")
Back to Journals » Journal of Blood Medicine » Volume 16
Utilization of Fetal Hemoglobin Parameters in Predicting Clinical Severity of Sickle Cell Disease: Retrospective Study From a Tanzanian Cohort
Authors Haji HM , Urio F, Nkya S, Chamba C, Nasser A, Lyimo M, Ally M , Mawalla W, Jonathan A, Kidenya B , Mutagonda R , Chirande L , Ruggajo P, Ambrose E , Metta E , Balandya E , Makani J
Received 3 October 2024
Accepted for publication 7 May 2025
Published 19 July 2025 Volume 2025:16 Pages 321—330
DOI https://doi.org/10.2147/JBM.S493425
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Hadiya M Haji,1,2 Florence Urio,1,3,4 Siana Nkya,3,4 Clara Chamba,2 Ahlam Nasser,2 Magdalena Lyimo,2,3 Mwashungi Ally,1– 3 William Mawalla,2 Agnes Jonathan,1,3 Benson Kidenya,1,5 Ritah Mutagonda,1,6 Lulu Chirande,1,7 Paschal Ruggajo,1,8 Emmanuela Ambrose,1,3,9 Emmy Metta,1,10 Emmanuel Balandya,1,3 Julie Makani1– 3
1Sickle Pan-African Research Consortium (SPARCO) - Tanzania, Dar es Salaam, Tanzania; 2Department of Haematology and Blood Transfusion, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania; 3Sickle Cell Program, Department of Hematology and Blood Transfusion, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania; 4Department of Biochemistry and Molecular Biology, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania; 5Department of Biochemistry and Molecular Biology, Catholic University of Health and Allied Sciences, Mwanza, Tanzania; 6Department of Clinical Pharmacy and Pharmacology, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania; 7Department of Paediatrics and Child Health, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania; 8Medical College, Aga Khan University, Dar es Salaam, Tanzania; 9Department of Pediatrics, Bugando Medical Centre, Mwanza, Tanzania; 10Department of Behavioral Sciences, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania
Correspondence: Hadiya M Haji, Department of Hematology and Blood Transfusion, Muhimbili University of Health and Allied Sciences, P.O. Box: 65001, Dar es salaam, Tanzania, Tel +255 777753436, Email [email protected]
Background: Fetal hemoglobin (HbF) is found at a measurable amount in red blood cells (RBCs) called F cells. High fetal hemoglobin (HbF) levels are linked with milder forms of sickle cell disease (SCD). However, some patients with high HbF levels still have severe symptoms. This variability has been associated with HbF per F cell (HbF/F cell) concentration; thus, it is hypothesized that high HbF/F cell (≥ 10 pg) is crucial in determining SCD disease severity rather than the overall HbF and F cell levels. This study assessed the utility of these three HbF parameters as predictors of SCD clinical events in Tanzania.
Methods: A retrospective cohort study was conducted at Muhimbili University of Health and Allied Sciences, involving 92 SCD individuals aged ≥ 6 years, not on hydroxyurea, between September 2022 and February 2023. Data was collected from the Sickle Pan-African Research Consortium (SPARCO)-Tanzania registry. HbF/F cell was calculated as: HbF/F cell = (HbF% × MCH pg)/F cell%. STATA version 15 was used to analyze the association between HbF parameters and clinical events measured by ordinal logistic regression. A p-value < 0.05 was considered statistically significant.
Results: Of the 92 SCD participants, the median age was 16 (IQR: 10– 21) years, 53 (57.6%) were below 18 years, and males were 48 (52.2%). Eighty-two patients (89.1%) had HbF/F cells below 10pg. Males had significantly higher HbF/F cell levels with a median of 6.4 (IQR: 4.3– 9.5) pg compared to females 5.3 (IQR: 3.5– 6.5) pg (p-value = 0.004). Although, we did not observe a statistically significant association between HbF/F cell with clinical parameters, increased HbF and F cell percentages correlated with reduced odds of multiple blood transfusions by 11% (p-value = 0.016) and 3% (p-value = 0.020), respectively.
Conclusion: In this cohort, HbF and F cell levels remain important predictors of disease severity, as higher levels predicted reduced requirement for multiple blood transfusions in SCD patients, while HbF/F cells did not correlate with SCD clinical events.
