II (RR820A, Takara) in a 2.1 Real-Time PCR System (Bio-Rad, USA) according to the manufacturer’s instructions. The PCR results were quantified via a 2−ΔΔCt method, and the relative mRNA expression of the target genes, including NLRP3, GSDMD, and RAGE, was normalized to that of the housekeeping gene (GAPDH) and expressed as a fold change relative to the control group. The sequences of the utilized primers are listed in Table 1.
 |
Table 1 The Primer Sequences of NLRP3, GSDMD, RAGE, and GAPDH |
Immunofluorescence Staining
The rats were transcardially perfused with 4% paraformaldehyde under deep anesthesia at 24 hours after reperfusion. Their brains were rapidly extracted, soaked in 4% paraformaldehyde for 48 hours, and then cut into 5-μm-thick continuous coronal brain slices. Double immunofluorescence staining was employed to identify the specific cell populations that exhibited NeuN expression in each rat’s brain and to illustrate the presence of NLRP3 and GSDMD in the neurons. The paraffin sections were heated for 1 hour (55–60°C), dewaxed in xylene, and rehydrated in PBS. Following antigen retrieval in Tris/EDTA buffer (pH 9.0) and blocking with 10% goat serum at 37°C for 1 hour, the slices were incubated overnight at 4°C with the following primary antibodies: anti-NLRP3 (1:200, Cat No: PA5-79740, Thermo Fisher, USA), anti-GSDMD (1:100, Cat No: AB219800, Abcam, USA), and anti-NeuN (1:100, Cat No: 66836-1-Ig, Proteintech, USA). On the subsequent day, after being washed with PBS, the sections were incubated with Alexa Fluor 594 goat anti-rabbit IgG (1:200, Cat No. SA00013-4, Proteintech, China) and Alexa Fluor 488 goat anti-mouse IgG (1:200, Cat No. SA00013-1, Proteintech, China) in the dark at 37°C for 1 hour. Sections were washed in PBS and then sealed with the DAPI mounting buffer (Cat No. S2110, Solarbio, China). All the images were acquired using a microscope (Olympus BX63, Japan).The data was assessed based on the count of positive cells and the intensity of immunofluorescence. Five images were taken from each rat. The observations were conducted across five independent batches. The ImageJ software was used to analyze the positive signal of the target protein (GSDMD or NLRP3 positive cells per 0.1mm2 cell).
Electron Microscopy
Tissues (1 × 1×1 mm) were dissected from the cerebral ischemic penumbra and then successively fixed in 2.5% glutaraldehyde and 1% osmium tetroxide. Following dehydration and insertion, the tissues were cut into 70-nm slices. The samples were observed and scanned using an H7500 Transmission Electron Microscope (Hitachi, Japan). Three regions were examined in the brain samples of each rat. The observations were conducted across three independent batches. The pyroptosis pores of GSDMD with a diameter of 10–14 nm30 were observed by transmission electron microscopy.
Immunohistochemistry (IHC)
IHC analysis was performed to determine the RAGE expression at 24 hours after reperfusion. Briefly, the rats’ brains were fixed, dehydrated, and trimmed according to the same method used in the H&E assay and then embedded in paraffin prior to slicing into 5-μm-thick sections. The endogenous peroxides were blocked using 3% H2O2 for 10 min and the nonspecific antigens were then blocked with 5% goat serum for 1 hour at 37°C. The sections were incubated with the primary antibody against RAGE (1:200, ab37647, Abcam, UK) overnight at 4°C. Brain sections were washed three times with PBS and subsequently incubated with the secondary antibody (Cat No. SP-0022, Bioss, China) for 60 minutes at 37°C. Then, the brain sections were incubated with the horseradish peroxidase-conjugated streptavidin working solution (Cat No. SP-0022, Bioss, China) for 60 min at 37°C. After being washed with PBS, the slices were treated with a DAB substrate (Cat No. ZLI-9017, Zhongshan, China). Images were acquired using a microscope (Olympus BX63, Japan). Five images were taken from each rat. The observations were conducted across five independent batches. The integrated optical density (IOD) of the RAGE protein in the sections was measured using Image J software (National Institutes of Health, Bethesda, MD, USA). The average IOD of each group was then calculated and compared.
Statistical Analysis
Following previous research guidelines and setting a significance criterion at α=0.05 and power at 0.80, the minimum required sample size with this effect size for a WilcoxonMann Whitney test between two groups was determined to be N=3.31
Normality was evaluated using the Shapiro–Wilk test and variance homogeneity was assessed by the Levene test within the group. The results of TTC staining, Western blot, qRT-PCR, immunofluorescence staining, and immunohistochemical staining experiments were presented as mean ± SD, and one-way ANOVA followed by Tukey’s analysis was used to analyze differences between the groups. The Zea-Longa’s scores were presented as medians±interquartile range and Kruskal–Wallis test was used to analyze differences between the groups. Detailed information on the statistical tests is presented in Supplemental Table 2. All statistical analyses were conducted using GraphPad Prism software 8.0 (GraphPad Software, USA), with P<0.05 deemed statistically significant. To maintain the objectivity of the research, the authors were blinded to the experimental protocol and did not have access to the statistical calculations during the execution of experiments.
Results
Obesity Induced by Leptin Receptor Knockout Aggravated Cerebral Ischemic Injury in Rats After MCAO
We subjected the Lepr−/− obese rats and WT littermates to MCAO and then compared the differences in their neurological function scores, infarct volumes, and neuronal morphological changes in the ischemic penumbra area of cerebral cortex (Figure 1). The results indicated significantly elevated neurological function scores neuronal injury, and increased infarct volume in both WT littermates and Lepr−/− obese rats in the MCAO group when compared with the Sham group (P < 0.05). When compared with WT MCAO group, Lepr−/− MCAO group showed aggravated neuronal injury and further increased infarct volume, which was 1.53 times that of the WT MCAO group. (P < 0.05), but no significant difference in neural function scores (P > 0.05) (Figure 1A). The above results indicate that compared to the WT littermate, Lepr−/− obese rats exhibited more severe neural injury after cerebral ischemia.
