")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 18
Advances in Mendelian Randomization Studies of Obesity Over the Past Decade: Uncovering Key Genetic Mechanisms
Authors Lu X , Ji L, Chen D, Lian X, Yuan M
Received 17 March 2025
Accepted for publication 3 July 2025
Published 17 July 2025 Volume 2025:18 Pages 2399—2415
DOI https://doi.org/10.2147/DMSO.S528669
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Liang Wang
Xinyue Lu,1,2 Lianhong Ji,1,2 Dong Chen,2 Xiaoyang Lian,2 Mengqian Yuan1,2
1Affiliated Hospital of Nanjing University of Chinese Medicine, Nanjing, 210029, People’s Republic of China; 2Jiangsu Province Hospital of Chinese Medicine, Nanjing, 210029, People’s Republic of China
Correspondence: Mengqian Yuan, Affiliated Hospital of Nanjing University of Chinese Medicine, 155 Hanzhong Road, Nanjing, 210029, People’s Republic of China, Email [email protected]
Abstract: Obesity is a major global public health issue linked to a wide range of chronic diseases. Understanding its complex causal pathways requires robust analytical methods. Mendelian randomization (MR), which employs genetic variants as instrumental variables, effectively addresses confounding and reverse causation and has become a key tool in obesity research. This review summarizes the development of MR methodologies, from single-sample to multivariable, mediation, and time-series models, and highlights key findings from the past decade. MR studies have revealed causal associations between obesity and nine major disease categories, including cardiovascular, metabolic, cancer, psychiatric, respiratory, renal, reproductive, musculoskeletal, and dermatological disorders. Obesity influences disease risk through mechanisms involving energy metabolism, hormonal regulation, and inflammation, with heterogeneity by age, sex, and fat distribution. Key genes such as MC4R, LEPR, FTO, and FGF21 have been identified as potential therapeutic targets. Current challenges include instrument strength, pleiotropy, population stratification, and the external validity of GWAS data. Future research that integrates multi-ancestry GWAS, functional validation, and multi-omics approaches may further enhance the utility of Mendelian randomization. MR provides a robust genetic framework for elucidating obesity’s causal effects and informing targeted interventions and personalized treatment strategies.
Keywords: Mendelian randomization, obesity, genetic variation, research progress
Introduction
With rapid economic development in China, obesity has become a major public health concern, with more than half of adults classified as overweight or obese.1 The World Health Organization (WHO) defines obesity as excessive fat accumulation that may impair health,2 and it constitutes a key component of metabolic syndrome.3 Obesity can be quantified using indicators such as body mass index (BMI), waist circumference, and waist-to-hip ratio. According to WHO BMI classifications, overweight is defined as a BMI of 25.0–29.9 kg/m², obesity as ≥30 kg/m², and severe obesity as ≥40 kg/m².4 The development of obesity is influenced by a complex interplay of genetic factors, environmental exposures, and gene-environment interactions. Although numerous clinical studies have demonstrated that obesity significantly increases the risk of metabolic diseases, cardiovascular diseases, musculoskeletal disorders, psychiatric conditions, and various cancers,5 the causal nature of these associations remains unclear due to potential confounding and reverse causation.
To address these limitations, researchers have increasingly adopted Mendelian randomization (MR), a method that uses genetic variants associated with obesity to infer causal effects on disease outcomes. Often described as a “natural randomized controlled trial (RCT)”, MR leverages the random allocation of genetic variants at conception, providing stable and unbiased exposure measures. This minimizes confounding and reverse causation, enabling more robust causal inference. The application of MR in obesity research has evolved significantly, progressing from single-sample analyses to multi-sample approaches and integrating mediation analysis and temporal modeling. These methodological advances allow researchers to explore the causal pathways linking obesity to various diseases under diverse contexts. This review systematically examines the methodological evolution of MR over the past decade and highlights key genetic discoveries in obesity research, aiming to provide a genetic foundation for the development of targeted therapies for obesity and its related conditions.
Principles and Methodology of Mendelian Randomization
Mendelian randomization (MR) is a statistical approach based on large-scale genome-wide association study (GWAS) data, widely used in epidemiology to infer causal relationships between exposures and disease outcomes. Originally proposed by Katan in 1986,6 MR leverages Mendel’s second law of inheritance, using genetic variants as instrumental variables (IVs). Since alleles are randomly allocated at conception and are not altered by disease processes or environmental and lifestyle factors, MR minimizes bias from confounding and reverse causation.7 Single nucleotide polymorphisms (SNPs) are commonly used as IVs,8 enabling estimation of causal effects through their associations with both exposures and outcomes.
MR relies on three core assumptions: (1) Relevance — IVs must be strongly associated with the exposure; (2) Independence — IVs must be independent of confounders of the exposure-outcome relationship; and (3) Exclusion restriction — IVs affect the outcome only through the exposure.9,10 (Figure 1) These assumptions ensure that MR can robustly estimate causal effects for modifiable risk factors and address limitations of traditional observational studies. Over the past two decades, MR has evolved from its initial use in identifying disease risk factors to broader applications, including elucidating drug mechanisms.11
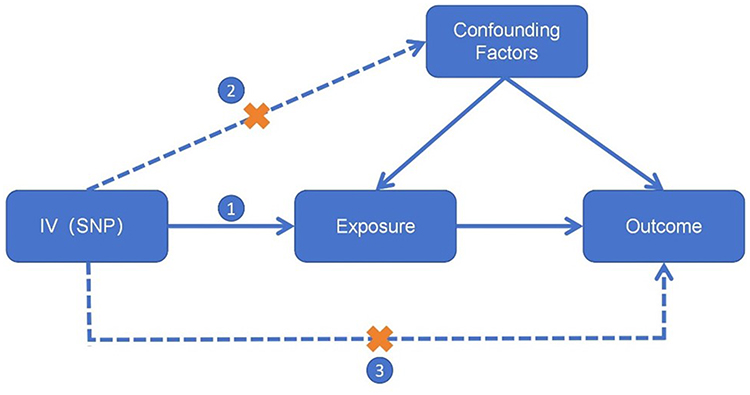 |
Figure 1 Mendelian randomization core hypothesis diagram. Abbreviations IV, instrumental variable; SNP, single nucleotide polymorphism. Notes: The core assumptions of Mendelian randomization include the correlation assumption(①), independence assumption(②), and exclusivity assumption(③). “X” on Path ② indicates that the genetic variant should be independent of all confounding factors. “X” on Path ③ indicates that the genetic variant must influence the outcome only indirectly through the exposure, and not through any direct pathway. |
To address different research needs, several MR extensions have been developed: Single-sample MR, the traditional form, estimates causal effects using genetic, exposure, and outcome data from the same cohort via two-stage least squares (2SLS) regression.12 However, it requires large sample sizes, which may not always be available.13 Two-sample MR addresses this limitation by using separate GWAS summary statistics for the exposure and outcome from independent samples, with SNPs serving as the link between them.14 This increases statistical power but requires careful matching of populations to avoid bias from population stratification.15
Bidirectional MR helps infer causal direction when genetic variants may influence both variables, making it difficult to disentangle cause and effect. It uses independent IVs for each trait to assess bidirectional relationships.7 For instance, if A causes B, IVs for A should influence both A and B, while IVs for B should not influence A.
Multivariable MR (MVMR) allows simultaneous evaluation of multiple exposures and their direct effects on an outcome.16 It requires a sufficient number of independent SNPs and adherence to additional assumptions to account for horizontal pleiotropy and confounding. Mediation MR (MMR) extends MR to decompose total effects into direct and indirect effects mediated through an intermediate variable,16 providing insights into causal pathways and potential intervention targets when direct modification of the exposure is difficult.
Time-varying MR (TVMR) is an emerging approach that examines how causal effects of exposures on outcomes change over time.17 It overcomes the limitations of traditional MR, which estimates lifetime average effects, and helps identify critical intervention windows. However, TVMR faces challenges such as age-dependent genetic effects and increased susceptibility to confounding in intergenerational studies.
The MR analytical process typically includes defining datasets, selecting IVs, testing IV assumptions, conducting MR analyses, and performing sensitivity analyses with visualizations.18 The availability of large GWAS resources, such as the UK Biobank and GWAS Catalog, has greatly enhanced MR research. Common analytical methods include inverse-variance weighted (IVW) estimation and Wald ratio, supplemented with sensitivity analyses such as MR-Egger regression,19 funnel plots, and Q-statistics to detect and correct for pleiotropy.20 Combining allele-score and mode-based approaches further improves causal inference accuracy.
Current Research Status of Mendelian Randomization in the Field of Obesity
Exploring the Biological Mechanisms of Obesity Etiology Using Mendelian Randomization
From 2014 to 2024, over 20 studies have systematically revealed multiple influencing factors of obesity through Mendelian randomization methods. As early as 2014, when Mendelian randomization had not yet been widely applied, researchers used MR to find that fetal protein-A from the liver increased the probability of obesity.21 In 2015, Helle et al demonstrated that the lactase persistence gene leads to obesity by increasing milk intake, although it does not directly cause obesity.22 Subsequent studies, however, proved that lactase persistence is positively correlated with obesity, suggesting that increasing milk intake in the target population may lead to obesity;23 thus, the specific impact of the lactase persistence gene on obesity requires further research. In 2017, studies from both China and abroad indicated that the methylation level of the ATP4A gene and DNA methylation at certain gene loci in blood cells may affect BMI.24 Additionally, Böckerman’s research showed that individuals with higher education levels have a significantly reduced likelihood of obesity.25 In 2018, Tao’s study indicated that larger maternal hip circumference is associated with heavier birth weights in infants, emphasizing the importance of preventing excessive maternal weight.26 In 2019, Mulugeta found that severe depression can lead to obesity through variations in the MC4R gene,27 and subsequent research indicated that severe depression can also trigger obesity by activating the HPA axis. In the same year, Fu discovered that elevated homocysteine levels increase the risk of obesity,28 expanding the application scope of MR research. This suggests that MR can be used not only to study influencing factors but also to include any gene-determined substances such as proteins and metabolites in MR research.