Keywords: sickle cell, F cell, fetal hemoglobin per F cell, clinical parameters, disease severity
Introduction
Sickle cell disease (SCD) results from a mutation in codon 6 of the β-globin gene, replacing Adenine (A) with Thymine (T) and causing the substitution of glutamic acid by valine amino acid. This leads to the formation of abnormal hemoglobin S (HbS), replacing normal adult hemoglobin A (HbA). Under low oxygen tension, HbS polymerizes, leading to red blood cells (RBCs) sickling. The rate and extent of polymerization depend on the duration and severity of deoxygenation. This leads to vaso occlusive crisis which is characterized by pain episodes and end organ failure.1–3 Vaso-occlusion in sickle cell disease (SCD) results from blocked blood vessels, thrombosis, and fat embolization, leading to hypoxia, ischemia, and inflammation. The combination of these factors makes SCD pain uniquely severe.4
F cells, a subset of RBCs, contain measurable amounts of fetal hemoglobin (HbF), a key modifier of SCD complications. HbF levels and intracellular HbS concentration influence the polymerization process. HbF and its hybrid form (α2βSγ) have high oxygen affinity, inhibiting HbS polymerization a critical factor in SCD pathophysiology. By reducing intracellular HbS concentration, HbF decreases the likelihood of sickling, serving as a protective disease modifier.1–3,5,6 HbF production in SCD individuals varies due to multiple factors, primarily genetics. The Arab-Indian and Senegal haplotypes are linked to higher HbF levels (20–30%), while the Bantu haplotype, common in Africans, including Tanzanians, is associated with lower HbF production.7,8 Different levels of HbF are associated with varied clinical presentation, with higher levels linked to milder disease manifestations.3,6 Furthermore, the definition of a high HbF level varies, with some studies setting the cutoff at ≥10% and others as low as 5.4%. Nevertheless, higher HbF levels, regardless of the threshold, have been linked to lower mortality.6 However, individuals with SCD exhibit variable levels and distributions of HbF in their RBCs, leading to diverse symptoms. Clinical presentations vary widely among individuals, ranging from asymptomatic to severe disease.1,6 While the association between HbF and clinical manifestations has traditionally focused on absolute HbF levels, there are reports of patients with high HbF levels still developing severe disease. Additionally, the response to hydroxyurea, a medication inducing HbF production, varies among patients, with some responding to treatment and some not, possibly due to uneven distribution of HbF in treated patients.1,3 Therefore, it is suggested that the distribution of HbF among F-cells is more crucial in predicting disease severity in SCD. It has been hypothesized that the high HbF/F cell (≥10pg) is associated with milder disease in SCD, rather than the overall percentage of HbF and F cell since the later do not inform the number of F-cells with high enough HbF concentration to protect against polymerization.9 The therapeutic impact of HbF/F cell levels may vary across populations due to genetic variations such as sickle cell haplotypes, environmental variances such as infection, geographical background, and biological factors such as age and sex.3,10
Despite studies in Asia, Europe and America associating overall HbF levels with clinical presentations of SCD,1 little research has been done on the concentration of HbF within F cell (HbF/F cell) as a predictor of the clinical severity of SCD. Hypothetical modelling predicted that Hb F/F cell is the most critical element in the pathophysiology of sickle cell anemia.3 A study in Uganda suggested that high HbF levels are associated with milder disease.11 In Tanzania, a baseline study explored the variability of HbF and HbF/F cell among SCD patients but did not investigate their association with clinical presentations.12 Therefore, this study aimed to evaluate the utility of HbF, F cells and Hb F/F cell as predictors of the severity of clinical presentation of SCD in patients of Tanzanian origin.
Materials and Methods
This retrospective cohort study was conducted at Muhimbili University of Health and Allied Sciences (MUHAS) between September 2022 and February 2023. It involved a secondary analysis of data from 92 individuals with SCD, stored at MUHAS in the SPARCO-Tanzania registry. The patients, who attended at Muhimbili National Hospital, Amana Regional Hospital, and Temeke Regional Hospital, were included in the parent study through serial sampling. Our study was nested within an ongoing parent study titled
Comparative Study of Sickle Cell Disease (SCD) Modifiers in Ghana, Nigeria, and Tanzania: Investigation of Fetal Hemoglobin Parameters and Clinical Manifestations.
Inclusion Criteria
Tanzanian individuals confirmed to have SCD through Hemoglobin Electrophoresis or High-Performance Liquid Chromatography (HPLC) showing HbS >70%, with HbF levels measured at or above the age of 6 years, and with complete data in the SPARCO-Tanzania database were included. Individuals with SCD who were not in a steady state or who were on hydroxyurea therapy were excluded. All 92 participants with complete data in the SPARCO-Tanzania database were enrolled in the study.