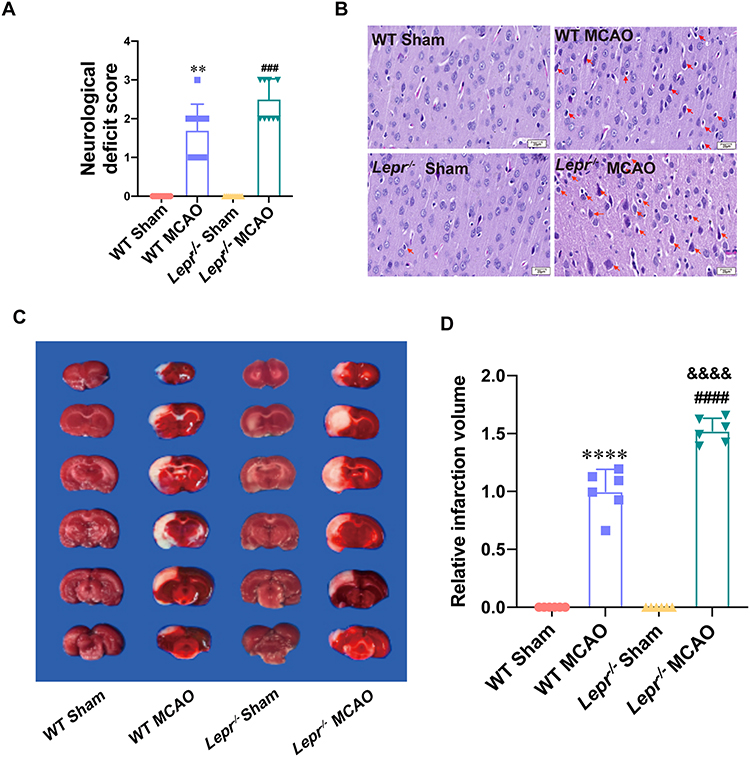 |
Figure 1 More severe neuronal damage after MCAO in Lepr−/− obesity rats. (A) Zea-Longa score of Lepr−/− obesity rats or litter of wild rats in each group. (n = 10 in each group, power value = 0.99, KW = 35.75, P < 0.0001). Data are presented as medians±interquartile range. Data were analyzed using the Kruskal–Wallis test. (B) Representative images of HE staining in the cortex penumbra region, with the abnormality neurons indicated through the red arrows (scale bar = 20 μm). (n = 5 in each group). (C and D) Representative TTC staining (C) and statistical comparison of relative infarction volume (D) in each group (n = 6 in each group, power value = 0.97, F(3, 20) = 290.7, P < 0.0001). Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s analysis. **P < 0.01, ****P < 0.0001 vs WT Sham group, ###P < 0.001, ####P < 0.0001 vs Lepr−/− Sham group, &&&&P < 0.0001 vs WT MCAO group. |
Obesity Induced by Leptin Receptor Knockout Exacerbates Neuronal Pyroptosis in Rats After MCAO
We compared the neuronal pyroptosis differences of the WT littermates and Lepr−/− obese rats after cerebral ischemia-reperfusion. Western blot result showed that compared with the WT Sham group, the expressions of pyroptosis-related proteins GSDMD and N-terminal GSDMD in the WT MCAO group significantly increased, which were 2.35 times and 3.05 times that of the WT Sham group, respectively. While the expressions of GSDMD and N-terminal GSDMD in the Lepr−/− MCAO group were 3.03 times and 4.32 times that of the Lepr−/− Sham group, respectively. Moreover, when compared with the WT MCAO group, the expressions of GSDMD and N-terminal GSDMD in Lepr−/− MCAO group were further elevated, which were 1.5 times and 1.54 times that of the WT MCAO group, respectively (P<0.05) (Figure 2A). The trend in terms of the GSDMD mRNA expression was consistent with that of the Western blot results, which the value in WT MCAO group was 2.13 times that in the WT Sham group, the value in the Lepr−/− MCAO group was 3.89 times that in the Lepr−/− Sham group, and the value in Lepr−/− MCAO group was 2.29 times that in the WT MCAO group (P<0.05) (Figure 2B). Immunofluorescence staining revealed that the value of GSDMD positive cells in the WT MCAO group was 13.8 times that in the WT Sham group, and the value in the Lepr−/− MCAO group was 16.89 times that in the Lepr−/− Sham group. Additionally, the neuronal GSDMD fluorescence intensity in Lepr−/− MCAO group was further increased, which was 2.2 times when compared with the WT MCAO group (P<0.05) (Figure 2C and D). Transmission electron microscopy was used to observe the pores in the neuronal cell membrane in the ischemic penumbra cortex. The results revealed that when compared with the WT MCAO group, the Lepr−/− MCAO group had more GSDMD pores in the neuronal cell membrane (Figure 3). These findings indicate that when compared with WT littermates, Lepr−/− obese rats exhibit more pronounced neuronal pyroptosis following cerebral ischemia.
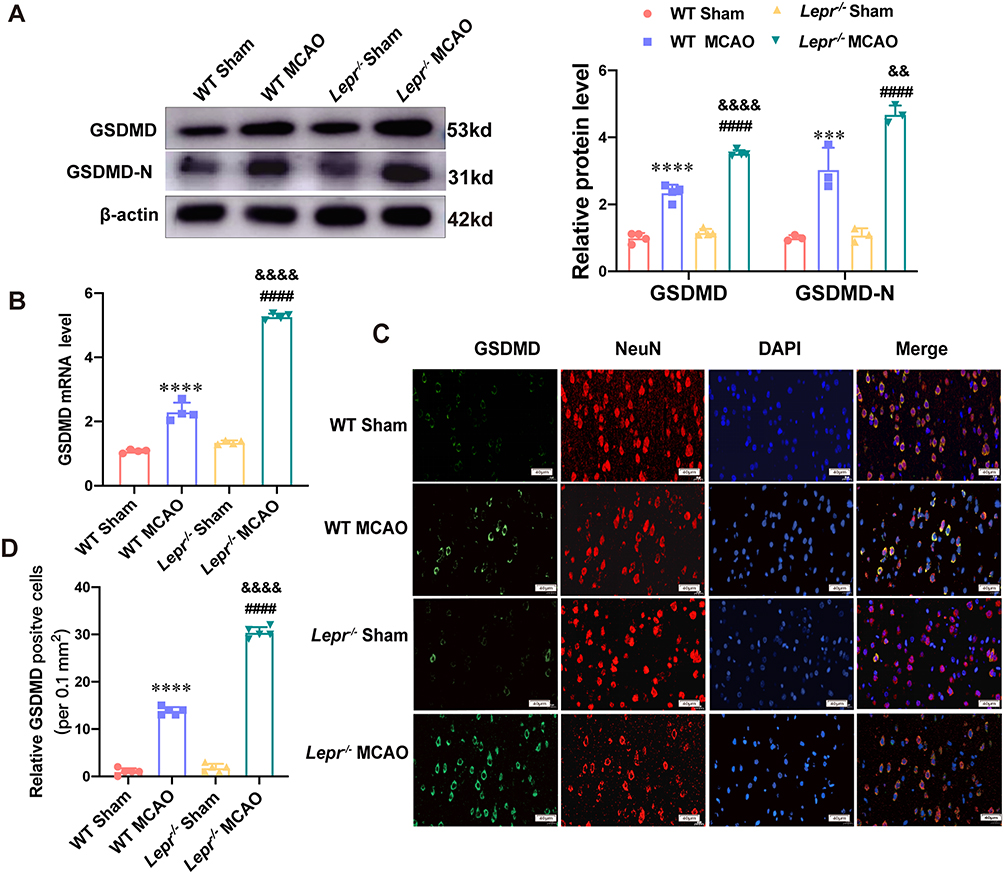 |
Figure 2 More severe neuronal pyroptosis after MCAO in Lepr−/− obesity rats. (A) Protein expression of GSDMD (n = 4 in each group power value = 0.94, F (3, 12) = 178.6, P < 0.001) and N-terminal GSDMD (n = 3 in each group, power value = 0.82, F (3, 8) = 69, P < 0.001) in the cortex penumbra region in each group. (B) qRT-PCR results showed the mRNA expression of GSDMD in the cortex penumbra region in each group. (n = 4 in each group, power value = 0.81, F (3, 12) = 592, P < 0.001). (C and D) Representative Immunofluorescence staining (C) and statistical comparison of Relative GSDMD positive cells (D) in each group (scale bar = 40 μm) (n = 5 in each group, power value = 0.91, F (3, 16) = 1180, P < 0.0001). All values are mean ± SD. One-way ANOVA followed by Tukey’s analysis. ****P < 0.0001 vs WT Sham group, ####P < 0.0001 vs Lepr−/− Sham group,&&&&P < 0.0001, &&P < 0.01 vs WT MCAO group. |
 |
Figure 3 Representative transmission electron microscopy images of neurons, with GSDMD pores on neuronal membrane highlighted with red arrows. (scale bar is 5 μm in left and 2 μm in right) (n = 3 in each group). |
Obesity Induced by Leptin Receptor Knockout Amplifies NLRP3 Inflammasome Activation in Rats After MCAO
The NLRP3 inflammasome is an upstream signal for neuronal pyroptosis. Therefore, this study further explored whether over activation of NLRP3 aggravates neuronal pyroptosis in the Lepr−/− obese rats after cerebral ischemia. Western blot results revealed that compared with the WT Sham group, the expression of NLRP3, Pro-caspase-1, Cleaved caspase-1, Mature IL-18, and Mature IL-1β in WT MCAO group were significantly elevated, which were 2.65 times, 1.82 times, 2.05 times, 1.95 times and 1.96 times that of the WT sham group, respectively. While the the expression of NLRP3, Pro-caspase-1, Cleaved caspase-1, Mature IL-18, and Mature IL-1β in Lepr−/− MCAO group were 2.16 times, 2.22 times, 2.78 times, 2.18 times and 2.17 times that of the Lepr−/− Sham group, respectively. Moreover, when compared with the WT MCAO group, the Lepr−/− MCAO group exhibited a further increase in the expression of NLRP3 (P<0.05), Pro-caspase-1, Cleaved caspase-1, Mature IL-18, and Mature IL-1β, which were 1.15 times, 1.39 times, 1.47 times, 1.38 times and 1.37 times that of WT MCAO group, respectively. (P<0.05) (Figure 4A). The qRT-PCR results indicated that the value in WT MCAO group was 2.08 times that in the WT Sham group and the value in the Lepr−/− MCAO group was 3.23 times that in the Lepr−/− Sham group. Additionally, when compared with the WT MCAO group, the Lepr−/− MCAO group displayed a significant increase in the NLRP3 mRNA levels, which was 2.16 times that of the WT MCAO group (Figure 4B) (P<0.05). Immunofluorescence staining revealed a notable increase in NLRP3-positive neurons in the ischemic penumbra cortex of the WT MCAO group which was 13.8 times that of the WT Sham group. While the value in the Lepr−/− MCAO group was 31.6 times that in the Lepr−/− Sham group. Furthermore, the Lepr−/− MCAO group exhibited a further elevation in the NLRP3 fluorescence intensity in the cortical penumbra neurons which was 2.29 times when compared with the WT MCAO group. (P<0.05) (Figure 4C and D). These results suggest that the NLRP3 inflammasome is over activated in Lepr−/− obese rats following cerebral ischemia, aggravating neuronal pyroptosis.