Since 2021, there has been an explosive growth in research literature utilizing Mendelian Randomization (MR) to explore the mechanisms of obesity. Dennis’s study indicates that a high-fat, low-carbohydrate diet may increase the risk of obesity by 29%,29 while a 2023 article suggests that an increase in noodle intake can lead to the same result.30 Research by Shaza and Pang Yao identifies eight proteins that may influence obesity levels: ITIH3, LRP11, SCAMP3, NUDT5, OGN, EFEMP1, TXNDC15, and PRDX6.31,32 Qian Xu et al revealed a bidirectional causal relationship between gut microbiota (such as Akkermansia and Bifidobacterium) and obesity,33 with subsequent studies indicating that these may contribute to obesity through metabolites and inflammatory factors.34 A study focused on children showed that Akkermansia has a protective effect against childhood obesity,35 suggesting that its impact mechanisms may differ across age groups and warrant further investigation. In 2022, Loukas et al found that the intake of DHA and Omega-3 can reduce the risk of obesity.36 Yuqian Li demonstrated that the methylation of the SOCS3 gene is associated with obesity.37 Yuefeng Yu explored the relationship between sleep and visceral fat using both linear and nonlinear MR,38 and the following year, Yannis used the same method to study the effects of sleep on metabolic syndrome.39 Bryony also investigated the relationship between sleep and obesity characteristics, with their findings indicating that insufficient sleep can lead to the accumulation of visceral fat (VAT), promoting obesity, particularly central obesity.40 Kim et al studied the relationship between smoking and obesity-related characteristics,41 and the following year, Germán similarly researched this relationship.42 Unsurprisingly, their results indicated that smoking can lead to an increase in visceral fat, thereby causing obesity. Stefan identified causal associations between the DBNDD1 and ERAP1 genes and obesity.43 Wang and Li found that specific metabolites in plasma (such as arachidonic acid, arginine, and glycine) and peripheral cells (such as platelets and CD14+ monocytes) are significantly associated with obesity.34,44 Overall, the Mendelian Randomization method in obesity research has gradually evolved from single-variable studies to multi-sample, multi-variable, and comprehensive factor analyses, encompassing genetic, metabolic, dietary, behavioral, gut microbiota, and socioeconomic dimensions, leading to a more comprehensive understanding of the complex etiology of obesity (Table 1).
 |
Table 1 Summary of Exploring the Biological Mechanisms of Obesity Using Mendelian Randomization |
Investigating the causal relationship between obesity and its related diseases through Mendelian randomization
Cardiovascular Diseases
Over the past decade, Mendelian randomization (MR) studies have systematically elucidated the causal relationship between obesity and cardiovascular diseases (CVD). Early research focused on key genetic variants such as FTO, MC4R, and TMEM18, demonstrating that elevated body mass index (BMI) significantly increases the risk of heart failure, ischemic stroke, and deep vein thrombosis (DVT). For instance, Sara et al employed a genetic risk score approach and found that each one-unit increase in BMI markedly raised the risk of heart failure.45 Subsequently, Klovaite and Anette’s team identified specific variants (FTO rs9939609, MC4R rs17782313, GNPDA2 rs10938397, BDNF rs10767664) that contribute to ischemic heart disease by modulating lipid metabolism and blood pressure.46,47 Since 2016, increasing attention has been directed toward the impact of obesity on hemodynamics and coagulation pathways, revealing strong associations with peripheral artery disease and venous thromboembolism (VTE).48,49 Yuan et al further highlighted that central obesity exerts a greater influence on VTE risk compared to overall obesity.50 Moreover, Susanna and Benjamin’s teams proposed that inflammatory pathways may serve as key mediators linking obesity to aortic valve stenosis.51,52
Post-2020, MR studies have adopted more refined approaches, investigating the impact of obesity on microvascular function and incorporating early-life factors. Francesco et al reported that variants in FTO (rs9939609), MC4R (rs17782313), and GNPDA2 (rs10938397) are associated with elevated urinary albumin-to-creatinine ratios, indicating microvascular damage.53 Research from 2021 suggested that childhood obesity-induced gene expression changes (eg, ADCY3 rs4077678) can have long-lasting effects on the risk of atrial fibrillation, stroke, and atherosclerosis in adulthood.54–56 Recent studies from 2023 to 2024 have further validated the role of these obesity-associated genes in post-stroke sequelae and heart failure, underscoring the importance of early prevention and intervention.57–60
Collectively, MR studies consistently demonstrate that obesity increases the risk of various cardiovascular diseases through multiple pathways, including alterations in metabolism, blood pressure regulation, lipid profiles, and inflammatory responses. BMI-related genes such as FTO, MC4R, and GNPDA2 play central roles in these processes. These advances provide a robust genetic foundation for understanding cardiovascular pathology and support obesity prevention as a key strategy in cardiovascular disease management.
Metabolic Diseases
Obesity exerts significant effects on a wide range of metabolic diseases through multiple biological pathways. As early as 2017, Censin et al demonstrated that genetic variants associated with childhood obesity and insulin sensitivity—including FTO (rs1421085), MC4R (rs6567160), GNPDA2 (rs13130484), ADCY3 (rs11676272), TMEM18 (rs4854349), and SEC16B (rs543874)—increase the risk of type 1 diabetes.61 Subsequent studies expanded to other common metabolic diseases, such as gout, hypertension, and type 2 diabetes (T2D). For example, Susanna et al reported that obesity-related variants (FTO rs9939609, among others) may contribute to gout by promoting fat accumulation and dysregulating uric acid metabolism.62 Lee et al confirmed a significant causal relationship between BMI-associated genetic variants and the development of hypertension.63 Moreover, studies by Tao and Tom et al revealed that these genetic variants not only directly increase the risk of T2D but may also indirectly contribute to coronary heart disease.64,65
Since 2023, research has increasingly focused on integrating mechanistic insights. Yuan et al identified that variants in FTO (rs9941349), TMEM18 (rs4854344), BDNF (rs571312), GNPDA2 (rs6752378), and SEC16B (rs7138803) significantly increase T2D risk by influencing lipid metabolism, chronic inflammation, and insulin resistance.66 In addition, these pathways appear to contribute to diabetic complications such as diabetic retinopathy and gestational hypertension.67 The long-term impact of childhood obesity has also been repeatedly validated; recent studies suggest that childhood obesity may increase the risk of preeclampsia in adult women via sex hormone regulatory pathways.
MR studies clearly demonstrate that multiple BMI-associated genetic variants systematically increase the risk of metabolic diseases—including diabetes, hypertension, and gout—through mechanisms involving metabolic dysregulation, inflammation, and hormonal imbalances. These findings further highlight the critical importance of early-life interventions to prevent the long-term metabolic consequences of obesity.
Cancer
Since the first report in 2009 linking FTO gene variant (rs9939609) to lung cancer risk,68 the relationship between obesity and cancer has been progressively elucidated. Early studies focused on variants in FTO, MC4R, and GNPDA2, which were associated with elevated risks of lung cancer, esophageal adenocarcinoma, and colorectal cancer. Aaron et al demonstrated that obesity-related gene variants promote gastrointestinal cancers by influencing gastroesophageal reflux, inflammatory pathways, and gut microbiota, with a more pronounced effect of elevated BMI on colorectal cancer risk in women.69,70 Caroline et al further revealed sex-specific mechanisms underlying the association between obesity and colorectal cancer.71 Similarly, Suzanne et al reported that these variants also increase the risk of non-high-grade serous ovarian cancer (non-HGSC).72
Since 2021, research has expanded to both hormone-related and non-hormone-related cancers. Muktar’s team showed that obesity-associated variants promote risks of multiple myeloma and endometrial cancer through insulin resistance and inflammation pathways, whereas in certain contexts (eg, breast cancer), altered estrogen signaling may confer a protective effect.73 Mathew et al confirmed the involvement of these obesity-driven pathways in gastrointestinal cancers.74 In 2022, Wu et al identified associations of FTO (rs9941349), BDNF (rs571312), and GNPDA2 (rs6752378) variants with pancreatic cancer risk, potentially mediated by obesity-induced CRP-driven inflammatory pathways. Abao’s 2023 findings further highlighted that variants in AKT1 (rs2498804), IL-6 (rs1800795), and TNF (rs1800629) may regulate PI3K-AKT and JAK/STAT3 signaling in obesity-induced gastric cancer progression.75
Recent studies have also emphasized the differential impact of fat distribution on cancer risk. Bai et al reported that body fat percentage is a better predictor of non-small cell lung cancer than BMI,76 while Xu et al found that visceral and hepatic fat are stronger predictors of primary liver cancer than general obesity. Moreover, in certain cancers, obesity-related variants may exert protective effects. Wu et al demonstrated that HOXC4/5/6 (rs10876528), TBX15 (rs1106529), and GDF5 (rs224333)—which regulate fat metabolism and immune responses—may lower Hodgkin lymphoma risk.77 Hasnat et al found that in premenopausal women, these variants may reduce breast cancer risk through hormone-regulation pathways;78 however, other studies reported that obesity significantly increases postmenopausal breast cancer risk,79 underscoring a menopausal status-dependent effect.