Data Collection
Socio-demographic and clinical data were collected from the SPARCO-Tanzania database, and Case Report Form (CRF) was filled out during the ongoing parental study. Socio-demographic data included age, sex and residence, and clinical data constituted the clinical presentation, history of painful crises, blood transfusions, febrile illnesses, acute chest syndrome, dactylitis, hospital admissions and blood counts. Blood Counts were measured by an automated haematology analyser (Sysmex XT 2000i Kobe, Japan), HbF levels were measured by HPLC machine Variant II (Biorad, Hercules, CA, USA), and the percentage of F cells was measured by flow cytometry (Becton Dickinson).
Measurement of Variables
The study focuses on clinical parameters, such as painful crises, blood transfusions, febrile illnesses, dactylitis, priapism, acute chest syndrome, and hospital admissions, as the outcome variables. These recurrent events serve as dependent variables. The independent variables, that predict fetal hemoglobin (HbF) parameters, include age and sex. Additionally, intermediate variables, such as HbF (%), F cells (%) and HbF per F cell (pg), are considered predictors of the outcome, influencing the occurrence of the clinical parameters.
Data Management and Statistical Analysis
In this study, the HbF and F cells level were categorized as low, intermediate and high, based on the distribution of the HbF and F cell levels of our study participants. The cut-off points for low, intermediate and high HbF levels used were ≤2.3%, 2.4%–7.1% and ≥7.2% respectively. The cut-off points for low, intermediate and high F cell levels were ≤13.2%, 13.3%–32.4% and ≥32.5% respectively. The cut-off points of 2.3% and 7.1% for HbF as well as 13,2% and 32.4% for F cells represent the 25th and 75th percentiles, respectively, in the distribution of these parameters in our study population. HbF/F cell was categorized as low and high whereby, the cut-off levels for low and high HbF/ F cell concentrations were (<10 pg) and (≥10 pg), respectively.3 Since there was no throughput method for obtaining the ratio of HbF to F cell levels, it was then calculated using the following formula: Hb F/F cell = (MCH (pg) × HbF %)/F cell %. However, this method assumes that each F-cell contains an equal amount of HbF.2,3
Recorded clinical parameters such as febrile, priapism, pain crises, hospital admission, dactylitis and blood transfusion were observed in their frequencies of occurrence in a lifetime. The occurrence and frequency of clinical events were self-reported by participants or participant parent/guardian whereby; the specific asked clinical parameter (recurrent event) was graded according to their number of occurrences ie Noted by “No” for no occurrence, “<5” occurrence of less than five times, “5–10” occurrence of five to ten times and “>10” occurrence of more than ten times in a life time. The “recurrent events” are clinical parameters that measure the severity of the disease by their frequency of occurrence in a specific time (such as per year) to a SCD individual.13 Since the occurrence of recurrent events was observed in a fixed lifetime during the study period, it was difficulty to categorize individuals of different age groups and length of lifetime into one group of severe or mild disease. Therefore, they were narrated as observed.
The data from the SPARCO-Tanzania registry are accessible within Tanzania. The data was collected on a Microsoft Excel spreadsheet and later exported to STATA version 15 for analysis. Categorical variables were summarized as frequencies and percentages, while continuous variables were summarized using median and interquartile ranges. The Mann–Whitney test was used to compare the fetal hemoglobin levels between the different social demographic groups. Since HbF parameters (levels of HbF (%), F cells (%) and HbF/ F cell (pg)) have collinearity, we only used univariable ordinal logistic regression to assess the crude association between fetal hemoglobin parameters (predictors) and the frequency of the clinical parameters (outcomes) such as febrile, priapism, pain crises, hospital admission, dactylitis and blood transfusion. Crude odds ratios with their 95% confidence interval (CI) were computed and a p-value of less than 0.05 was considered statistically significant in determining the strength of these associations.