 |
Figure 4 Excessive activation of NLRP3 inflammasome after MCAO in Lepr−/− obesity rats. (A) Protein expression of NLRP3 (n = 3 in each group, power value = 0.99, F (3, 8) = 139, P < 0.001), Pro-caspase1 (n = 4 in each group, power value = 0.92, F (3, 12) = 92.23, P < 0.001), Cleaved caspase1 (n = 4 in each group, power value = 0.91, F (3, 12) =584.8, P <0.001), Mature IL-18 (n = 4 in each group, power value = 0.93, F (3, 12) = 172.1, P < 0.001) and Mature IL-1β (n = 4 in each group, power value = 0.99, F (3, 12) = 229.2, P < 0.001) in the cortex penumbra region in each group. (B) qRT-PCR results showed the mRNA expression of NLRP3 in the cortex penumbra region in each group. (n = 4 in each group, power value = 0.81, F (3, 12) =108.1, P < 0.001). (C and D) Representative Immunofluorescence staining (C) and statistical comparison of Relative NLRP3 positive cells (D) in each group (scale bar = 40 μm) (n = 5 in each group, power value = 0.91, F (3, 16) =1398, P < 0.0001). Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s analysis. ****P < 0.0001, **P < 0.01, *P < 0.05 vs WT Sham group, ####P < 0.0001 vs Lepr−/− Sham group, &&&&P < 0.0001, &P < 0.05 vs WT MCAO group. |
Inhibition of NLRP3 Inflammasome Improved Neuronal Pyroptosis in Lepr−/− Obese Rats After MCAO
To further validate whether cerebral ischemia in Lepr−/− obese rats induces neuronal pyroptosis via NLRP3, the NLRP3 inhibitor MCC950 was administered to the Lepr−/− obese rats 1 hour prior to MCAO, so as to observe whether there is a decrease in neuronal pyroptosis. As shown in Figure 5A, the neurological function scores decreased (P<0.05), and improved the neuronal injury (Figure 5B). Western blot results indicated a significant decrease in the expressions of pyroptosis-related proteins GSDMD and GSDMD-N, Pro-caspase-1, Cleaved caspase-1, Mature IL-18, and Mature IL-1β in the MCC950+Lepr−/− MCAO, which were 0.22 times, 0.52 times, 0.48 times, 0.34 times, 0.3 times and 0.28 times that of the DMSO+Lepr−/− MCAO group, respectively (P<0.05) (Figure 5C). These results indicate that inhibiting NLRP3 can alleviate neuronal pyroptosis in Lepr−/− obese rats after MCAO.
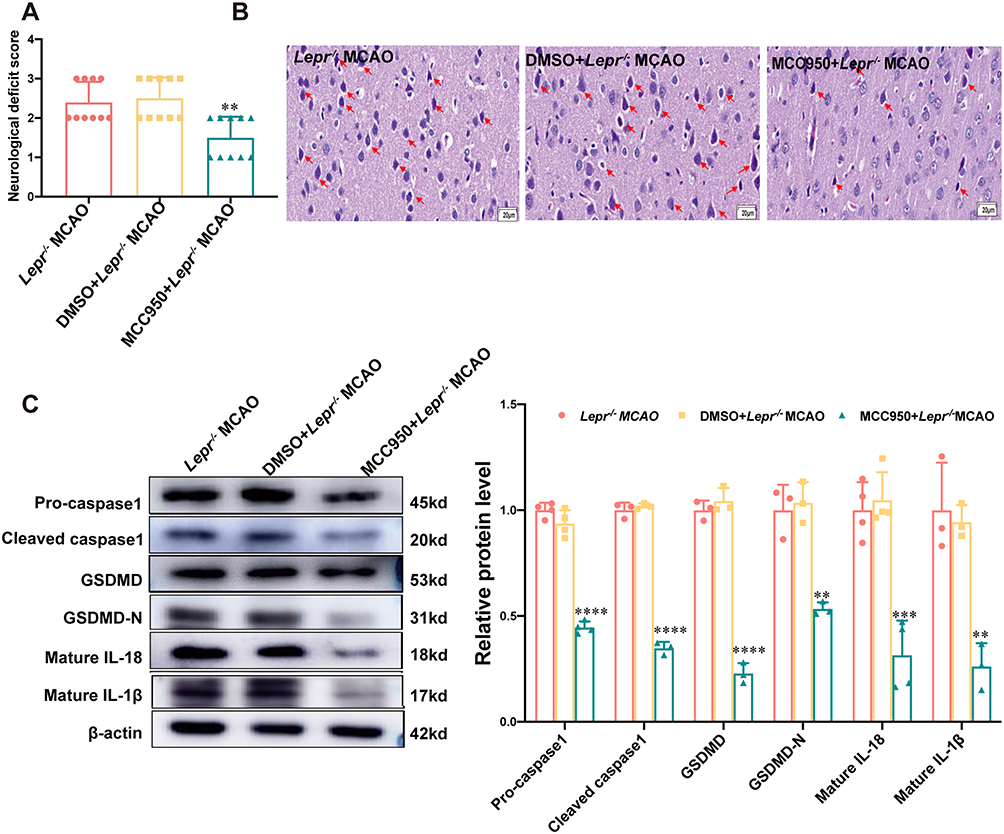 |
Figure 5 Inhibition of NLRP3 reversed neuronal pyroptosis after MCAO in Lepr−/− obesity rats. (A) Zea-Longa score of Lepr−/− obesity rats in each group. (n = 10 in each group, power value = 0.87, KW = 12.567, P = 0.0019). Data are presented as medians±interquartile range. Data were analyzed using the Kruskal–Wallis test. (B) Representative images of HE staining in the cortex penumbra region, with the abnormality neurons indicated through the red arrows (scale bar = 20 μm). (n = 5 in each group). (C) Protein expression of Pro-caspase1 (n = 4 in each group, power value = 0.94, F (2, 9) =188.4, P < 0.001), Cleaved caspase1 (n = 3 in each group, power value = 0.93, F (2, 6) = 554, P < 0.001), GSDMD (n = 3 in each group, power value = 0.86, F (2, 6) =230.2, P < 0.001), N-terminal GSDMD (n = 3 in each group, power value = 0.99, F (2, 6) =28.27, P < 0.001), Mature IL-18 (n = 4 in each group, power value = 0.92, F (2, 9) =27.7, P < 0.001) and Mature IL-1β (n = 3 in each group, power value = 0.97, F (2, 6) = 22.17, P < 0.001) in the cortex penumbra region in each group. Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s analysis. ****P < 0.0001, ***P < 0.001, **P < 0.01 vs DMSO+ Lepr−/− MCAO. |
Leptin Receptor Knockout Over-Activated the AGEs/RAGE Signaling Pathway in Rats After Cerebral Ischemia
The changes in the AGEs/RAGE expression were observed. Western blot results showed that when compared with the WT Sham group, the expressions of AGEs and RAGE proteins in Lepr−/− Sham group exhibited increased, which were 2.12 times and 1.66 times that of the WT Sham group, respectively. (P<0.05). Moreover, the value of AGEs and RAGE proteins expression in the WT MCAO group was 2.86 times and 2.03 times that in the WT Sham group, respectively and the value in the Lepr−/− MCAO group was 2.07 times and 1.66 times that in the Lepr−/− Sham group, respectively. (P<0.05). When compared with the WT MCAO group, the Lepr−/− MCAO group demonstrated a further increase in the expressions of AGEs and RAGE protein, which were 1.53 times and 1.25 times that of the WT MCAO group, respectively. (P<0.05) (Figure 6A). The qRT-PCR results indicated that the value of RAGE mRNA expression in WT MCAO group was 4.75 times that in the WT Sham group, the value in the Lepr−/− MCAO group was 3.04 times that in the Lepr−/− Sham group, and the value in Lepr−/− MCAO group was 1.36 times that in the WT MCAO group) (P<0.05) (Figure 6B). The immunohistochemical staining results revealed that the IOD value of RAGE protein expression in the WT MCAO group was 3.77 times that in the WT Sham group, the IOD value in the Lepr−/− MCAO group was 2.05 times that in the Lepr−/− Sham group, and the IOD value in Lepr−/− MCAO group was 1.32 times that in the WT MCAO group, respectively. (P<0.05) (Figure 6C and D). These findings suggest that MCAO can activate the AGEs/RAGE signaling pathway, and in Lepr−/− obese rats, this pathway is further activated.