A large body of MR evidence now supports a causal relationship between obesity and multiple cancers. Mechanistically, this involves dysregulated lipid metabolism, inflammatory activation, hormonal axis alterations, and gut microbiota changes. The impact of obesity varies by cancer type, organ specificity, and individual characteristics, suggesting that cancer prevention strategies should account for sex, fat distribution, and physiological stage to enable more precise, subtype-specific interventions.
Psychiatric Disorders
Obesity is intricately linked to various psychiatric disorders through complex genetic and metabolic pathways. Early studies indicated that obesity-related genes may influence brain structure and function. Stéphanie et al reported that the VEGFA (rs6905288) variant may impair angiogenesis and cerebral blood flow, leading to reduced gray matter volume in individuals with abdominal obesity.80 Additionally, Maria et al found that variants in FTO (rs9939609), MC4R (rs17782313), BDNF (rs10767664), and GNPDA2 (rs10938397) may increase depression risk by altering fat metabolism and inflammatory pathways.81 Since 2021, research has expanded to include neurodegenerative diseases such as dementia and Alzheimer’s disease. Anwar et al reported that obesity may accelerate brain aging,82 while Zhuang et al identified that specific genes such as CARTPT promote the progression from obesity to Alzheimer’s disease.83
Emerging evidence also highlights the significant impact of obesity on child mental health. Ding et al revealed that several genes involved in neurodevelopment, metabolism, and synaptic plasticity—DCC (rs11663824), NEGR1 (rs2815752), NCAM1 (rs7106434), INO80E (rs6556833)—may mediate the risk of attention-deficit/hyperactivity disorder (ADHD), depression, and autism spectrum disorders.84 Amanda and Thais et al further confirmed that these risks are more pronounced among children with higher BMI.85,86 More recently, in 2023, Chen et al demonstrated that obesity-related variants such as POMC (rs713586) may increase the risk of schizophrenia by modulating energy metabolism and neurotransmitter systems,87 providing deeper insights into the biological connections between obesity and severe psychiatric disorders.
Overall, obesity increases the risk of various psychiatric disorders through mechanisms involving brain metabolism, inflammation, and neurodevelopmental pathways. The long-term negative impact of childhood obesity on future mental health underscores the importance of early intervention and personalized prevention strategies for psychiatric disorders.
Respiratory Diseases
Between 2019 and 2024, Mendelian randomization (MR) studies have progressively revealed strong causal links between obesity and a variety of respiratory diseases. In 2019, variants in FTO (rs9939609), MC4R (rs17782313), and BDNF (rs10767664) were found to increase asthma risk through pathways involving lipid metabolism and inflammation.88 Subsequent studies further explored airway structure and immune regulation, identifying variants in ERBB3 (rs4759229), COL16A1 (rs6681149), UNC13D (rs111365807), SMAD3 (rs79071878), and FOXA3 (rs8103278) as contributors to airway remodeling processes, particularly in non-allergic adult asthma.89–91
Obesity has also been shown to adversely affect lung function. Nicole et al demonstrated that obesity-related genes impair lung function through inflammatory, metabolic, and neuroregulatory mechanisms, leading to airway obstruction (eg, reduced FEV₁/FVC ratio) and small airway damage (eg, reduced FEF25–75%).92 In children, obesity-related systemic inflammation—mediated by factors such as IL-6 and adiponectin—has also been linked to an increased risk of asthma.93
In recent years, research has expanded to other respiratory diseases. Feng et al reported that obesity-related variants (FTO, GCKR, GNPDA2) are associated with chronic obstructive pulmonary disease (COPD), pneumonia, pulmonary embolism, and idiopathic pulmonary fibrosis. Other groups have further shown that these variants, through their effects on BMI, influence the risk of obstructive sleep apnea and acute respiratory distress syndrome (ARDS).
Overall, obesity systematically increases the risk of respiratory diseases via mechanisms involving inflammation, energy metabolism imbalance, and airway remodeling. MR studies provide robust genetic evidence supporting these causal relationships, highlighting the importance of weight control—especially in childhood—for the prevention of chronic respiratory diseases.
Kidney Disease
Although research in this area has emerged more recently, Mendelian randomization (MR) studies have begun to clarify how obesity adversely affects renal health through mechanisms involving metabolic dysregulation, inflammation, and glomerular hyperfiltration. In 2021, Yuan et al reported that variants in FTO (rs9939609), MC4R (rs17782313), SH2B1 (rs7498665), SLC2A9 (rs6449213), and IL-6 (rs1800795) increase the risk of nephrolithiasis via pathways related to energy and uric acid metabolism.94 Yang et al further demonstrated that these genes are also associated with an elevated risk of chronic kidney disease (CKD), with obesity-related renal damage being particularly pronounced during adulthood.95 However, Nguyen et al argued that after adjusting for blood pressure and glycemic control, obesity may not be a direct causal factor for CKD.96 In diabetic nephropathy, Lu et al found that variants in FTO (rs17817449), MC4R (rs6567160), BDNF (rs6265), SEC16B (rs574367), and PAX6 (rs652722) contribute to metabolic abnormalities and inflammatory responses that trigger glomerular hyperfiltration and structural kidney damage, with women showing greater susceptibility.97
Recent studies have also explored the link between obesity and immune-related kidney diseases. In 2024, Dong et al reported that obesity may increase the risk of IgA nephropathy through pathways involving sodium channel regulation. Similarly, Zhang et al identified that variants in SCNN1B and SCNN1G mediate the risk trajectory from childhood obesity to diabetic nephropathy in adulthood.98
In summary, obesity increases the risk of various renal diseases through pathways involving energy metabolism abnormalities, inflammation, and elevated glomerular pressure. The long-term impact of childhood obesity on renal health should not be underestimated, and effective weight management remains a key strategy for kidney disease prevention.
Reproductive Health
Mendelian randomization (MR) studies have clarified that multiple BMI-associated genetic variants influence reproductive health through mechanisms involving metabolic dysregulation, hormonal imbalance, and abnormal fat distribution. In 2019, Brower et al identified that variants in FTO (rs1421085), MC4R (rs6567160), SEC16B (rs543874), GNPDA2 (rs10938397), PCSK1 (rs261967), BDNF (rs7103411), and MAP2K5 (rs2241420) may increase the risk of polycystic ovary syndrome (PCOS) via insulin resistance and hormonal pathways.99 Subsequent studies revealed that central obesity has a stronger association with PCOS,100 maintaining a BMI between 20–25 kg/m² supports normal reproductive function,101 and variants such as GIPR (rs11672660) and PROX1 (rs3767846) may further mediate the adverse effects of childhood and adult obesity on PCOS risk.102 In 2022, Samvida et al reported that ZNF664 (rs11057429) is associated with other female reproductive abnormalities, including menorrhagia and preeclampsia.103 Pubertal development disorders have also gained attention. In 2024, Fang et al found that variants in LIN28B (rs7759938), MKRN3 (rs530324838), and DLK1 (rs1802710) may influence the timing of puberty onset in females by modulating the hypothalamic-pituitary-gonadal (HPG) axis. Zhou et al further demonstrated that INSR (rs1051690) increases endometrial cancer risk through enhanced estrogen exposure and insulin resistance.
In men, studies by Rao and Wan et al showed that variants in FTO (rs1421085), MC4R (rs6567160), SHBG (rs6258), LEPR (rs1137101), CYP19A1 (rs2470152), and IRS1 (rs2943650) affect energy metabolism, sex hormone conversion, and sperm quality, thereby increasing the risk of benign prostatic hyperplasia and infertility.104,105
Collectively, these findings suggest that obesity systematically disrupts both male and female reproductive health through multiple pathways, affecting processes from pubertal development and endocrine balance to reproductive system disorders and reproductive cancers. These insights underscore the critical role of obesity prevention and management in safeguarding reproductive health.
Bone and Joint Health
In recent years, researchers have increasingly applied Mendelian randomization (MR) to investigate the impact of obesity on bone and joint health. Early work by Kalliope et al demonstrated that variants in FTO (rs8044769), MC4R (rs6567160), LEPR (rs1137101), and INSR (rs1051690) significantly increase the risk of osteoarthritis through pathways involving BMI regulation and inflammation.106 Subsequent studies highlighted the role of visceral fat in contributing to cartilage wear and joint loading. In 2021, Zhou et al reported that related genetic variants also increase the risk of intervertebral disc degeneration and lumbar spondylosis, potentially via mechanisms involving cartilage metabolism disruption and chronic inflammation.107
The long-term impact of childhood obesity has also been explored. Cao et al found that early-life obesity increases the risk of adult knee and hip joint degeneration through fat accumulation pathways.108 Since 2023, research has expanded into bone density regulation. Victoria’s team identified that variants in SPO3 (rs13204965, rs13194508), IQCH (rs3743347), HOXC4 (rs894738), and HOXC5 (rs4759320) may reduce bone density through Wnt signaling and bone formation pathways.109
Recent studies further clarified the dualistic nature of obesity’s effects on bone density. He et al proposed that while increased BMI enhances bone loading and formation, fat-induced inflammation may stimulate osteoclast activity, thereby exacerbating osteoporosis. Ying et al confirmed that childhood obesity elevates long-term osteoporosis risk via metabolic alterations.110 Additionally, Chen et al observed that in individuals with low BMI, variants related to fat metabolism and cartilage formation increase the risk of temporomandibular joint disorders (TMJD).111
Collectively, MR studies reveal a bidirectional effect of obesity on bone and joint health: increased body weight may enhance bone density, but adipose tissue-driven chronic inflammation and metabolic dysfunction can promote osteoporosis and joint degeneration. These insights underscore the importance of optimizing body composition and fat distribution for maintaining bone health.