Ethical Consideration
The Study Adheres to the Ethical Guidelines Set Forth by the Declaration of Helsinki
Ethical approvals for SPARCO-Tanzania and the parental study “Comparative study of Sickle Cell Disease (SCD) modifiers in Ghana, Nigeria and Tanzania; investigation of HbF parameters and clinical manifestation” were obtained from the MUHAS Research Ethics Committee (numbers MUHAS-REC-12-2020-453 and MUHAS-REC- 01-2021-463, respectively). For the parental study, all participants provided written informed consent. Each eligible parent/guardian was given a thorough description of the study and its objective, as well as assurance that his or her decision to participate or not participate would have no bearing on his or her child’s care. Participants could change their minds and leave the study at any time. There were no additional patients enrolled in the current study. Only collected data from SPARCO -Tanzania registry and through the ongoing parental study (Comparative Study of Sickle Cell Disease (SCD) Modifiers in Ghana, Nigeria, and Tanzania: Investigation of Fetal Hemoglobin Parameters and Clinical Manifestations) has been used within Tanzania. The informed consent from the participants for the current study was not obtained, and the requirement for consent was waived by an ethic committee (Institutional Review Board) for the current data analysis. To ensure confidentiality, data has been kept on computers and protected by passwords.
Results
Among the 92 SCD patients recruited, 53 (57.6%) were children under 18 years of age, and 39 (42.4%) were adults. The median age was 16 (IQR: 10–21) years, and males were 48 (52.2%). The majority of the patients 87 (96.4%) resided in Dar es Salaam (Table 1).
 |
Table 1 Social Demographic Characteristics of the Study Participants (N = 92) |
Of the 92 patients, 69 (75.0%), had low and intermediate HbF levels (levels <7.2%), with a median level of 4.5% (IQR: 2.3–7.2). Similarly, the majority of the patients 72/92 (78.3%), had F cell levels that fell within the low and intermediate range (levels <32.5%), with a median of 21.6% (IQR: 13.2–32.5). Consequently, a significant proportion of the study participants 82 (89.1%) was found to have low levels of HbF/F cell, with a median of 5.6 (IQR: 4.0–8.0) pg (Table 2).
 |
Table 2 Levels of Fetal Hemoglobin Parameters of the Study Participants (N=92) |
Among the 92 study participants, 76 (82.6%) and 75 (81.5%) reported had more than 10 episodes of pain crises and febrile illnesses, respectively, while 50 (54.3%), 18 (19.6%) and 15 (16.3%) of the study participants reported less than 5 episodes of blood transfusion, hospital admissions and dactylitis, respectively, in their lifetime (Table 3).
 |
Table 3 The Occurrence of Clinical Parameters (Recurrent Events) in the Study Participants (N=92) |
Males had significantly higher HbF/F cell levels compared to females, median 6.4 (IQR: 4.3–9.5) pg versus 5.3 (IQR: 3.5–6.5) pg; p-value = 0.004). However, there was no significant difference in HbF and F cell levels between male and female. Also, there were no significant differences in HbF, F cells, or HbF/F cell levels between the different age groups (Table 4).
 |
Table 4 Comparison of Medians of Hb F, F Cells and Hb F per F Cells Levels Between Patients with Different Socio Demographic Characteristics |
The HbF and F cell levels showed significant association with the requirement of multiple blood transfusions in sickle cell patients. As the unit of HbF levels increased, it significantly decreased the odds of receiving multiple blood transfusions in all combined groups by 11% (OR: 0.89; 95% CI: 0.802–0.968; p-value = 0.016) (Figure 1). Again, as the unit of F cells increased, it significantly reduced the odds of receiving multiple blood transfusions in all combined groups by 3% (OR: 0.971; 95% CI: 0.947–0.995; p-value = 0.020) (Figure 2). However, there was no significant association between the concentration of HbF/F cell and clinical parameters (frequency of clinical events) (Figure 3).
 |
Figure 1 Forest plot showing the association between fetal hemoglobin level and clinical parameters (recurrent events). |
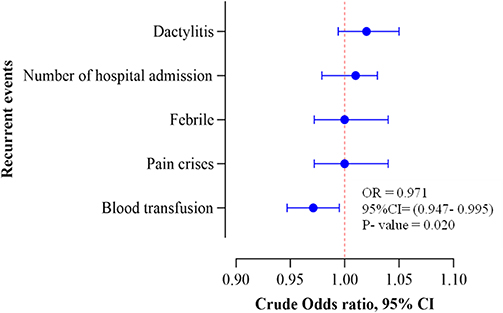 |
Figure 2 Forest plot showing the association between F cell level and clinical param Seters (recurrent events). |
 |
Figure 3 Forest plot showing the association between Hb F/ F cell level and clinical parameters (recurrent events). |
Discussion
This retrospective study aimed to assess the link between HbF, F cells, and HbF/F cell levels and clinical parameters in Tanzanian SCD patients. We observed that as the percentage of HbF and F cells increased, the risk of receiving multiple blood transfusions was significantly reduced. However, HbF/ F cell levels were not associated with sickle cell clinical events in this cohort.