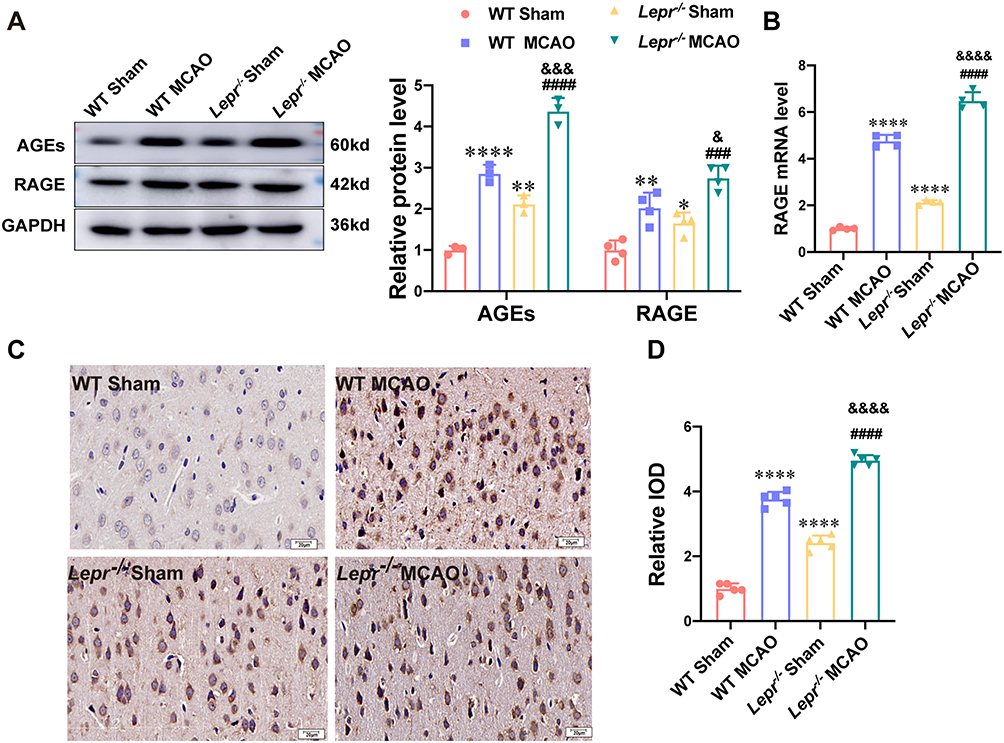 |
Figure 6 AGEs/RAGE signaling pathway was over-activated after MCAO in Lepr−/− obesity rats. (A) Protein expression of AGEs (n = 3 in each group, power value = 0.85, F (3, 8) =121.3, P < 0.001) and RAGE (n = 4 in each group, power value = 0.96, F (3, 12) =25.29, P < 0.001) in the cortex penumbra region in each group. (B) qRT-PCR results showed the mRNA expression of RAGE in the cortex penumbra region in each group. (n = 4 in each group, power value = 0.83, F (3, 12) = 449.2, P < 0.001). (C and D) Representative immunohistochemical staining of RAGE (C) and Relative IOD (D) in each group (scale bar = 20 μm) (n = 5 in each group, power value = 0.91, F (3, 16) =395.1, P < 0.001). Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s analysis. ****P < 0.0001, **P < 0.01,*P < 0.05 vs WT Sham group, ####P < 0.0001,###P < 0.001 vs Lepr−/− Sham group,&&&&P < 0.0001,&&&P < 0.001, &P < 0.05 vs WT MCAO group. |
Inhibition of AGEs/RAGE Signaling Pathway Improved the Neuronal Pyroptosis Mediated by NLRP3 in Lepr−/− Obese Rats After MCAO
The AGEs/RAGE signaling pathway inhibitor FPS-ZM1 was administered to Lepr−/− obese rats to investigate whether the neuronal pyroptosis was alleviated. Compared with the Lepr−/− MCAO + DMSO group, FPS-ZM1 reduced the neurological function score (P<0.05) (Figure 7A) and improved the neuronal injury (Figure 7B). Western blot results demonstrated that, compared with the Lepr−/− MCAO + DMSO group, the Lepr−/− MCAO + FPS-ZM1 group exhibited significantly reduced expression of NLRP3, Pro-caspase-1, Cleaved caspase-1, Mature IL-18, and Mature IL-1β, which were 0.41 times, 0.42 times, 0.48 times, 0.3 times and 0.53 times that of the Lepr−/− MCAO + DMSO group, respectively. (P<0.05) (Figure 7C). The expressions of the pyroptosis-related proteins GSDMD and GSDMD-N were also significantly decreased in the Lepr−/− MCAO + FPS-ZM1 group, which were 0.34 times and 0.59 times that of the Lepr−/− MCAO + DMSO group, respectively. (P<0.05) (Figure 7D). The immunofluorescence staining results demonstrated that, compared with the Lepr−/− MCAO + DMSO group, the Lepr−/− MCAO + FPS-ZM1 group exhibited reduced fluorescence intensity of NLRP3 (Figure 8A and B) and pyroptosis-related protein GSDMD (Figure 8C and D) in the ischemic cortical penumbra, which were 0.38 times and 0.49 times that of the Lepr−/− MCAO + DMSO group, respectively. (P<0.05). These results indicate that inhibiting the AGEs/RAGE signaling pathway can alleviate neuronal pyroptosis in Lepr−/− obese rats after cerebral ischemia-reperfusion through suppressing the activation of NLRP3.