Skin Diseases
Obesity has been causally linked to a variety of skin diseases, with Mendelian randomization (MR) studies providing strong genetic support for these associations. In 2017, Milena et al identified that variants in FTO (rs1558902), KCNK3 (rs11126666), POC5 (rs2112347), GRP (rs7243357), and ERBB4 (rs7599312) may elevate the risk of multiple sclerosis through neuroinflammatory and immune regulatory pathways.112 Notably, the long-term impact of childhood obesity on this disease is particularly pronounced and may involve disruptions in vitamin D metabolism.113
Subsequent research has focused on more common skin conditions. Ashley et al reported that variants in MC4R (rs7138803), LEPR (rs1137101), and IL-6R (rs2228145) significantly increase the risk of psoriasis through activation of inflammatory signaling, with higher susceptibility observed in individuals with childhood obesity.114 In 2020, Yik et al further demonstrated that the TNF (rs1800629) variant promotes the development of atopic dermatitis.115 More recently, in 2024, Huang et al found that obesity may indirectly increase the risk of pressure ulcers by inducing type 2 diabetes, suggesting a potential link between obesity and impaired skin barrier function.116
Overall, obesity increases the risk of various skin diseases through mechanisms involving chronic inflammation, metabolic dysregulation, and immune system imbalance. MR studies have been instrumental in elucidating these causal pathways, underscoring the importance of weight management in the prevention of skin-related disorders.
Discussion
In recent years, Mendelian randomization (MR) studies have provided substantial causal evidence, further deepening our understanding of the mechanisms underlying obesity. Initially, MR studies focused on the effects of single genes; however, with the diversification of analytical strategies and expansion of data sources, MR has now widely integrated proteomic, metabolomic, behavioral, dietary, and gut microbiome data. This enables the development of multivariable models to assess the systemic impact of obesity. MR has been extensively applied to investigate causal relationships between obesity and various diseases, including cardiovascular disease, metabolic disorders, and cancer, with a marked increase in publications since 2021 (Figure 2). Findings indicate that the pathogenic effects of obesity vary by age, sex, and physiological status. These studies confirm that the development of obesity and its related diseases involves complex polygenic regulatory networks and multiple signaling pathways across physiological systems such as energy balance, neuroendocrine regulation, glucose metabolism, lipid metabolism, inflammatory response, and immune modulation.
 |
Figure 2 A summary chart of the relationship between obesity and various diseases through genetics. Abbreviations: IPF, Idiopathic pulmonary fibrosis; HTN, Hypertension; GH, Gestational Hypertension; LADA, Latent Autoimmune Diabetes in Adults; CKD, Chronic Kidney Disease; IgA, immunoglobulin A; TMD, Temporomandibular Disorder; AD, Alzheimer’s Disease; ADHD, Attention Deficit Hyperactivity Disorder; ASD, Autism Spectrum Disorder; SCZ, Schizophrenia; IHD, Ischemic Heart Disease; AF, Atrial Fibrillation; BC, Breast Cancer; PC, Prostate Cancer; PCOS, polycystic ovary syndrome; HMB, Heavy Menstrual Bleeding; EC, Endometrial Cancer; BPH, Benign Prostatic Hyperplasia. Notes: Created in BioRender. Shen, A. (2025) https://BioRender.com/gja148q. |
Key polymorphisms and aberrant expression of certain genes serve as the molecular basis for imbalances in energy intake and expenditure, insulin signaling impairment, and abnormal lipid accumulation. Neuroendocrine pathways regulating appetite and energy balance play a central role in obesity susceptibility. MC4R, located in the hypothalamic paraventricular nucleus, suppresses appetite and promotes energy expenditure via α-MSH-mediated activation of the G_s protein and cAMP/PKA signaling pathway.117 LEPR mediates leptin signaling, activating the JAK2/STAT3 pathway, inhibiting orexigenic NPY/AgRP neurons, and activating anorexigenic POMC/α-MSH neurons, thereby enhancing MC4R-mediated anorexic effects.118 The BDNF/NTRK2 pathway, via PI3K/Akt and MAPK/ERK signaling, mediates satiety and modulates reward responses, linking obesity to emotional disorders.119
In terms of insulin and glucose metabolism, genes such as GIPR, FGF21, and PPARGC1A regulate insulin sensitivity and hepatic glucose-lipid homeostasis. GIPR activation enhances insulin responsiveness in adipose tissue and improves systemic insulin sensitivity.120 FGF21, through binding to FGFR1c/β-Klotho, activates ERK1/2 signaling to promote glucose uptake in adipose tissue and modulate gluconeogenesis and lipid metabolism in the liver.121 PPARGC1A regulates mitochondrial biogenesis and oxidative phosphorylation, enhancing metabolic efficiency in skeletal muscle and liver.122
Regarding lipid metabolism and adipogenesis, FTO regulates mRNA demethylation and influences preadipocyte differentiation, with high expression associated with adipose tissue expansion.123 Insulin-induced activation of SREBF1 promotes fatty acid and triglyceride synthesis, exacerbating fat accumulation.124 ACACB downregulation impairs β-oxidation of fatty acids, enhancing fat storage. The CREBRF variant is strongly linked to obesity in Polynesian populations, highlighting inter-population differences in energy storage regulation.125
Chronic low-grade inflammation is another critical mechanism in obesity-related metabolic dysregulation. TNF-α activates NF-κB and MAPK pathways, inducing expression of IL-6 and other cytokines, thereby inhibiting IRS1 activity and promoting insulin resistance.126 IL-6 also induces CRP expression in the liver via JAK/STAT3 signaling.127 Additionally, TLR4 activation in obesity triggers macrophage infiltration in adipose tissue, amplifying local inflammation and exacerbating insulin resistance.128
Notably, genes such as TRAPPC9, BCL2A1, and TNNI3K, although not traditionally classified within metabolic pathways, indirectly influence obesity-related organ damage by modulating NF-κB activation, apoptosis inhibition, and cardiac energy metabolism. TRAPPC9 in particular may bridge neurodevelopment and metabolic signaling, offering new avenues for exploring the “neuro-metabolic” axis.
Several obesity-related molecular targets have advanced into clinical application. MC4R agonists, GIPR/GLP-1R dual agonists, and FGF21 analogs represent cutting-edge therapeutic targets. Setmelanotide, an MC4R agonist, is the first approved treatment for congenital POMC or LEPR deficiency-related obesity, significantly reducing appetite and promoting weight loss.129 Dual GLP-1R/GIPR agonists, such as Tirzepatide, enhance insulin secretion and improve metabolic outcomes and weight management.130 FGF21 analogs (eg, Pegbelfermin, Efruxifermin) have shown promise in reducing hepatic steatosis and improving metabolic profiles in NAFLD/NASH patients with obesity.131 ACCβ (ACACB) inhibitors promote fatty acid β-oxidation and contribute to weight loss in NAFLD/NASH, though gastrointestinal side effects remain a concern.132
By contrast, targets such as FTO, SREBP-1c, ADCY3, and SH2B1 remain in early-stage development. While FTO inhibitors demonstrate anti-obesity effects in animal models, challenges related to central delivery and off-target toxicity must be addressed.133 SREBP-1c inhibitors, such as Fatostatin, suppress lipogenesis-related gene expression in vitro, but in vivo selectivity and toxicity require further optimization.134 Other promising targets, such as Leptin, BDNF, and CREBRF, present substantial drug development challenges. Leptin therapy is limited by central leptin resistance,135 and BDNF delivery is hindered by poor blood-brain barrier penetration.136 CREBRF, a transcription factor, lacks suitable binding pockets for small molecules, necessitating RNA-based or protein-protein interaction targeting approaches.137 Anti-TNF-α and anti-IL-6R therapies improve insulin sensitivity in animal models but carry infection risks and poor long-term tolerability in humans. Despite these challenges, MR continues to provide valuable genetic insights that support target prioritization for obesity therapeutics.
It is important to note that even within the same gene, different genetic variants may affect disease development through distinct biological pathways. Future research should therefore further explore the potential associations between multiple variants within the same gene and various diseases, providing a stronger genetic basis for identifying novel therapeutic targets for obesity and its related comorbidities. Moreover, childhood obesity exerts long-term adverse effects on the risk of multiple adult diseases, emphasizing the need for early intervention during childhood and adolescence. Efforts from both societal and family levels are essential to prevent the long-term health consequences of obesity in adulthood. Additionally, future research should focus on the differential effects of fat distribution and composition on obesity-related outcomes, enabling a more refined understanding of obesity pathophysiology.