The study found that 75.0%, 78.3%, and 89.1% of participants had HbF, F cell, and Hb F/F cell levels below the thresholds (≥7.2%, ≥32.5%, and ≥10 pg, respectively), and most of these participants 87 (96.4%) live in Dar es Salaam.
This is consistent with the previous two studies conducted in Tanzania, both of which involved participants from Dar es Salaam and reported lower median HbF levels among SCD patients compared to their British counterparts. Notably, none of the patients from either Tanzania or Britain were on hydroxyurea.14,15 Genetic factors, such as SNPs in the β-globin gene cluster, variations in genes like BCL11A, and environmental factors like hypoxia, infections, and inflammation, contribute to HbF regulation.3,14–16 The higher SCD burden in African populations, coupled with conditions like malaria and the Bantu sickle cell haplotype, which is associated with lower HbF levels, highlights differences in HbF production and regulation between populations.3,12,14–17
Our study found that males had significantly higher median HbF/F cell levels than females, contradicting previous studies that reported higher HbF and F cell levels in females.18,19 These findings suggest complex genetic influences on HbF levels and can be due to X-linked polymorphisms.1,12,18–23 These discrepancies may be due to differences in sample sizes and participants’ ages. Our smaller sample size (92 participants) and younger median age (16 years) compared to sample size of 222 with a median age of 20 years in a study done in Tanzania and 150 participants with a mean age of 7.3 ± 3.6 years in a study done in Nigeria could influence the results.18,19 The influence of puberty hormones in females might also contribute, although age-related trends in HbF levels beyond age 10 are inconsistent.21,24 Additionally, differences in study methodologies, such as our retrospective cohort study versus others’ descriptive cross-sectional designs, can affect the generalizability and findings of the research. In our study, a smaller sample size due to incomplete socio-demographic data and potential recall bias from asking participants about lifetime clinical events limited the depth of our analysis within the available timeframe. These constraints might influence the generalizability of our results and the accuracy of the relationship between exposure and outcomes, especially when compared to cross-sectional studies that capture data at a single point in time and may not face the same limitations.
This study found no statistically significant differences in HbF, F cells, and HbF/F cell levels across age groups. While levels seemed lower in adults (≥18 years) than in children, the predominance of participants under 18 (median age: 16) may explain this phenomenon. HbF levels generally decline with age and become less stable after 10 years, aligning with findings from a Nigerian study.25
When we examined the clinical parameters in our study participants, 82.6% and 81.5% reported experiencing more than 10 times pain crises and febrile illness, respectively, and 54.3% had experienced fewer than 5 episodes of blood transfusion in their lifetime. These results are consistent with the findings of a study done in Tanzania that found the main reason for admission among adults was pain, while, in children, it was fever and severe anemia.26,27 This suggests that geographical factors, environmental influences such as infections like malaria prevalent in Tanzania, and the Bantu sickle cell haplotype common among Africans can increase the occurrence of clinical manifestations of SCD.3,12,14–17
Our study investigated the relationship between Hb F parameters (Hb F, Hb F/F cell, and F cells) and clinical events. We found a significant reduction in the risk of multiple blood transfusions with increased percentages of HbF and F cells. This aligns with the study in Uganda which reported fewer hospital admissions, transfusions, and severe pain episodes in individuals with Hb F levels ≥10%.11 A study conducted in Los Angeles, USA, also noted a reduction in recurrent events in SCD patients with HbF levels of 20%.13 A study done in Boston, USA, highlighted that protective cells increase with Hb F levels, with significant protection starting at 10% HbF.3
In our study, Hb F/F cell levels ≥10 pg were associated with reduced dactylitis, febrile illness, pain crises, and transfusions although they were not statistically significant, possibly due to the generally low Hb F/F cell levels (median 5.63 pg, IQR: 4.00 −7.98 pg) observed in 89.1% of participants. This means the Hb F in the F cells of these patients was relatively low and less protective against polymerization, as suggested in the Boston study that at least 10 pg of Hb F/F cell is needed to prevent Hb S polymerization when oxygen saturation is low.3 However, Hb F concentration is unevenly distributed among F cells. For instance, with an Hb F level of 20%, only 1% to 24% of cells might have protective Hb F levels (~10 pg). Lower overall Hb F levels result in fewer or no protected cells. Specifically, 15%, 25%, and 70% of red blood cells (RBCs) are protected at 10%, 20%, and 30% Hb F levels, respectively, while a 5% Hb F level offers no clinical protection.3 Therefore, even participants with high Hb F levels in our study might have had fewer than 10% protected cells, explaining their frequent clinical events. As it was described in previous studies that the HbF per F-cell calculation used assumes that the distribution of HbF per F-cell is uniform, which is almost always incorrect, and that the characteristics of this distribution are likely a better predictor of clinical severity.2,3 These findings highlight the need to consider both the overall Hb F level and the Hb F/F cell concentration when assessing disease severity in SCD patients, as even high Hb F levels can result in severe manifestations if the concentration of Hb F distributed in each F cell is insufficient to prevent Hb S polymerization.