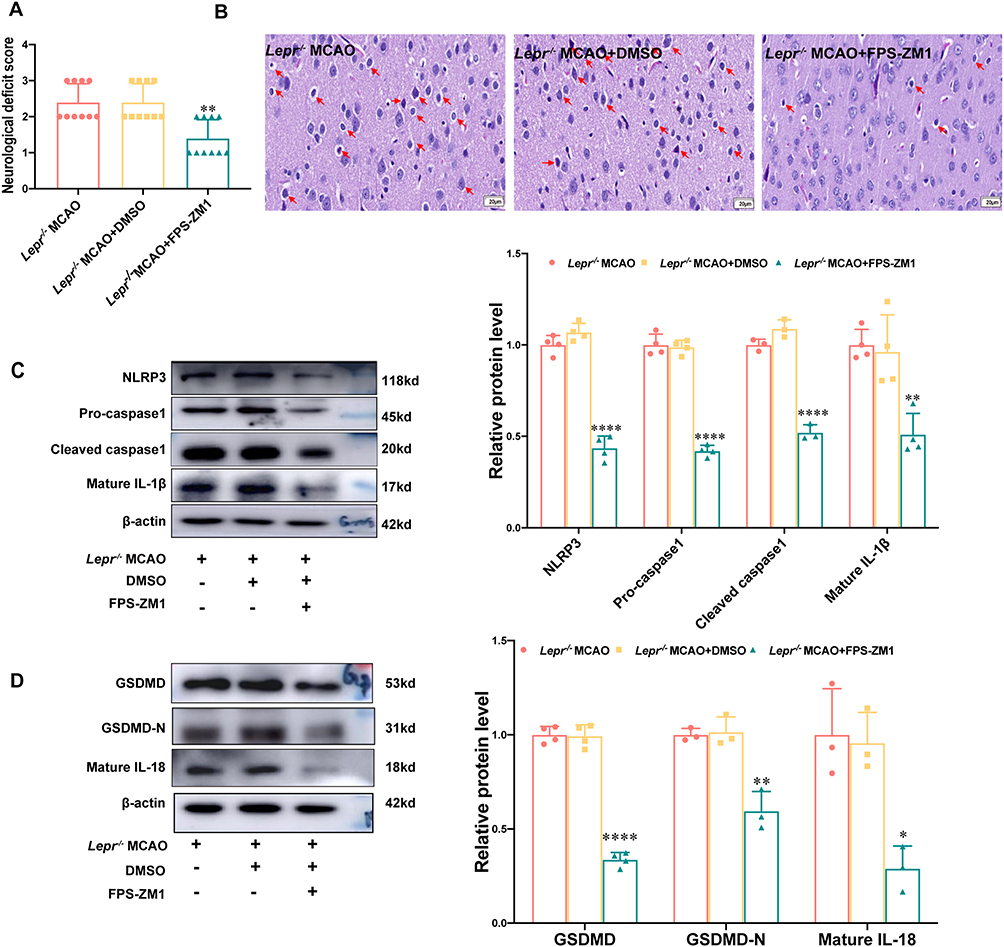 |
Figure 7 FPS-ZM1 improved the neuronal pyroptosis mediated by NLRP3 in Lepr−/− obese rats after MCAO. (A) Zea-Longa score of Lepr−/− obesity rats in each group. (n = 10 in each group, power value = 0.89, KW = 13.596, P = 0.0011). Data are presented as medians±interquartile range. Data were analyzed using the Kruskal–Wallis test. (B) Representative images of HE staining in the cortex penumbra region, with the abnormality neurons indicated through the red arrows (scale bar = 20 μm). (n = 5 in each group). (C and D) Protein expression of NLRP3 (n = 4 in each group, power value = 0.99, F (2, 9) =153.7, P < 0.001), Pro-caspase1 (n = 4 in each group, power value = 0.91, F (2, 9) =224.9, P < 0.001), Cleaved caspase1 (n = 3 in each group, power value = 0.96, F (2, 6) =160.5, P < 0.001), Mature IL-1β (n = 4 in each group, power value = 0.94, F (2, 9) =14.45, P = 0.0016) (C) and GSDMD (n = 4 in each group, power value = 0.97, F (2, 9) =248.6, P < 0.00), N-terminal GSDMD (n = 3 in each group, power value = 0.97, F (2, 6) =27.43, P < 0.001), Mature IL-18 (n = 3 in each group, power value = 0.93, F (2, 6) =9.062, P < 0.001) (D) in the cortex penumbra region in each group. Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s analysis. ****P < 0.0001,***P < 0.001,**P < 0.01,*P < 0.05 vs Lepr−/− MCAO+DMSO group. |
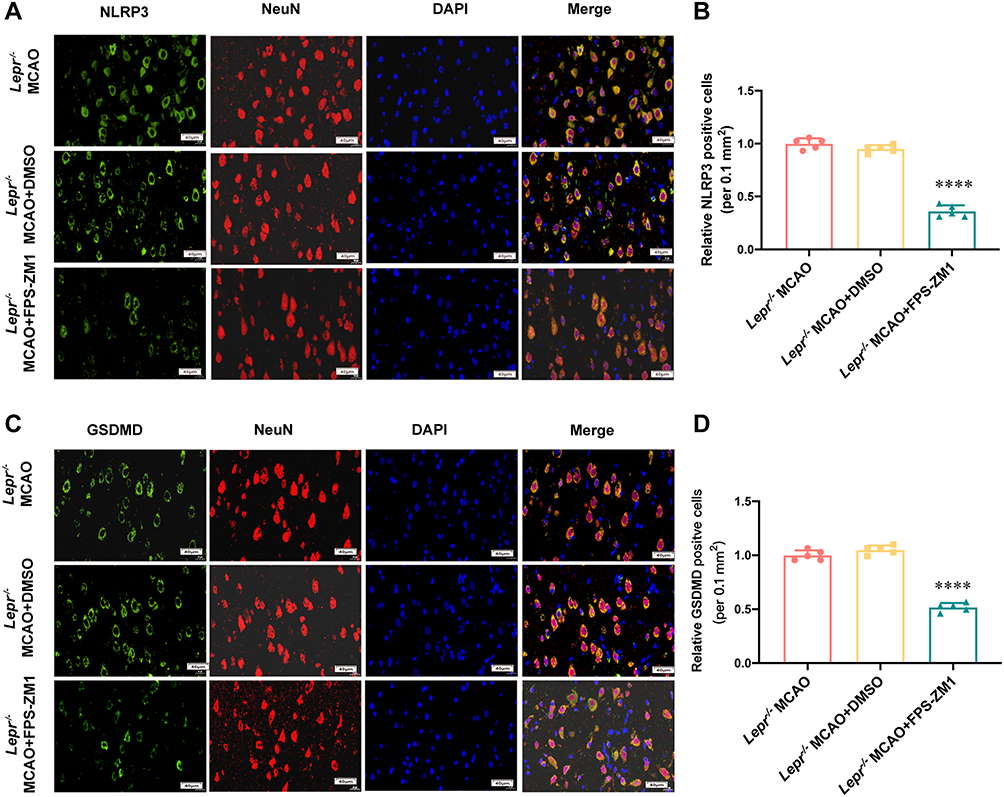 |
Figure 8 FPS-ZM1 inhibited the expression of NLRP3 and GSDMD in Lepr−/− obesity rats after MCAO. (A and B) Representative Immunofluorescence staining (A) and statistical comparison of Relative NLRP3 positive cells (B) in each group (scale bar = 40 μm) (n = 5 in each group, power value = 0.81, F (2,12) =269.8, P < 0.0001). (C and D) Representative Immunofluorescence staining (C) and statistical comparison of Relative GSDMD positive cells (D) in each group (scale bar = 40 μm) (n = 5 in each group, power value = 0.84, F (2, 12) =239.5, P < 0.0001). Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s analysis. ****P < 0.0001 vs Lepr−/− MCAO+DMSO group. |
Discussion
Ischemic stroke is the leading cause of both death and disability.28,29 Obesity is an independent risk factor for cerebral ischemia.32 Indeed, obese individuals exhibit a higher incidence of ischemic stroke.33,34 However, there is ongoing debate regarding whether obesity exacerbates neuronal damage after cerebral ischemia and the specific mechanisms underlying such damage. Our study revealed that Lepr−/− obese rats showed more severe infarct volume after cerebral ischemia compared with their wild-type littermates. The AGEs/RAGE signaling pathway is implicated in the exacerbation of cerebral ischemic injury via modulating the neuronal pyroptosis mediated by NLRP3.
In the present study, Lepr−/− rats were chosen as the research subjects, as generated using CRISPR/Cas9 technology, with a 298 base pair deletion and the insertion of four base pairs in the fourth exon of the leptin receptor gene in SD rats, thereby resulting in the knockout of the Lepr gene (Lepr−/−). In other studies, animal models of obesity are mainly induced via a high-fat diet or genetic modification.35 High-fat diet-induced obesity models exhibit species differences, and even among male SD rats, only some of them become obese, with approximately half exhibiting dietary resistance.36 Zucker rats, a widely used genetic obesity model, result from the hybridization of Merck3M rats and Sherman rats,37 have a complex genetic background, including a missense mutation in the leptin receptor gene, and develop both impaired glucose tolerance and insulin resistance at three months of age.38–40 Another commonly used obesity and diabetes model is the db/db mouse, wherein the severity varies with the genetic background, with B6 background mice having longer survival but transiently elevated blood glucose levels.41 When compared with other obesity models, Lepr−/− obese rats exhibit stable phenotypes, with obesity appearing in early life and glucose and lipids metabolic disturbances during adulthood.42 Previous experiments have confirmed that Lepr−/− rats represent an ideal and reliable obesity model.