Despite the substantial progress made by MR in elucidating causal relationships between obesity and a wide range of diseases, several limitations remain when applying MR to explore the mechanisms of obesity. First, the validity of MR analysis depends critically on several core assumptions and the robustness of methodological design. The strength of instrumental variables (IVs) is a key determinant of the reliability of causal inference. Weak IVs can introduce substantial bias, resulting in estimates regressing toward the null or yielding misleading conclusions. To mitigate this issue, three-sample designs and partially Bayesian-weighted methods have been proposed to improve estimation efficiency and robustness.138
Even with sufficiently strong IVs, MR analyses remain susceptible to horizontal pleiotropy, where genetic variants affect the outcome through pathways unrelated to the exposure of interest. The recently developed MR-LDP model incorporates linkage disequilibrium (LD) structure information to better capture pleiotropic effects and enhance adaptability to complex genetic backgrounds.139 Population stratification is another often underestimated but critical source of bias. In populations with systematic differences in allele frequencies, MR analyses may yield spurious associations even when no true causal relationship exists.140 To identify and control for this bias, methods such as the negative control outcome approach—introducing outcomes theoretically unaffected by the exposure—have been employed.141 Moreover, the MR-Twin approach, which compares siblings within the same family, can effectively account for population structure, environmental confounding, and generational effects without relying on external adjustments, offering clear advantages in controlling for population stratification.142
Limitations inherent to GWAS data—the foundational data for MR—also affect the generalizability and accuracy of MR inferences. Most GWAS studies are based on European populations, which limits the external validity of results and hampers their generalization to other ethnic groups. Additionally, some GWAS-identified SNPs lack clear biological functional annotations. If such variants are used as IVs, they may introduce pleiotropic bias, undermining the credibility of causal inferences.143 Several strategies have been proposed to address these limitations: first, expanding GWAS sample diversity by including multi-ancestry data to enhance the external validity of MR results; second, prioritizing SNPs with known regulatory functions or expression quantitative trait loci (eQTL) evidence to reduce pleiotropy;143 and third, leveraging family-based data to improve causal inference accuracy and robustness by controlling for confounding factors.142
A second major limitation is that MR is inherently restricted to using genetic variants as IVs, making it difficult to directly assess non-genetic factors such as environmental exposures and lifestyle behaviors, thereby limiting its application in fully elucidating the multifactorial etiology of obesity. Finally, obesity is a complex condition involving interactions across the genome, transcriptome, metabolome, and other biological layers. Its effective treatment typically requires multidisciplinary interventions and combined therapeutic strategies; MR alone cannot provide sufficient clinical guidance. Therefore, it is essential to rigorously assess the core assumptions of MR when constructing models and to complement MR analyses with thorough heterogeneity and sensitivity assessments, as well as other research approaches—such as randomized controlled trials and observational studies—to enhance the precision and reliability of causal inferences.
Conclusion
This review summarizes recent advances in the application of Mendelian randomization (MR) in obesity research, highlighting both methodological developments and its role in causal inference across multiple disease domains. By utilizing genetic variants as instrumental variables, MR effectively controls for confounding bias and has elucidated the causal relationships between obesity and various systemic diseases—including cardiovascular, metabolic, and cancer-related conditions. Additionally, MR studies emphasize the modulatory effects of childhood obesity, sex differences, and fat distribution on disease risk. Key biological pathways and genes, such as MC4R, LEPR, FTO, FGF21, and GCKR, have been identified, providing a genetic foundation and potential therapeutic targets for the treatment of obesity and its complications. Despite ongoing challenges such as instrument selection and pleiotropy, MR remains a vital tool for advancing precision medicine and informing public health interventions. Future integration with multi-omics and interdisciplinary approaches will further deepen our understanding of obesity mechanisms and support more effective intervention strategies.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research is partially supported by National Natural Science Foundation of China, 81904290.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Pan XF, Wang L, Pan A. Epidemiology and determinants of obesity in China. Lancet Diabetes Endocrinol. 2021;9(6):373–392. doi:10.1016/S2213-8587(21)00045-0
2. World Health Organization (WHO). Obesity: preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ Tech Rep Ser. 2000;894:i–xii,1–253.
3. Després JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444(7121):881–887. doi:10.1038/nature05488
4. Boutari C, Mantzoros CS. A 2022 update on the epidemiology of obesity and a call to action: as its twin COVID-19 pandemic appears to be receding, the obesity and dysmetabolism pandemic continues to rage on. Metab Clin Exp. 2022;133. doi:10.1016/j.metabol.2022.155217
5. Blüher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol. 2019;15(5):288–298. doi:10.1038/s41574-019-0176-8
6. Katan MB. Apolipoprotein E isoforms, serum cholesterol, and cancer. Lancet. 1986;1(8479):507–508. doi:10.1016/s0140-6736(86)92972-7
7. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–R98. doi:10.1093/hmg/ddu328
8. Davey Smith G, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease?*. Int J Epidemiol. 2003;32(1):1–22. doi:10.1093/ije/dyg070
9. Davies NM, Holmes MV, Smith GD. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. doi:10.1136/bmj.k601
10. Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. JAMA. 2017;318(19):1925–1926. doi:10.1001/jama.2017.17219
11. Luo Q, Zhang S, Yang Q, Deng Y, Yi H, Li X. Causal factors for osteoarthritis risk revealed by mendelian randomization analysis. Aging Clin Exp Res. 2024;36(1):176. doi:10.1007/s40520-024-02812-9
12. Haycock PC, Burgess S, Wade KH, Bowden J, Relton C, Davey smith G. Best (but oft-forgotten) practices: the design, analysis, and interpretation of Mendelian randomization studies. Am J Clin Nutr. 2016;103(4):965–978. doi:10.3945/ajcn.115.118216
13. Cornish AJ, Tomlinson IPM, Houlston RS. Mendelian randomisation: a powerful and inexpensive method for identifying and excluding non-genetic risk factors for colorectal cancer. Mol Aspects Med. 2019;69:41–47. doi:10.1016/j.mam.2019.01.002
14. Hartwig FP, Davies NM, Hemani G, Davey Smith G. Two-sample Mendelian randomization: avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int J Epidemiol. 2016;45(6):1717–1726. doi:10.1093/ije/dyx028
15. Nguyen K, Mitchell BD. A guide to understanding Mendelian randomization studies. Arthritis Care Res. 2024. doi:10.1002/acr.25400
16. Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;181(4):251–260. doi:10.1093/aje/kwu283
17. Tian H, Burgess S. Estimation of time-varying causal effects with multivariable Mendelian randomization: some cautionary notes. Int J Epidemiol. 2023;52(3):846–857. doi:10.1093/ije/dyac240
18. Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. 2015;30(7):543–552. doi:10.1007/s10654-015-0011-z
19. Greenland S. An introduction to instrumental variables for epidemiologists. Int J Epidemiol. 2018;47(1):358. doi:10.1093/ije/dyx275
20. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi:10.1093/ije/dyv080
21. Thakkinstian A, Chailurkit L, Warodomwichit D, et al. Causal relationship between body mass index and fetuin-A level in the asian population: a bidirectional Mendelian randomization study. Clin Endocrinol. 2014;81(2):197–203. doi:10.1111/cen.12303
22. Bergholdt HKM, Nordestgaard BG, Ellervik C. Milk intake is not associated with low risk of diabetes or overweight-obesity: a Mendelian randomization study in 97,811 Danish individuals. Am J Clin Nutr. 2015;102(2):487–496. doi:10.3945/ajcn.114.105049
23. Hartwig FP, Horta BL, Smith GD, de Mola CL, Victora CG. Association of lactase persistence genotype with milk consumption, obesity and blood pressure: a Mendelian randomization study in the 1982 Pelotas (Brazil) Birth Cohort, with a systematic review and meta-analysis. Int J Epidemiol. 2016;45(5):1573–1587. doi:10.1093/ije/dyw074
24. Mendelson MM, Marioni RE, Joehanes R, et al. Association of body mass index with DNA methylation and gene expression in blood cells and relations to cardiometabolic disease: a Mendelian randomization approach. PLoS Med. 2017;14(1):e1002215. doi:10.1371/journal.pmed.1002215