There were several limitations to this study. The study was retrospective in nature, and this may have led to possible recall bias on the frequency of clinical events. We also calculated the HbF/F cell manually which could have introduced errors. Additionally, the focus on a single region (Dar es Salaam) limited the generalizability of the results. We recommend more extensive prospective studies involving diverse regions of Tanzania and utilizing high-throughput techniques for better control of the confounding factors as well as more accurate and efficient measurement of HbF/F cell.
Conclusion
In this cohort, HbF and F cell levels remain important predictors of disease severity, as higher levels predicted reduced requirement for multiple blood transfusions in SCD patients, while HbF/F cells did not correlate with SCD clinical events. Furthermore, patients with SCD in Tanzania generally had low levels of HbF parameters, indicating the potential usefulness of treatments aimed at boosting F cell and HbF levels, such as Hydroxyurea therapy. We recommend to invest in expanding F cell and HbF levels by doing pharmacogenetic studies for personalized medicine, such as optimizing hydroxyurea dosage.
Acknowledgment
We express gratitude to all staff at Muhimbili the University of Health and Allied Sciences, Muhimbili National Hospital, Department of Haematology and Blood Transfusion, and SPARCO Tanzania for their valuable support throughout every stage of this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was funded by the Sickle Pan African Research Consortium (SPARCO)-Tanzania, grant number U01HL156853, awarded by the NIH-NHLBI, and the International Centre for Genetic Engineering and Biotechnology (ICGEB), project no. CRP/TZA19-01.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Akinsheye I, Alsultan A, Solovieff N, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118(1):19–27. doi:10.1182/blood-2011-03-325258
2. Maier-redelsperger BM, Noguchi CT, De MM, et al. Variation in fetal hemoglobin parameters and predicted hemoglobin s polymerization in sickle cell children in the first two years of life: Parisian prospective study on sickle cell disease. Am J Hematol. 1994;84(9):3182–3188. doi:10.1182/blood.V84.9.3182.3182
3. Steinberg MH, Chui DHK, Dover GJ, Sebastiani P, Alsultan A, Hopkins J. Perspectives Fetal hemoglobin in sickle cell anemia: a glass half full? 4 by. Am Soc Hematol. doi:10.1182/blood
4. Darbari DS, Sheehan VA, Ballas SK. The vaso-occlusive pain crisis in sickle cell disease: definition, pathophysiology, and management. Eur J Haematol. 2020;105(3):237–246. doi:10.1111/ejh.13430
5. Buchanan GR. “Packaging” of fetal hemoglobin in sickle cell anemia. Am Soc Hematol. 2013;161(1):3–14.
6. Ak B. Fetal hemoglobin level and severity of sickle cell anemia the influence of fetal hemoglobin on clinical and hematological variables of children and adolescents with sickle cell anemia in Basra, Southern Iraq. 2015;Vol 7.