We first observed whether Lepr−/− induced obese rats would aggravate cerebral ischemic injury. The results showed that compared with WT littermates, there were obvious aggravation of nerve injury and increased cerebral infarction volume after brain ischemia in Lepr−/− obese rats. Liang et al investigated dysbiosis of the gut microbiota in high-fat diet-induced obese C57 mice and found an increased infarct volume and higher serum inflammatory cytokine levels following MCAO.43 Another study showed that SD rats fed with a high-fat diet for four weeks exhibited higher brain tissue water content and increased infarct volume after MCAO, which were accompanied by increased oxidative stress and apoptosis in the brain tissues.44,45 Emilio Rodríguez-Castro et al found that obese patients in the acute phase of stroke (24 hours after stroke) had stronger inflammatory response and more severe damage. Our results are consistent with the above conclusions. However, some studies have found that obese patients have a better prognosis of cerebral ischemia than non-obese patients, that is, the “obesity paradox”. For example, Emilio Rodríguez-Castro et al further studied the prognosis of obese patients at 1 week and 3 months after cerebral ischemia. Compared with the non-obese patients, the anti-inflammatory response in obese patients was enhanced, offsetting its early inflammatory damage, and the prognosis was improved, thereby reducing the mortality of stroke.4 Wi-Sun Ryu et al studied the association between BMI and long-term mortality after ischemic stroke, and found that the overweight and obese patients’ prognosis was good, probably because they were more likely to receive doctor’s care in clinical practice than underweight people.46 However, these studies are retrospective, and potential influencing factors may confuse the reliability of the results and have certain limitations.
We further observed whether NLRP3-mediated pyroptosis is involved in exacerbating cerebral ischemic injury in Lepr−/− obese rats. We detected the expression of the pyroptosis marker protein GSDMD at both the protein and mRNA levels. Our results showed that following MCAO, the Lepr−/− obese rats exhibited a further increase in the expression of GSDMD and GSDMD-N when compared with the WT littermates. Transmission electron microscopy also revealed more pores in the neurons after cerebral ischemia in Lepr−/− obese rats. Furthermore, we examined the expressions of various pyroptosis-related proteins (Pro-caspase-1 and Cleaved caspase-1) and inflammatory factors (IL-18 and IL-1β) in Lepr−/− obese rats and their WT littermates following cerebral ischemia. The results indicated that the related expression changes were consistent with those of GSDMD. These findings suggest that the mechanism underlying the aggravated neuronal damage seen after MCAO in Lepr−/− obese rats may be related to pyroptosis. Pyroptosis induced by Caspase-1 activation by NLRP3 inflammasome is a classical pathway of pyroptosis. Some studies have demonstrated the association between NLRP3 and the development of many diseases, including stroke, type 2 diabetes, and Alzheimer’s disease.47–49 However, it has not been retrieved whether NLRP3 exacerbates cerebral ischemic injury in obese rats by activating pyroptosis. We further detected the NLRP3 expression changes at both the protein and mRNA levels. The results showed that, when compared with the WT littermates, the Lepr−/− obese rats exhibited an increasing trend in NLRP3 expression, while following cerebral ischemia, there was further activation of the NLRP3 inflammasome. Similarly, immunofluorescence staining showed that when compared with the WT littermates, the Lepr−/− obese rats showed higher fluorescence intensity of NLRP3 in the neurons in the cortical penumbra after cerebral ischemia. The above results suggest that cerebral ischemia can lead to NLRP3 hyperactivation in Lepr−/− obese rats.
When using NLRP3 inhibitors, we observed that the neurological damage was reduced after cerebral ischemia in Lepr−/− obese rats, and the expressions of the related proteins GSDMD, GSDMD-N, Pro-caspase-1, Cleaved caspase-1, as well as the inflammatory factors mature IL-18 and mature IL-1β, were decreased in the Lepr−/− obese rats. This suggests that, in Lepr−/− obese rats, NLRP3 activation-induced pyroptosis may play a crucial role in the exacerbation of cerebral ischemic injury. Further study using an NLRP3 agonist to mimic the effect NLRP3 would provide further evidence to illustrate the role of NLRP3 in the process. Li et al found that anxiety- and depression-like behaviors in db/db obese diabetic mice were associated with the increased expression of NLRP3 in the hippocampal tissue, and inhibiting NLRP3 activation could improve the cognitive behavioral abnormalities in db/db mice.49 In a study of hepatic ischemia-reperfusion in db/db mice, it was found that leptin receptor deficiency exacerbates liver ischemia-reperfusion injury by increasing reactive oxygen production and activating the NLRP3 inflammasome, and inhibiting NLRP3 expression can alleviate liver ischemia-reperfusion injury.50 Our research findings are consistent with the results mentioned above.
AGEs are irreversible polymers formed through a non-enzymatic reaction between reducing sugars, such as glucose, and the free amino groups found on lipids, proteins, and nucleic acids, which involves a series of molecular rearrangements. RAGE, the receptor for AGEs, belongs to the cell surface immunoglobulin superfamily of multiligand receptor members and is located on the cell membrane. The AGEs/RAGE signaling pathway is a cellular inflammatory pathway that stimulates oxidative stress, thereby participating in the pathogenesis of diseases such as diabetes, cardiovascular disorders, neuroinflammation, and neurodegenerative diseases.51 Recent studies have identified elevated expression levels of AGEs and RAGE proteins in the plasma and adipose tissues of obese patients.19 In comparison to wild-type mice, the db/db obese diabetic mouse model, as characterized by leptin receptor deficiency, exhibits the increased expression of both AGEs and RAGE proteins in cardiac tissue.19 In cell experiments, it has been found that AGEs induce the production of inflammatory mediators in adipocytes and macrophages via binding to RAGE, contributing to the development of obesity-related complications.19
In this study, Lepr−/− obese rats, as an animal model of obesity, exhibited an increase in the expressions of AGEs, RAGE proteins and RAGE mRNA in the cortical region when compared with WT littermates. When both WT littermates and Lepr−/− obese rats were subjected to MCAO, a more pronounced increase in the expressions of AGEs, RAGE proteins, and RAGE mRNA was observed in the cortical penumbra neurons of the Lepr−/− obese rats. Immunohistochemical staining of the RAGE proteins showed consistent trends with the Western Blot and qRT-PCR results. This confirms the elevated levels of AGEs and RAGE in the brain tissues of Lepr−/− obese rats, and it is also indicative of the excessive activation of the AGEs/RAGE signaling pathway during cerebral ischemia. Previous research in kidney diseases has demonstrated that prolonged administration of D-ribose can induce NLRP3 activation in renal podocytes via the AGEs/RAGE signaling pathway, thereby mediating podocyte pyroptosis.52 In chronic kidney disease, long-term exposure to AGEs induces the generation of reactive oxygen species through binding to RAGE, activating NLRP3, releasing inflammatory factors, and thus, mediating kidney damage.52 Shen et al found that the administration of the RAGE-specific inhibitor FPS-ZM1 reduces the post-MCAO inflammatory responses seen in SD rats, providing protection against cerebral ischemic injury.15 Similarly, Ren et al found that treatment with FPS-ZM1 suppresses the expression of various inflammatory factors through targeting the AGEs/RAGE signaling pathway, exerting neuroprotective effects against the hippocampal damage induced by liver ischemia-reperfusion.53
In this study, Lepr−/− rats were administered FPS-ZM1 to inhibit the AGEs/RAGE signaling pathway following cerebral ischemia. The related results revealed a reduction in cerebral ischemic injury in the Lepr−/− rats, as manifested by the decreased neurological function scores and a decrease in degenerated and necrotic neurons in the cortical penumbra. Western blot analysis showed that the increased expressions of NLRP3 and pyroptosis-related proteins GSDMD, GSDMD-N, Pro-caspase1, Cleaved caspase1, as well as cytokines IL-18 and IL-1β in the neurons within the penumbra region were inhibited after MCAO. Simultaneously, the fluorescence intensity of NLRP3 and GSDMD in the cortical penumbra neurons was also reduced. The above results suggest that the AGEs/RAGE signaling pathway aggravates cerebral ischemic injury in Lepr−/− obese rats by regulating NLRP3-mediated pyroptosis. As to mechanisms inducing NLRP3 activation, Ajoolabady et al54 found that the reduction or disruption of chloride ion efflux, can lead to changes in intracellular osmotic pressure and membrane potential, which in turn activated the NLRP3 inflammasome to induce pyroptosis. Therefore, the change of the status of chloride ion channels may be an important factor in the activation of NLRP3 during cerebral ischemia.