25. Böckerman P, Viinikainen J, Pulkki-Råback L, et al. Does higher education protect against obesity? Evidence using Mendelian randomization. Prev Med. 2017;101:195–198. doi:10.1016/j.ypmed.2017.06.015
26. Huang T, Geng T. Maternal central obesity and birth size–A Mendelian randomization analysis. Diabetes. 2018;67. doi:10.2337/db18-1430-P
27. Mulugeta A, Zhou A, Vimaleswaran KS, Dickson C, Hyppönen E. Depression increases the genetic susceptibility to high body mass index: evidence from UK Biobank. Depress Anxiety. 2019;36(12):1154–1162. doi:10.1002/da.22963
28. Fu L, nan LY, Luo D, Deng S, Hu YQ. Plausible relationship between homocysteine and obesity risk via MTHFR gene: a meta-analysis of 38,317 individuals implementing Mendelian randomization. Diabetes Metab Syndr Obes. 2019;12:1201–1212. doi:10.2147/DMSO.S205379
29. Freuer D, Meisinger C, Linseisen J. Causal relationship between dietary macronutrient composition and anthropometric measures: a bidirectional two-sample Mendelian randomization analysis. Clin Nutr. 2021;40(6):4120–4131. doi:10.1016/j.clnu.2021.01.047
30. Park S, Liu M. A positive causal relationship between noodle intake and metabolic syndrome: a two-sample Mendelian randomization study. Nutrients. 2023;15(9):2091. doi:10.3390/nu15092091
31. Zaghlool SB, Sharma S, Molnar M, et al. Revealing the role of the human blood plasma proteome in obesity using genetic drivers. Nat Commun. 2021;12(1):1279. doi:10.1038/s41467-021-21542-4
32. Yao P, Iona A, Kartsonaki C, et al. Conventional and genetic associations of adiposity with 1463 proteins in relatively lean Chinese adults. Eur J Epidemiol. 2023;38(10):1089–1103. doi:10.1007/s10654-023-01038-9
33. Xu Q, Zhang SS, Wang RR, et al. Mendelian Randomization analysis reveals causal effects of the human gut microbiota on abdominal obesity. J Nutr. 2021;151(6):1401–1406. doi:10.1093/jn/nxab025
34. Li Y, Wang X, Zhang Z, Shi L, Cheng L, Zhang X. Effect of the gut microbiome, plasma metabolome, peripheral cells, and inflammatory cytokines on obesity: a bidirectional two-sample Mendelian randomization study and mediation analysis. Front Immunol. 2024;15:1348347. doi:10.3389/fimmu.2024.1348347
35. Li Q, Gao J, Luo J, Lin D, Wu X. Mendelian randomization analyses support causal relationship between gut microbiota and childhood obesity. Front Pediatr. 2023;11:1229236. doi:10.3389/fped.2023.1229236
36. Zagkos L, Dib MJ, Pinto R, et al. Associations of genetically predicted fatty acid levels across the phenome: a mendelian randomisation study. PLoS Med. 2022;19(12):e1004141. doi:10.1371/journal.pmed.1004141
37. Li Y, Liu X, Tu R, Hou J, Zhuang G. Mendelian randomization analysis of the association of SOCS3 methylation with abdominal obesity. Nutrients. 2022;14(18):3824. doi:10.3390/nu14183824
38. Yu Y, Chen Y, Zhang H, et al. Sleep duration and visceral adipose tissue: linear and nonlinear Mendelian randomization analyses. J Clin Endocrinol Metab. 2022;107(11):2992–2999. doi:10.1210/clinem/dgac551
39. Liang YY, Chen J, Peng M, et al. Association between sleep duration and metabolic syndrome: linear and nonlinear Mendelian randomization analyses. J Transl Med. 2023;21(1):90. doi:10.1186/s12967-023-03920-2
40. Hayes BL, Vabistsevits M, Martin RM, Lawlor DA, Richmond RC, Robinson T. Establishing causal relationships between sleep and adiposity traits using Mendelian randomization. Obesity. 2023;31(3):861–870. doi:10.1002/oby.23668
41. Park S, Kim SG, Lee S, et al. Causal effects from tobacco smoking initiation on obesity-related traits: a Mendelian randomization study. Int J Obes Lond. 2023;47(12):1232–1238. doi:10.1038/s41366-023-01371-9
42. Carrasquilla GD, García-Ureña M, Romero-Lado MJ, Kilpeläinen TO. Estimating causality between smoking and abdominal obesity by Mendelian randomization. Addiction. 2024;119(6):1024–1034. doi:10.1111/add.16454
43. Konigorski S, Janke J, Patone G, et al. Identification of novel genes whose expression in adipose tissue affects body fat mass and distribution: an RNA-Seq and Mendelian randomization study. Eur J Hum Genet. 2024;32(9):1127–1135. doi:10.1038/s41431-022-01161-3
44. Wang Z, Yang Q. The causal relationship between human blood metabolites and the risk of visceral obesity: a mendelian randomization analysis. Lipids Health Dis. 2024;23(1):39. doi:10.1186/s12944-024-02035-x
45. Hägg S, Fall T, Ploner A, et al. Adiposity as a cause of cardiovascular disease: a Mendelian randomization study. Int J Epidemiol. 2015;44(2):578–586. doi:10.1093/ije/dyv094
46. Varbo A, Benn M, Smith GD, Timpson NJ, Tybjaerg-Hansen A, Nordestgaard BG. Remnant cholesterol, low-density lipoprotein cholesterol, and blood pressure as mediators from obesity to ischemic heart disease. Circ Res. 2015;116(4):665–673. doi:10.1161/CIRCRESAHA.116.304846
47. Klovaite J, Benn M, Nordestgaard BG. Obesity as a causal risk factor for deep venous thrombosis: a Mendelian randomization study. J Intern Med. 2015;277(5):573–584. doi:10.1111/joim.12299
48. Huang Y, Xu M, Xie L, et al. Obesity and peripheral arterial disease: a Mendelian randomization analysis. Atherosclerosis. 2016;247:218–224. doi:10.1016/j.atherosclerosis.2015.12.034
49. Lindström S, Germain M, Crous-Bou M, et al. Assessing the causal relationship between obesity and venous thromboembolism through a Mendelian randomization study. Hum Genet. 2017;136(7):897–902. doi:10.1007/s00439-017-1811-x
50. Yuan S, Bruzelius M, Xiong Y, Håkansson N, Åkesson A, Larsson SC. Overall and abdominal obesity in relation to venous thromboembolism. J Thromb Haemost. 2021;19(2):460–469. doi:10.1111/jth.15168
51. Adams B, Jacocks L, Guo H. Higher BMI is linked to an increased risk of heart attacks in European adults: a Mendelian randomisation study. BMC Cardiovasc Disord. 2020;20(1):258. doi:10.1186/s12872-020-01542-w
52. Larsson SC, Bäck M, Rees JMB, Mason AM, Burgess S. Body mass index and body composition in relation to 14 cardiovascular conditions in UK Biobank: a Mendelian randomization study. Eur Heart J. 2020;41(2):221–226. doi:10.1093/eurheartj/ehz388
53. Casanova F, Wood AR, Yaghootkar H, et al. Provides evidence that adiposity and dyslipidemia lead to lower Urinary Albumin-to-Creatinine Ratio, a marker of microvascular function. Diabetes. 2020;69(5):1072–1082. doi:10.2337/db19-0862
54. Power GM, Tyrrell J, Frayling TM, Davey Smith G, Richardson TG. Mendelian randomization analyses suggest childhood body size indirectly influences end points from across the cardiovascular disease spectrum through adult body size. J Am Heart Assoc. 2021;10(17):e021503. doi:10.1161/JAHA.121.021503
55. Zou XL, Wang S, Wang LY, et al. Childhood obesity and risk of stroke: a Mendelian randomisation analysis. Front Genet. 2021;12:727475. doi:10.3389/fgene.2021.727475
56. Chen W, Yao D, Yan H, Wang M, Pan Y. Genetically predicted childhood obesity and adult atrial fibrillation: a mendelian randomization study. Nutr Metab Cardiovasc Dis. 2022;32(4):1019–1026. doi:10.1016/j.numecd.2021.12.001
57. Trichia E, Malden DE, Jin D, et al. Independent relevance of adiposity measures to coronary heart disease risk among 0.5 million adults in UK Biobank. Int J Epidemiol. 2023;52(6):1836–1844. doi:10.1093/ije/dyad143
58. Lempesis IG, Varrias D, Sagris M, et al. Obesity and peripheral artery disease: current evidence and controversies. Curr Obes Rep. 2023;12(3):264–279. doi:10.1007/s13679-023-00510-7
59. Daghlas I, Gill D. Mechanisms of hypercoagulability driving stroke risk in obesity: a Mendelian randomization study. Neurology. 2024;103(1):e209431. doi:10.1212/WNL.0000000000209431
60. Benn M, Marott SCW, Tybjærg-Hansen A, Nordestgaard BG. Obesity increases heart failure incidence and mortality: observational and Mendelian randomization studies totalling over 1 million individuals. Cardiovasc Res. 2023;118(18):3576–3585. doi:10.1093/cvr/cvab368
61. Censin JC, Nowak C, Cooper N, Bergsten P, Todd JA, Fall T. Childhood adiposity and risk of type 1 diabetes: a Mendelian randomization study. PLoS Med. 2017;14(8):e1002362. doi:10.1371/journal.pmed.1002362
62. Larsson SC, Burgess S, Michaëlsson K. Genetic association between adiposity and gout: a Mendelian randomization study. Rheumatology. 2018;57(12):2145–2148. doi:10.1093/rheumatology/key229
63. Lee MR, Lim YH, Hong YC. Causal association of body mass index with hypertension using a Mendelian randomization design. Medicine. 2018;97(30):e11252. doi:10.1097/MD.0000000000011252
64. Richardson TG, Mykkänen J, Pahkala K, et al. Evaluating the direct effects of childhood adiposity on adult systemic metabolism: a multivariable Mendelian randomization analysis. Int J Epidemiol. 2021;50(5):1580–1592. doi:10.1093/ije/dyab051
65. Wang T, Zhang R, Ma X, et al. Causal association of overall obesity and abdominal obesity with type 2 diabetes: a Mendelian randomization analysis. Obesity. 2018;26(5):934–942. doi:10.1002/oby.22167
66. Yuan S, Merino J, Larsson SC. Causal factors underlying diabetes risk informed by Mendelian randomisation analysis: evidence, opportunities and challenges. Diabetologia. 2023;66(5):800–812. doi:10.1007/s00125-023-05879-7
67. Hu B, He X, Li F, Sun Y, Sun J, Feng L. Childhood obesity and hypertension in pregnancy: a two-sample Mendelian randomization analysis. J Hypertens. 2023;41(7):1152–1158. doi:10.1097/HJH.0000000000003442