7. Martin HS. Fetal hemoglobin in sickle cell anemia: the Arab-Indian haplotype and new therapeutic agents. Am J Hematol. 1–30. doi:10.1002/ajh.24872
8. Powars D, Hiti A. Sickle cell anemia: βs gene cluster haplotypes as genetic markers for severe disease expression. Am J Dis Child. 1993;147(11):1197–1202. doi:10.1001/archpedi.1993.02160350071011
9. Stuart MJ, Nagel RL, Jefferson T. Sickle-cell disease. Lancet. 2004;364:1343–1360. doi:10.1016/S0140-6736(04)17192-4
10. Thein SL, Menzel S, Lathrop M, Garner C. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Genet. 2009;18(R2):R216–R223. doi:10.1093/hmg/ddp401
11. Mpalampa L, Ndugwa CM, Ddungu H, Idro R. Foetal haemoglobin and disease severity in sickle cell anaemia patients in Kampala, Uganda. BMC Blood Disord. 2012;12. doi:10.1186/1471-2326-12-11
12. Urio F, Lyimo M, Mtatiro SN, Cox SE, p MB, Makani J. High prevalence of individuals with low concentration of fetal hemoglobin in F-cells in sickle cell anemia in Tanzania. Am J Hematol. 2016;91(8):E323–E324. doi:10.1002/ajh.24390
13. Chan LS. Is there a threshold of fetal hemoglobin that ameliorates morbidity in sickle cell anemia ? Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia ? Bloodjournal Hematologylibrary Org by. 2014;63.doi: 10.1182/blood.V63.4.921.bloodjournal634921.
14. Makani J, Menzel S, Nkya S, et al. Genetics of fetal hemoglobin in Tanzanian and British patients with sickle cell anemia. Blood. 2011;117(4):1390–1392. doi:10.1182/blood-2010-08-302703
15. Mtatiro SN, Singh T, Rooks H, et al. Genome wide association study of fetal hemoglobin in sickle cell Anemia in Tanzania. PLoS One. 2014;9(11):7–14. doi:10.1371/journal.pone.0111464
16. Proc H, Meeting I, Wainscoat JS. Thalassaemia the origin OF mutant ß-globin genes in human populations. Acta Haemat. 1987;78(158):154–158. doi:10.1159/000205867
17. Nkya S, Mwita L, Mgaya J, et al. Identifying genetic variants and pathways associated with extreme levels of fetal hemoglobin in sickle cell disease in Tanzania. BMC Med Genet. 2020;21(1). doi:10.1186/s12881-020-01059-1
18. Urio F, Haematol BJ, Ackaouy A. - Urio - F cell numbers are associated with an X‐linked genetic polymorphism and correlate with.pdf. Ann Int Conference. 2020;2020(191):888–896. doi:10.1111/bjh.17102
19. Adeodu OO, Akinlosotu MA, Adegoke SA, Oseni SBA. Foetal haemoglobin and disease severity in Nigerian children with sickle cell anaemia. Mediterr J Hematol Infect Dis. 2017;9(1):1–8. doi:10.4084/mjhid.2017.063
20. Rutland PC, Pembrey ME, Davies T. The estimation of fetal haemoglobin in healthy adults by radioimmunoassay. Br J Haematol. 1983;53(4):673–682. doi:10.1111/j.1365-2141.1983.tb07319.x
21. Olufemi AE, Sola OB, Oluwaseyi BE, Ajani RA, Olusoji MO, Olubunmi HR. Hemoglobin F level in different hemoglobin variants. Korean J Hematol. 2011;46118–46122. doi:10.5045/kjh.2011.46.2.118
22. Thein SL, Menzel S. Discovering the genetics underlying foetal haemoglobin production in adults. Br J Haematol. 2009;145(4):455–467. doi:10.1111/j.1365-2141.2009.07650.x
23. Urio F, Nkya S, Mgaya J, et al. Gender effect on production and enrichment of F cell numbers in sickle cell disease patients in Tanzania. Am J Hematol. 2023:2–4. doi:10.1002/ajh.26914.
24. Maude GH, Hayes RJ, Serjeant GR. The haematology of steady state homozygous sickle cell disease: interrelationships between haematological indices. Br J Haematol. 1987;66(4):549–558. doi:10.1111/j.1365-2141.1987.tb01343.x
25. Olaniyi J, Arinola A, Arinola AB. Foetal haemoglobin (HbF) status in adult sickle cell anaemia patients in Ibadan, Nigeria. Ann Ibadan Postgrad Med. 2011;8(1):30–33. doi:10.4314/aipm.v8i1.63955
26. Makani J, Tluway F, Makubi A, et al. A ten year review of the sickle cell program in Muhimbili National Hospital, Tanzania. BMC Hematol. 2018;18(1). doi:10.1186/s12878-018-0125-0
27. Melchionda F, Oncology P, Spreafico F, et al. A novel WT1 mutation in familial Wilms tumor. Pediatr Blood Cancer. 2013;60:1388–1389. doi:10.1002/pbc
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.