We specifically investigated the role of the AGEs/RAGE/NLRP3 signaling pathway in the induction of pyroptosis during ischemic brain injury in Lepr−/− obese rats. Whereas, there are still some limitations in the present study. Firstly, it has been shown that AGEs/RAGE signaling pathway is also related to oxidative stress damage. For example, AGEs significantly increased MDA production and decreased GSH content and and SOD activity in rat primary microglia.55 Inhibiting AGEs/RAGE signaling pathway can effectively reduce oxidative stress in the kidneys of rats with high sugar-fat diet.56 Secondly, in the present study, we only examined the level of IL-18 and IL-1β due to their close association with pyroptosis. However, some studies57–59 have shown that the activation of the AGEs/RAGE pathway generally leads to the upregulation of pro-inflammatory cytokines, such as IL-6, CRP, and TNF-α which in turn amplifies the inflammatory response and further activates the NLRP3 inflammasome, exacerbating neuronal pyroptosis. In cell cultures, AGEs triggered the transformation of bone marrow-derived macrophages into the M1 phenotype.60 At last, the lack of detection of blood-brain barrier was also a limitation in the present study. Studies have shown that ischemia-induced pyroptosis can lead to the destruction of the blood-brain barrier (BBB), and inhibiting pyroptosis may improve BBB integrity.61,62 Therefore, further study focusing on assessing the oxidative stress markers, the inflammatory cytokines and macrophage polarization and the BBB integrity would provide more information for a better understanding of the effect of AGEs/RAGE/NLRP3 signaling pathway in regulating NLRP3-mediated neuronal pyroptosis in ischemic injury in obese patients.
Conclusions
In conclusion, the present study investigated for the first time the mechanism of exacerbated cerebral ischemic injury in Lepr−/− obese rats, which served as an obese animal model. The findings demonstrate that Lepr−/− obese rats exhibit overactivation of the AGEs/RAGE signaling pathway after cerebral ischemia, followed by neuronal pyroptosis mediated by NLRP3 inflammasome, which aggravates the cerebral ischemia-reperfusion injury.
Data Sharing Statement
Data will be made available on request. The original data generated in the study are included in the article; further inquiries can be directed to the corresponding author.
Ethics Approval
The study was conducted in accordance with the Declaration of Helsinki, and all animal experiments were reviewed and approved by the Laboratory Animal Ethical and Welfare Committee of Hebei Medical University (Approval No. IACUC-Hebmu–2020013). All experiments were conducted in compliance with the ARRIVE guidelines.
Acknowledgments
Ling Zhao and Shichao Li are co-first authors for this study. The authors wish to thank all participants for supporting this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81771253, 81971228 and 82072531) and the Natural Science Foundation of Hebei Province (Nos. H2019206702, H2021206160 and H2024206037). All experiments were conducted in compliance with the ARRIVE guidelines.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Heuschmann PU, Kircher J, Nowe T, et al. Control of main risk factors after ischaemic stroke across Europe: data from the stroke-specific module of the EUROASPIRE III survey. Europ J Prevent Cardiol. 2015;22:1354–1362. doi:10.1177/2047487314546825
2. Xie T, Yang R, Zhang X, et al. Fecal microbiota transplantation alleviated cerebral ischemia reperfusion injury in obese rats. Tohoku J Exp Med. 2022;259:49–55. doi:10.1620/tjem.2022.J094
3. Yu Z, Zheng L, Geng Y, et al. FTO alleviates cerebral ischemia/reperfusion-induced neuroinflammation by decreasing cGAS mRNA stability in an m6A-dependent manner. Cell. Signalling. 2023;109:110751. doi:10.1016/j.cellsig.2023.110751
4. Rodríguez-Castro E, Rodríguez-Yáñez M, Arias-Rivas S, et al. Obesity paradox in ischemic stroke: clinical and molecular insights. Translat Stroke Res. 2019;10:639–649. doi:10.1007/s12975-019-00695-x
5. Forlivesi S, Cappellari M, Bonetti B. Obesity paradox and stroke: a narrative review. Eating Weight Disord. 2021;26:417–423. doi:10.1007/s40519-020-00876-w
6. Lee SH, Jung JM, Park MH. Obesity paradox and stroke outcomes according to stroke subtype: a propensity score-matched analysis. Int J Obesity. 2023;47:669–676. doi:10.1038/s41366-023-01318-0
7. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42:245–254. doi:10.1016/j.tibs.2016.10.004
8. Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi:10.1038/nature15514
9. Sun R, Peng M, Xu P, et al. Low-density lipoprotein receptor (LDLR) regulates NLRP3-mediated neuronal pyroptosis following cerebral ischemia/reperfusion injury. J Neuroinflammation. 2020;17:330. doi:10.1186/s12974-020-01988-x
10. Liu J, Zheng J, Xu Y, et al. Enriched environment attenuates pyroptosis to improve functional recovery after cerebral ischemia/reperfusion injury. Front Aging Neurosci. 2021;13:717644. doi:10.3389/fnagi.2021.717644
11. Li C, Xu MM, Wang K, et al. Macrophage polarization and meta-inflammation. Translat Res. 2018;191:29–44. doi:10.1016/j.trsl.2017.10.004
12. Ying W, Fu W, Lee YS, et al. The role of macrophages in obesity-associated islet inflammation and β-cell abnormalities. Nat Rev Endocrinol. 2020;16:81–90. doi:10.1038/s41574-019-0286-3
13. Wani K, AlHarthi H, Alghamdi A, et al. Role of NLRP3 inflammasome activation in obesity-mediated metabolic disorders. Int J Environ Res Public Health. 2021;19:18. doi:10.3390/ijerph18020511
14. Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature Med. 2011;17:179–188. doi:10.1038/nm.2279
15. Shen L, Zhang T, Yang Y, et al. FPS-ZM1 alleviates neuroinflammation in focal cerebral ischemia rats via blocking ligand/RAGE/DIAPH1 pathway. ACS Chem Neurosci. 2021;12:63–78. doi:10.1021/acschemneuro.0c00530
16. Son S, Hwang I, Han SH, et al. Advanced glycation end products impair NLRP3 inflammasome-mediated innate immune responses in macrophages. J Biol Chem. 2017;292:20437–20448. doi:10.1074/jbc.M117.806307
17. Ma L, Carter RJ, Morton AJ, et al. RAGE is expressed in pyramidal cells of the hippocampus following moderate hypoxic-ischemic brain injury in rats. Brain Res. 2003;966:167–174. doi:10.1016/s0006-8993(02)04149-5
18. Reddy VP, Obrenovich ME, Atwood CS, et al. Involvement of Maillard reactions in Alzheimer disease. Neurotox Res. 2002;4:191–209. doi:10.1080/1029840290007321
19. Plante E, Menaouar A, Danalache BA, et al. Treatment with brain natriuretic peptide prevents the development of cardiac dysfunction in obese diabetic db/db mice. Diabetologia. 2014;57:1257–1267. doi:10.1007/s00125-014-3201-4
20. Wu G, Gu W, Cheng H, et al. Huangshan maofeng green tea extracts prevent obesity-associated metabolic disorders by maintaining homeostasis of gut microbiota and hepatic lipid classes in leptin receptor knockout rats. Foods. 2022;12:11. doi:10.3390/foods11192939
21. Yao F, Jiang DD, Guo WH, et al. FABP4 inhibitor attenuates inflammation and endoplasmic reticulum stress of islet in leptin receptor knockout rats. Eur Rev Med Pharmacol Sci. 2020;24:12808–12820. doi:10.26355/eurrev_202012_24182
22. Saleh MC, Connell BJ, Saleh TM. Estrogen may contribute to ischemic tolerance through modulation of cellular stress-related proteins. Neurosci Res. 2009;63:273–279. doi:10.1016/j.neures.2009.01.004
23. Li X, Yao M, Li L, et al. Aloe-emodin alleviates cerebral ischemia-reperfusion injury by regulating microglial polarization and pyroptosis through inhibition of NLRP3 inflammasome activation. Phytomedicine. 2024;129:155578. doi:10.1016/j.phymed.2024.155578