68. Brennan P, McKay J, Moore L, et al. Obesity and cancer: Mendelian randomization approach utilizing the FTO genotype. Int J Epidemiol. 2009;38(4):971–975. doi:10.1093/ije/dyp162
69. Thrift AP, Gong J, Peters U, et al. Mendelian randomization study of body mass index and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2015;24(7):1024–1031. doi:10.1158/1055-9965.EPI-14-1309
70. Thrift AP, Shaheen NJ, Gammon MD, et al. Obesity and risk of esophageal adenocarcinoma and Barrett’s esophagus: a Mendelian randomization study. J Natl Cancer Inst. 2014;106(11):dju252. doi:10.1093/jnci/dju252
71. Bull CJ, Bell JA, Murphy N, et al. Adiposity, metabolites, and colorectal cancer risk: Mendelian randomization study. BMC Med. 2020;18(1):396. doi:10.1186/s12916-020-01855-9
72. Dixon SC, Nagle CM, Thrift AP, et al. Adult body mass index and risk of ovarian cancer by subtype: a Mendelian randomization study. Int J Epidemiol. 2016;45(3):884–895. doi:10.1093/ije/dyw158
73. Ahmed M, Mulugeta A, Lee SH, Mäkinen VP, Boyle T, Hyppönen E. Adiposity and cancer: a Mendelian randomization analysis in the UK biobank. Int J Obes Lond. 2021;45(12):2657–2665. doi:10.1038/s41366-021-00942-y
74. Vithayathil M, Carter P, Kar S, Mason AM, Burgess S, Larsson SC. Body size and composition and risk of site-specific cancers in the UK Biobank and large international consortia: a mendelian randomisation study. PLoS Med. 2021;18(7):e1003706. doi:10.1371/journal.pmed.1003706
75. Xing A, Tong HHY, Liu S, Zhai X, Yu L, Li K. The causal association between obesity and gastric cancer and shared molecular signatures: a large-scale Mendelian randomization and multi-omics analysis. Front Oncol. 2023;13:1091958. doi:10.3389/fonc.2023.1091958
76. Xu FQ, Xu QY, Zhu ZJ, et al. Visceral and ectopic fat are more predictively associated with primary liver cancer than overall obesity from genetic sights: a Mendelian randomization study. Int J Cancer. 2024;154(3):530–537. doi:10.1002/ijc.34751
77. Wu L, Liao F, Guo X, Li N. The causal effect of adipose tissue on Hodgkin’s lymphoma: two-sample Mendelian randomization study and validation. Front Immunol. 2024;15:1400756. doi:10.3389/fimmu.2024.1400756
78. Amin HA, Kaewsri P, Yiorkas AM, Cooke H, Blakemore AI, Drenos F. Mendelian randomisation analyses of UK Biobank and published data suggest that increased adiposity lowers risk of breast and prostate cancer. Sci Rep. 2022;12(1):909. doi:10.1038/s41598-021-04401-6
79. Yao S. Causality inference of obesity and cancer risk by Mendelian randomization analysis: are we there yet? J Natl Cancer Inst. 2022;114(3):331–332. doi:10.1093/jnci/djab103
80. Debette S, Wolf C, Lambert JC, et al. Abdominal obesity and lower gray matter volume: a Mendelian randomization study. Neurobiol Aging. 2014;35(2):378–386. doi:10.1016/j.neurobiolaging.2013.07.022
81. Speed MS, Jefsen OH, Børglum AD, Speed D, Østergaard SD. Investigating the association between body fat and depression via Mendelian randomization. Transl Psychiatry. 2019;9(1):184. doi:10.1038/s41398-019-0516-4
82. Mulugeta A, Lumsden A, Hyppönen E. Unlocking the causal link of metabolically different adiposity subtypes with brain volumes and the risks of dementia and stroke: a Mendelian randomization study. Neurobiol Aging. 2021;102:161–169. doi:10.1016/j.neurobiolaging.2021.02.010
83. Zhuang QS, Meng L, Wang Z, Shen L, Ji HF. Associations between obesity and Alzheimer’s disease: multiple bioinformatic analyses. J Alzheimers Dis. 2021;80(1):271–281. doi:10.3233/JAD-201235
84. Ding H, Ouyang M, Wang J, et al. Shared genetics between classes of obesity and psychiatric disorders: a large-scale genome-wide cross-trait analysis. J Psychosomatic Res. 2022;162:111032. doi:10.1016/j.jpsychores.2022.111032
85. Hughes AM, Sanderson E, Morris T, et al. Body mass index and childhood symptoms of depression, anxiety, and attention-deficit hyperactivity disorder: a within-family Mendelian randomization study. Elife. 2022:
86. Jokela M, Laakasuo M. Obesity as a causal risk factor for depression: systematic review and meta-analysis of Mendelian randomization studies and implications for population mental health. J Psychiatr Res. 2023;163:86–92. doi:10.1016/j.jpsychires.2023.05.034
87. Chen W, Feng J, Jiang S, et al. Mendelian randomization analyses identify bidirectional causal relationships of obesity with psychiatric disorders. J Affect Disord. 2023;339:807–814. doi:10.1016/j.jad.2023.07.044
88. Xu S, Gilliland FD, Conti DV. Elucidation of causal direction between asthma and obesity: a bi-directional Mendelian randomization study. Int J Epidemiol. 2019;48(3):899–907. doi:10.1093/ije/dyz070
89. Sun YQ, Brumpton BM, Langhammer A, Chen Y, Kvaløy K, Mai XM. Adiposity and asthma in adults: a bidirectional Mendelian randomisation analysis of The HUNT Study. Thorax. 2020;75(3):202–208. doi:10.1136/thoraxjnl-2019-213678
90. Zhu Z, Guo Y, Shi H, et al. Shared genetic and experimental links between obesity-related traits and asthma subtypes in UK Biobank. J Allergy Clin Immunol. 2020;145(2):537–549. doi:10.1016/j.jaci.2019.09.035
91. Yin P, Tao S, Xing Z, et al. Association between visceral adipose tissue and asthma based on the NHANES and Mendelian randomization study. Postgrad Med J. 2024;100(1187):642–648. doi:10.1093/postmj/qgae031
92. Probst-Hensch N, Jeong A, Stolz D, et al. Causal effects of body mass index on airflow obstruction and forced mid-expiratory flow: a Mendelian randomization study taking interactions and age-specific instruments into consideration toward a life course perspective. Front Public Health. 2021;9:584955. doi:10.3389/fpubh.2021.584955
93. Han X, Zhu Z, Xiao Q, et al. Obesity-related biomarkers underlie a shared genetic architecture between childhood body mass index and childhood asthma. Commun Biol. 2022;5(1):1098. doi:10.1038/s42003-022-04070-9
94. Yuan S, Larsson SC. Assessing causal associations of obesity and diabetes with kidney stones using Mendelian randomization analysis. Mol Genet Metab. 2021;134(1–2):212–215. doi:10.1016/j.ymgme.2021.08.010
95. Li D, Zou Y. Causal effects of life course adiposity on chronic kidney disease: a Mendelian randomization study. Ann Palliat Med. 2021;10(10):10861–10869. doi:10.21037/apm-21-2528
96. Nguyen A, Khafagy R, Gao Y, et al. Association between obesity and chronic kidney disease: multivariable Mendelian randomization analysis and observational data from a Bariatric surgery cohort. Diabetes. 2023;72(4):496–510. doi:10.2337/db22-0696
97. Lu J, Liu X, Jiang S, et al. Body mass index and risk of diabetic nephropathy: a Mendelian randomization study. J Clin Endocrinol Metab. 2022;107(6):1599–1608. doi:10.1210/clinem/dgac057
98. Zhang H, Zhang Q, Song Y, et al. Separating the effects of life course adiposity on diabetic nephropathy: a comprehensive multivariable Mendelian randomization study. Front Endocrinol. 2024;15:1285872. doi:10.3389/fendo.2024.1285872
99. Brower MA, Hai Y, Jones MR, et al. Bidirectional Mendelian randomization to explore the causal relationships between body mass index and polycystic ovary syndrome. Hum Reprod. 2019;34(1):127–136. doi:10.1093/humrep/dey343
100. De Silva K, Demmer RT, Jönsson D, et al. Causality of anthropometric markers associated with polycystic ovarian syndrome: findings of a Mendelian randomization study. PLoS One. 2022;17(6):e0269191. doi:10.1371/journal.pone.0269191
101. Hernáez Á, Rogne T, Skåra KH, et al. Body mass index and subfertility: multivariable regression and Mendelian randomization analyses in the Norwegian mother, father and child cohort study. Hum Reprod. 2021;36(12):3141–3151. doi:10.1093/humrep/deab224
102. Liu Q, Zhu Z, Kraft P, Deng Q, Stener-Victorin E, Jiang X. Genomic correlation, shared loci, and causal relationship between obesity and polycystic ovary syndrome: a large-scale genome-wide cross-trait analysis. BMC Med. 2022;20(1):66. doi:10.1186/s12916-022-02238-y
103. Venkatesh SS, Ferreira T, Benonisdottir S, et al. Obesity and risk of female reproductive conditions: a Mendelian randomisation study. PLoS Med. 2022;19(2):e1003679. doi:10.1371/journal.pmed.1003679
104. Wan B, Ma N, Zhou Z, Lv C. Putative causal inference for the relationship between obesity and sex hormones in males: a bidirectional Mendelian randomization study. PeerJ. 2023;
105. Rao X, Xu Z, Zhang J, et al. The causal relationship between sarcopenic obesity factors and benign prostate hyperplasia. Front Endocrinol. 2023;14:1290639. doi:10.3389/fendo.2023.1290639
106. Panoutsopoulou K, Metrustry S, Doherty SA, et al. The effect of FTO variation on increased osteoarthritis risk is mediated through body mass index: a Mendelian randomisation study. Ann Rheum Dis. 2014;73(12):2082–2086. doi:10.1136/annrheumdis-2013-203772
107. Zhou J, Mi J, Peng Y, Han H, Liu Z. Causal associations of obesity with the intervertebral degeneration, low back pain, and sciatica: a two-sample Mendelian randomization study. Front Endocrinol. 2021;12:740200. doi:10.3389/fendo.2021.740200
108. Cao Z, Wu Y, Li Q, Li Y, Wu J. A causal relationship between childhood obesity and risk of osteoarthritis: results from a two-sample Mendelian randomization analysis. Ann Med. 2022;54(1):1636–1645. doi:10.1080/07853890.2022.2085883
109. Bland VL, Bea JW, Going SB, et al. Metabolically favorable adiposity and bone mineral density: a Mendelian randomization analysis. Obesity. 2023;31(1):267–278. doi:10.1002/oby.23604
110. Ying D, Ying M. Causal link between childhood obesity and adult osteoporosis. Medicine. 2024;103(36):e39548.
111. Chen X, Cheng Z, Xu J, Wang Q, Zhao Z, Jiang Q. Causal effects of life course adiposity on temporomandibular disorders: a Mendelian randomization study. J Oral Rehabil. 2024;51(2):278–286. doi:10.1111/joor.13607
112. Gianfrancesco MA, Glymour MM, Walter S, et al. Causal effect of genetic variants associated with body mass index on multiple sclerosis susceptibility. Am J Epidemiol. 2017;185(3):162–171. doi:10.1093/aje/kww120
113. Harroud A, Manousaki D, Butler-Laporte G, et al. The relative contributions of obesity, vitamin D, leptin, and adiponectin to multiple sclerosis risk: a Mendelian randomization mediation analysis. Mult Scler J. 2021;27(13):1994–2000. doi:10.1177/1352458521995484
114. Budu-Aggrey A, Brumpton B, Tyrrell J, et al. Evidence of a causal relationship between body mass index and psoriasis: a mendelian randomization study. PLoS Med. 2019;16(1):e1002739. doi:10.1371/journal.pmed.1002739
115. Yew YW, Loh M, Thng STG, Chambers JC. Investigating causal relationships between Body Mass Index and risk of atopic dermatitis: a Mendelian randomization analysis. Sci Rep. 2020;10(1):15279. doi:10.1038/s41598-020-72301-2
116. Huang C, Luo P, Zhu X, et al. Causal effect of obesity and adiposity distribution on the risk of pressure ulcers and potential mediation by type 2 diabetes mellitus: insights from multivariable mendelian randomization and mediation analysis. Arch Dermatol Res. 2024;316(8):550. doi:10.1007/s00403-024-03299-0
117. Matsumura S, Miyakita M, Miyamori H, et al. Stimulation of Gs signaling in MC4R cells by DREADD increases energy expenditure, suppressing food intake, and increasing locomotor activity in mice. Am J Physiol Endocrinol Metab. 2022. doi:10.1152/ajpendo.00439.2021
118. Wauman J, Tavernier J. Leptin receptor signaling: pathways to leptin resistance. Front Biosci. 2011;16:2771–2793. doi:10.2741/3885
119. Lizunov A, Sekste E, Lebedev AA, et al. Involvement of BDNF, NTRK2 and PI3K in the mechanisms of binge eating after psychogenic stressors in ontogenesis. Rev Clin Pharmacol Drug Ther. 2024. doi:10.17816/rcf625676
120. Furber EC, Hyatt K, Collins K, et al. GIPR agonism enhances TZD-induced insulin sensitivity in obese IR mice. Diabetes. 2023;73:292–305. doi:10.2337/db23-0172
121. Szczepańska E, Gietka-Czernel M. FGF21: a novel regulator of glucose and lipid metabolism and whole-body energy balance. Hormone Metab Res. 2022;54(4):203–211. doi:10.1055/a-1778-4159
122. Aljohani A, Khan MI, Bonneville A, et al. Hepatic stearoyl CoA desaturase 1 deficiency increases glucose uptake in adipose tissue partially through the PGC-1α–FGF21 axis in mice. J Biol Chem. 2019;294:19475–19485. doi:10.1074/jbc.RA119.009868
123. Yang Z, Yu G, Zhu X, Peng T, Lv Y. Critical roles of FTO-mediated mRNA m6A demethylation in regulating adipogenesis and lipid metabolism: implications in lipid metabolic disorders. Genes Dis. 2021;9:51–61. doi:10.1016/j.gendis.2021.01.005
124. Aksoy MO, Bilińska A, Stachowiak M, Flisikowska T, Szczerbal I. Deciphering the role of the SREBF1 gene in the transcriptional regulation of porcine adipogenesis using CRISPR/Cas9 editing. Int J Mol Sci. 2024;25. doi:10.3390/ijms252312677
125. Hanson RL, Safabakhsh S, Curtis JM, et al. Association of CREBRF variants with obesity and diabetes in Pacific Islanders from Guam and Saipan. Diabetologia. 2019;62(9):1647–1652. doi:10.1007/s00125-019-4932-z
126. Abdullah MY, AlQwaidi SD, Alshehri AM, et al. Obesity-induced inflammation and its role in the development of insulin resistance. J Healthcare Sci. 2024. doi:10.52533/johs.2024.41002
127. Cacciapaglia F, Perniola S, Stano S, et al. Modulation of IL-6 receptor/STAT3 downstream signaling in rheumatoid arthritis patients. Exp Mol Pathol. 2025;141:104951. doi:10.1016/j.yexmp.2024.104951
128. Hasan A, Akhter N, Al-Roub A, et al. TNF-α in combination with palmitate enhances IL-8 production via the MyD88- independent TLR4 signaling pathway: potential relevance to metabolic inflammation. Int J Mol Sci. 2019. doi:10.3390/ijms20174112
129. Wabitsch M, Farooqi S, Flück CE, et al. Natural history of obesity due to POMC, PCSK1, and LEPR deficiency and the impact of setmelanotide. J Endocr Soc. 2022;6(6):bvac057. doi:10.1210/jendso/bvac057
130. Campbell JE, Müller T, Finan B, DiMarchi R, Tschöp M, D’Alessio D. GIPR/GLP-1R dual agonist therapies for diabetes and weight loss-chemistry, physiology, and clinical applications. Cell Metab. 2023. doi:10.1016/j.cmet.2023.07.010
131. Puengel T, Tacke F. Efruxifermin, an investigational treatment for fibrotic or cirrhotic nonalcoholic steatohepatitis (NASH). Expert Opin Invest Drugs. 2023;32:451–461. doi:10.1080/13543784.2023.2230115
132. Attia SL, Softic S, Mouzaki M. Evolving role for pharmacotherapy in NAFLD/NASH. Clin Transl Sci. 2021;14(1):11–19. doi:10.1111/cts.12839
133. Dai W, Qiao X, Fang Y, et al. Epigenetics-targeted drugs: current paradigms and future challenges. Signal Transduct Target Ther. 2024;9(1):1–71. doi:10.1038/s41392-024-02039-0
134. Shao W, Machamer C, Espenshade P. Fatostatin blocks ER exit of SCAP but inhibits cell growth in a SCAP-independent manner. J Lipid Res. 2016;57:1564–1573. doi:10.1194/jlr.M069583
135. Obradović M, Sudar-Milovanovic E, Soskić S, et al. Leptin and obesity: role and clinical implication. Front Endocrinol. 2021:12. doi:10.3389/fendo.2021.585887
136. Pawar G, Parayath N, Sharma A, et al. Endonasal CNS delivery system for blood-brain barrier impermeant therapeutic oligonucleotides using heterotopic mucosal engrafting. Front Pharmacol. 2021:12. doi:10.3389/fphar.2021.660841
137. Kanshana JS, Mattila PE, Ewing M, et al. A murine model of the human CREBRFR457Q obesity-risk variant does not influence energy or glucose homeostasis in response to nutritional stress. PLoS One. 2021;16. doi:10.1371/journal.pone.0251895
138. Zhao Q, Chen Y, Wang J, Small DS. Powerful three-sample genome-wide design and robust statistical inference in summary-data Mendelian randomization. Int J Epidemiol. 2019. doi:10.1093/ije/dyz142
139. Cheng Q, Yang Y, Shi X, et al. MR-LDP: a two-sample Mendelian randomization for GWAS summary statistics accounting for linkage disequilibrium and horizontal pleiotropy. NAR Genom Bioinform. 2020. doi:10.1093/nargab/lqaa028
140. Hu X, Zhao J, Lin Z, et al. Mendelian randomization for causal inference accounting for pleiotropy and sample structure using genome-wide summary statistics. In:
141. Sanderson E, Richardson T, Hemani G, Smith GD. The use of negative control outcomes in Mendelian randomization to detect potential population stratification. Int J Epidemiol. 2021;50:1350–1361. doi:10.1093/ije/dyaa288
142. Lapierre N, Fu B, Turnbull S, Eskin E, Sankararaman S. Leveraging family data to design Mendelian randomization that is provably robust to population stratification. Genome Res. 2023;33:1032–1041. doi:10.1101/gr.277664.123
143. Mbutiwi F, Dessy T, Sylvestre M. Mendelian randomization: a review of methods for the prevention, assessment, and discussion of pleiotropy in studies using the fat mass and obesity-associated gene as an instrument for adiposity. Front Genetics. 2022;13. doi:10.3389/fgene.2022.803238
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Relationship Between the Ovarian Cyst and Depression: A Two-Sample Mendelian Randomization Study
Wen J, Zhou W, Lin Y
International Journal of Women's Health 2023, 15:1727-1732
Published Date: 10 November 2023