24. Yang F, Wang Z, Zhang JH, et al. Receptor for advanced glycation end-product antagonist reduces blood-brain barrier damage after intracerebral hemorrhage. Stroke. 2015;46:1328–1336. doi:10.1161/strokeaha.114.008336
25. Longa EZ, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi:10.1161/01.str.20.1.84
26. Shang S, Sun F, Zhu Y, et al. Sevoflurane preconditioning improves neuroinflammation in cerebral ischemia/reperfusion induced rats through ROS-NLRP3 pathway. Neurosci Lett. 2023;801:137164. doi:10.1016/j.neulet.2023.137164
27. Ward R, Li W, Abdul Y, et al. NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharmacol Res. 2019;142:237–250. doi:10.1016/j.phrs.2019.01.035
28. Campbell BCV, De Silva DA, Macleod MR, et al. Ischaemic stroke. Nature Reviews Disease Primers. 2019;5:70. doi:10.1038/s41572-019-0118-8
29. Stinear CM, Lang CE, Zeiler S, et al. Advances and challenges in stroke rehabilitation. Lancet Neurol. 2020;19:348–360. doi:10.1016/s1474-4422(19)30415-6
30. Yu P, Zhang X, Liu N, et al. Pyroptosis: mechanisms and diseases. Signal Transduct Target Therap. 2021;6:128. doi:10.1038/s41392-021-00507-5
31. Reid MM, Kautzmann MI, Andrew G, et al. NPD1 Plus RvD1 mediated ischemic stroke penumbra protection increases expression of pro-homeostatic microglial and astrocyte genes. Cell Mol Neurobiol. 2023;43:3555–3573. doi:10.1007/s10571-023-01363-3
32. Isozumi K. Obesity as a risk factor for cerebrovascular disease. Keio J Med. 2004;53:7–11. doi:10.2302/kjm.53.7
33. Osmond JM, Mintz JD, Dalton B, et al. Obesity increases blood pressure, cerebral vascular remodeling, and severity of stroke in the Zucker rat. Hypertension. 2009;53:381–386. doi:10.1161/hypertensionaha.108.124149
34. Deutsch C, Portik-Dobos V, Smith AD, et al. Diet-induced obesity causes cerebral vessel remodeling and increases the damage caused by ischemic stroke. Microvascular Res. 2009;78:100–106. doi:10.1016/j.mvr.2009.04.004
35. Haley MJ, Lawrence CB. Obesity and stroke: can we translate from rodents to patients? J Cereb Blood Flow Metab. 2016;36:2007–2021. doi:10.1177/0271678x16670411
36. Sreenan S, Sturis J, Pugh W, et al. Prevention of hyperglycemia in the Zucker diabetic fatty rat by treatment with metformin or troglitazone. A J Physiol. 1996;271:E742–7. doi:10.1152/ajpendo.1996.271.4.E742
37. Argilés JM. The obese Zucker rat: a choice for fat metabolism 1968-1988: twenty years of research on the insights of the Zucker mutation. Prog Lipid Res. 1989;28:53–66. doi:10.1016/0163-7827(89)90007-6
38. Bray GA, York DA. Hypothalamic and genetic obesity in experimental animals: an autonomic and endocrine hypothesis. Physiol Rev. 1979;59:719–809. doi:10.1152/physrev.1979.59.3.719
39. Bray GA. The Zucker-fatty rat: a review. Federation Proc. 1977;36:148–153.
40. Lutz TA, Woods SC. Overview of animal models of obesity. Curr Protocol Pharmacol. 2012. doi:10.1002/0471141755.ph0561s58
41. Coleman DL, Hummel KP. The influence of genetic background on the expression of the obese (Ob) gene in the mouse. Diabetologia. 1973;9:287–293. doi:10.1007/bf01221856
42. Bao D, Ma Y, Zhang X, et al. Preliminary characterization of a leptin receptor knockout rat created by CRISPR/Cas9 system. Sci Rep. 2015;5:15942. doi:10.1038/srep15942
43. Liang J, Zhang M, Wang H, et al. Cholestyramine resin administration alleviated cerebral ischemic injury in obese mice by improving gut dysbiosis and modulating the bile acid profile. Exp Neurol. 2023;359:114234. doi:10.1016/j.expneurol.2022.114234
44. Liu Z, Sanossian N, Starkman S, et al. Adiposity and outcome after ischemic stroke: obesity paradox for mortality and obesity parabola for favorable functional outcomes. Stroke. 2021;52:144–151. doi:10.1161/strokeaha.119.027900
45. Hornung V, Latz E. Critical functions of priming and lysosomal damage for NLRP3 activation. European J Immunol. 2010;40:620–623. doi:10.1002/eji.200940185
46. Ryu WS, Lee SH, Kim CK, et al. Body mass index, initial neurological severity and long-term mortality in ischemic stroke. Cerebrovas Dis. 2011;32:170–176. doi:10.1159/000328250
47. Liu SB, Mi WL, Wang YQ. Research progress on the NLRP3 inflammasome and its role in the central nervous system. Neurosci Bull. 2013;29:779–787. doi:10.1007/s12264-013-1328-9
48. Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi:10.1016/j.jneuroim.2006.11.014
49. W LC, Deng MZ, Gao ZJ, et al. Effects of compound K, a metabolite of ginsenosides, on memory and cognitive dysfunction in db/db mice involve the inhibition of ER stress and the NLRP3 inflammasome pathway. Food Funct. 2020;11:4416–4427. doi:10.1039/c9fo02602a
50. Sharma D, Kanneganti TD. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213:617–629. doi:10.1083/jcb.201602089
51. Gaens KH, Stehouwer CD, Schalkwijk CG. Advanced glycation endproducts and its receptor for advanced glycation endproducts in obesity. Curr Opin Lipidol. 2013;24:4–11. doi:10.1097/MOL.0b013e32835aea13
52. Yeh WJ, Yang HY, Pai MH, et al. Long-term administration of advanced glycation end-product stimulates the activation of NLRP3 inflammasome and sparking the development of renal injury. J Nutr Biochem. 2017;39:68–76. doi:10.1016/j.jnutbio.2016.09.014
53. Ren L, Yan H. Targeting AGEs-RAGE pathway inhibits inflammation and presents neuroprotective effect against hepatic ischemia-reperfusion induced hippocampus damage. Clin Res Hepatol Gastroenterol. 2022;46:101792. doi:10.1016/j.clinre.2021.101792
54. Ajoolabady A, Nattel S, Lip GYH, et al. Inflammasome signaling in atrial fibrillation: JACC state-of-the-art review. J Am Coll Cardiol. 2022;79:2349–2366. doi:10.1016/j.jacc.2022.03.379
55. Shen C, Ma Y, Zeng Z, et al. RAGE-specific inhibitor FPS-ZM1 attenuates AGEs-induced neuroinflammation and oxidative stress in rat primary microglia. Neurochem Res. 2017;42:2902–2911. doi:10.1007/s11064-017-2321-x
56. Francisqueti-Ferron FV, Ferron AJT, Altomare A, et al. Gamma-oryzanol reduces renal inflammation and oxidative stress by modulating AGEs/RAGE axis in animals submitted to high sugar-fat diet. J Brasileiro de Nefrol. 2021;43:460–469. doi:10.1590/2175-8239-jbn-2021-0002
57. Hu R, Wang MQ, Ni SH, et al. Salidroside ameliorates endothelial inflammation and oxidative stress by regulating the AMPK/NF-κB/NLRP3 signaling pathway in AGEs-induced HUVECs. Eur J Pharmacol. 2020;867:172797. doi:10.1016/j.ejphar.2019.172797
58. Jia HL, Lu CQ, Wang J, et al. [The Effect of Interference of NLRP3 with shRNA on AGEs-induced inflammatory response in myocardial cell]. Sichuan Da Xue Xue Bao Yi Xue Ban. 2019;50:7–12. Dutch
59. Cao X, Xia Y, Zeng M, et al. Caffeic acid inhibits the formation of advanced glycation end products (AGEs) and mitigates the ages-induced oxidative stress and inflammation reaction in human umbilical vein endothelial cells (HUVECs). Chem Biodivers. 2019;16:e1900174. doi:10.1002/cbdv.201900174
60. Huang HL, Kuo CS, Chang TY, et al. An oral absorbent, AST-120, restores vascular growth and blood flow in ischemic muscles in diabetic mice via modulation of macrophage transition. J Mol Cell Cardiol. 2021;155:99–110. doi:10.1016/j.yjmcc.2021.03.001
61. Zhou S, Li Y, Hong Y, et al. Puerarin protects against sepsis-associated encephalopathy by inhibiting NLRP3/Caspase-1/GSDMD pyroptosis pathway and reducing blood-brain barrier damage. Eur J Pharmacol. 2023;945:175616. doi:10.1016/j.ejphar.2023.175616
62. Liang Y, Song P, Chen W, et al. Inhibition of caspase-1 ameliorates ischemia-associated blood-brain barrier dysfunction and integrity by suppressing pyroptosis activation. Front Cell Neurosci. 2020;14:540669. doi:10.3389/fncel.2020.540669
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.